
Abstract
Reduced N-methyl-D-aspartate-receptor (NMDAR) signaling has been associated with schizophrenia, autism and intellectual disability. NMDAR-hypofunction is thought to contribute to social, cognitive and gamma (30–80 Hz) oscillatory abnormalities, phenotypes common to these disorders. However, circuit-level mechanisms underlying such deficits remain unclear. This study investigated the relationship between gamma synchrony, excitatory–inhibitory (E/I) signaling, and behavioral phenotypes in NMDA-NR1neo−/− mice, which have constitutively reduced expression of the obligate NR1 subunit to model disrupted developmental NMDAR function. Constitutive NMDAR-hypofunction caused a loss of E/I balance, with an increase in intrinsic pyramidal cell excitability and a selective disruption of parvalbumin-expressing interneurons. Disrupted E/I coupling was associated with deficits in auditory-evoked gamma signal-to-noise ratio (SNR). Gamma-band abnormalities predicted deficits in spatial working memory and social preference, linking cellular changes in E/I signaling to target behaviors. The GABAB-receptor agonist baclofen improved E/I balance, gamma-SNR and broadly reversed behavioral deficits. These data demonstrate a clinically relevant, highly translatable neural-activity-based biomarker for preclinical screening and therapeutic development across a broad range of disorders that share common endophenotypes and disrupted NMDA-receptor signaling.
Similar content being viewed by others


Introduction
Disruptions in the NMDA-receptor signaling pathway have been associated with several neurodevelopmental, neuropsychiatric diseases. Polymorphisms in the obligatory NR1 subunit of the NMDA receptor (GRIN1) have been associated with schizophrenia, including in a recent large study from the Consortium on the Genetics of Schizophrenia (COGS).1, 2, 3, 4 Seven proteins (GRIN2A, GRIN2B, GRIN3A, GRM1, GRM5, DRD1, DLG4) that directly interact with NR1 were linked to features of schizophrenia in the COGS study,4 including both NR2A and NR2B subunits of the NMDA-receptor, which have been previously associated with the disorder.5, 6 NR2A, NR2B and NR2C have also been associated with autism spectrum disorders (ASD), and differential splicing of NR1 was reported in a recent postmortem study of ASD.7, 8, 9, 10 Similarly, pathway analysis of gene associations in autism has implicated reduced NMDA-receptor-mediated neurotransmission.10 Recent work has also shown that reduced NMDA-receptor signaling is a key pathophysiological deficit in several transgenic mouse models of autism (SHANK3, NLGN1, NRXN1, FMRP1, MeCP2, DISC1, RELN), as well as rodents exposed in utero to immune challenge, a known autism risk factor.11, 12, 13, 14, 15, 16, 17, 18 Finally, deleterious mutations in NR1, NR2A and NR2B have been identified in large studies of subjects with intellectual disability (ID).19, 20 Taken together, these results indicate that disrupted NMDA-receptor signaling may be a molecular substrate common to a number of neurodevelopmental, neuropsychiatric disorders.
Schizophrenia, autism and ID share several treatment-resistant symptoms, including social and cognitive impairments, which may reflect common neural circuit insults. Neurophysiological studies of these clinical populations have demonstrated abnormal gamma frequency (30–80 Hz) neural synchrony,21, 22, 23, 24, 25, 26, 27 which is critical for cognitive and sensory functions including working memory and perceptual feature binding.28, 29, 30 Deficits in gamma synchrony have been linked to treatment-refractory social and cognitive symptoms in patients with schizophrenia25, 31, 32, 33 and ID,27 a relationship that has also been proposed in autism.34 Appropriate gamma-range synchronization is known to require precise excitatory–inhibitory (E/I) balance,35 deficits in which are thought to contribute to core symptoms in both schizophrenia36, 37 and autism.38 In addition, proper levels of GABAergic signaling, in particular that of parvalbumin-expressing (PV+) fast-spiking interneurons (FSI), are critical for gamma-band synchrony and cortical information processing.35, 39 As such, abnormalities in gamma synchrony may reflect shared circuit abnormalities across disorders and may provide a novel biomarker for preclinical therapeutic development to target treatment-resistant symptoms.40, 41
This study investigated the relationship between abnormalities in E/I signaling, gamma synchrony and behavioral function in NR1neo−/− mice, which were engineered to have constitutive, 85% downregulation of the obligate NR1 subunit of the NMDA receptor.42 Whereas previous studies have linked acute NMDAR blockade to several of these deficits, no studies have investigated the relationship between these phenotypes, gamma synchrony and E/I balance in transgenic mice that appropriately reflect the continuous NMDA-receptor dysfunction likely associated with schizophrenia, autism and ID. Likewise, to our knowledge, no studies have demonstrated the restoration of behavioral function and neural synchrony by pharmacologically normalizing E/I balance in such transgenic mouse models. As such, we present an activity-based approach to preclinical therapeutic screening for schizophrenia and related disorders. This approach provided a novel pharmacologic target for reversal of the treatment-resistant social and cognitive deficits seen in disorders associated with NMDAR-hypofunction.
Materials and methods
Animals
NR1 transgenic mice were bred and genotyped in house, as previously published.42, 43 Only adult male homozygous mice and wild-type (WT) littermates were used in this study. Animals were housed 4–5 per cage on a 12-h light/dark cycle in a temperature-controlled facility with food and water available ad libitum. After electrode implantation surgery, mice were housed individually. All protocols were approved by the University of Pennsylvania Institutional Animal Care and Use Committees.
Parvalbumin cell counts
PV immunocytochemistry was performed on frozen tissue sections, using a parvalbumin monoclonal antibody (MAB1572, Chemicon, Billerica, MA, USA, 1:1000) and an HRP-conjugated secondary antibody. Stereological cell counts were made in (1) prefrontal cortex and (2) a broad region of sensorimotor neocortex (layers I-VI from −0.82 to 1.54 mm relative to bregma AP). PV cell density was assessed by in situ hybridization using the same region of interest (ROI) as above. For details, see Supplemental Information.
Molecular biology
Western blotting was performed according to standard lab protocols, using 50 μg of left-hemisphere brain homogenate. The membrane was probed with anti-NMDAR1 (1:500, sc1468, Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-Parvalbumin (1:1000, MAB1572, Millipore, Billerica, MA, USA), anti-Calbindin (1:2000, AB1778, Millipore), anti-Calretinin (1:10 000, AB1550, Millipore), anti-GAD65 (1:500, ab75750, Abcam, Cambridge, MA, USA), anti-GAD67 (1:1000, 5305S, Cell Signaling Technology, Danvers, MA, USA) and anti-β-Actin (1:50 000, AB6276, Abcam), and the appropriate secondary antibodies. Given lower protein levels, expression of postsynaptic GABA-receptor subunits (GABRA1, GABRA2, GABBR1, GABBR2) was assessed using quantitative PCR (qPCR). For details, see Supplemental Information.
Whole-cell recordings
Whole-cell current clamp recordings were made in CA3 pyramidal neurons from acute coronal hippocampal slices (300 μm thickness) taken from adult mice (3–4 months). Evoked responses were triggered by constant-current pulses delivered at 0.2 Hz via a bipolar tungsten stimulation electrode positioned within 100 μm of the recorded cell. Baclofen was bath perfused at 10 μM. For details, see Supplemental Information.
In vivo electrophysiology
Adult animals (6.2±0.2 months) were anesthetized with isoflurane and underwent stereotaxic implantation of tripolar electrode assemblies (PlasticsOne, Roanoke, VA, USA) for nonanesthetized recording of auditory event-related potentials, as published.21, 43, 44, 45 Baseline and auditory-evoked electrophysiological signals were recorded following paired-click stimuli using low-impedance macroelectrodes placed in hippocampal CA3 and the ipsilateral frontal sinus. This differential recording configuration captures both early and late components of the auditory-evoked potential, including the acoustic brainstem response, mid-latency P20 (for example, human P50/M50) and N40 (for example, human N100/M100), as well as the late P2 and P3a peaks, as published,46, 47, 48 with strong analogy to human scalp electroechocardiogram (EEG).21, 49 Signal processing was performed using EEGLab in Matlab, as published.21, 50 For details, see Supplemental Information.
Behavior and pharmacology
Behavioral testing for social interactions (7.8±0.3 months), prepulse inhibition (8.2±0.3 months) and locomotor activity (7.5±0.4 months) was performed as published at the specified ages.45 Spatial memory was assessed using a spontaneous alternation T-maze paradigm (9.6±0.2 months), according to established protocols.51, 52 Baclofen, risperidone and L-838,417 were administered via intrapreitoneal (IP) injections 15–30 min before electrophysiological and behavioral testing. Baclofen and risperidone were dissolved in saline, whereas L-838,417 was dissolved in a 10% DMSO solution in saline. For details, see Supplemental Information.
Results
Reduced excitatory–inhibitory balance
To begin dissecting the circuit-level effects of constitutive NMDAR-hypofunction, we investigated cellular and molecular changes in excitatory and inhibitory populations. We first assessed the integrity of pre- and postsynaptic interneuron markers, given the evidence of GABAergic dysfunction in schizophrenia, autism and ID,53 as well as the importance of PV+ FSI for generation of gamma synchrony.24, 35 Cortical cell counts of immunolabeled FSI were reduced by 40–70% in NR1neo−/− mice across prefrontal and sensorimotor cortices (Figure 1a; main effect of group: F1,24=15.18, P=0.0007) and western blotting revealed a 50% reduction in PV expression (Figure 1b, Supplementary Figure S1; group × marker: F4,44=3.8, P<0.01; PV: P<0.001, Bonferroni post-test). However, group differences were not observed following in situ hybridization (Figure 1c; t10=0.45, P=0.7), indicating that the total number of FSI was not reduced. Postsynaptically, α2 subunit-containing GABAA-receptors, known to be enriched in the axon initial segment opposing FSI terminals,24, 54 were significantly upregulated in NR1 transgenic mice (Figure 1d; group: F1,28=8.1, P<0.01; P<0.05, post-test), as observed in schizophrenia.55 In contrast, there were no group differences in expression of GABAB-receptor subunits, expression of markers for non-FSI (calbindin, calretinin) and total interneuron populations (GAD65, GAD67). These findings indicate that NMDAR-hypofunction induces multiple, selective GABAergic deficits.

Constitutive NMDA-receptor hypofunction causes a selective disruption of parvalbumin-expressing (PV+), fast-spiking interneurons (FSI). (a) Immunoreactivity for FSI was significantly reduced in NR1neo−/− mice. (b) Protein expression for markers of GABA interneuron populations was assessed by western blot. Calbindin and calretinin are markers for nonfast-spiking interneurons, whereas GAD65 and GAD67 are expressed in all GABAergic interneurons. PV expression was significantly reduced in NR1neo−/− mice, whereas other proteins were unaffected. (c) No group differences were observed following in situ hybridization for PV mRNA, indicating that FSI were present, but disrupted, in NR1neo−/− mice. (d) Expression of postsynaptic GABAA- and GABAB-receptor subunits was measured by quantitative PCR (qPCR). The GABAA-receptor alpha-2 subunit was significantly upregulated in NR1neo−/− mice as seen in schizophrenia,55 whereas other subunits were unaffected. Figures show mean +/− s.e.m., *P<0.05, **P<0.01, ***P<0.001.
To determine the effect of NMDAR-hypofunction on excitatory signaling, we next examined membrane properties and synaptic events in hippocampal pyramidal cells using patch clamp. Despite genetic reduction in a major excitatory signaling pathway, the major effect of constitutive NMDA-receptor downregulation was an increase in intrinsic excitability, as measured by the slope of the spike frequency (I–F) curve in current clamp (WT: 0.023±0.002 Hz pA−1, NR1neo−/−: 0.085±0.002 Hz pA−1; F1,27=272.4, P<0.0001; see Figure 3). This neuronal hyperexcitability is consistent with reports of increased blood flow in limbic regions of patients with schizophrenia and autism.56, 57 Spontaneous and evoked excitatory postsynaptic currents (EPSCs) did not differ between groups. Together, these results demonstrate that constitutive NMDAR-hypofunction is sufficient to impair E/I balance, favoring excitation.
Reduced gamma signal-to-noise ratio
E/I balance and FSI function are integral for gamma oscillatory activity,35, 58 which is disrupted in several clinical populations with overlapping symptom domains.21, 22, 27 Consistent with circuit hyperexcitability, NR1neo−/− mice showed a broadband increase in spontaneous local field potential (LFP) power (Figures 2a and e; F1,34=87.64, P<0.0001), with groups differing most significantly at gamma frequencies (P<0.001, post-test). Conversely, evoked power was reduced in NR1neo−/− mice in response to auditory stimulation (Figures 2b and f; group × frequency: F2,34=9.18, P<0.001), selectively at gamma frequencies (P<0.001, post-test). These results mirror clinical findings using analogous recording and analysis methods, demonstrating elevated background LFP power (‘noise’) and reduced stimulus-evoked gamma-band activity (‘signal’).22, 27 Indeed, background and stimulus-evoked gamma-band responses were negatively correlated (R=−0.71, P<0.0001), yielding an underlying deficit in gamma signal-to-noise ratio (SNR). We therefore calculated SNR by dividing the gamma-band-evoked response by the background (pre-stimulus) level of gamma power. These data fit with a recent report that abnormalities in spontaneous gamma power serve as a biomarker for altered E/I balance.58

NMDAR-circuit disruption causes gamma-band deficits, which predict behavioral deficits. (a) Baseline, pre-stimulus power is plotted as a function of frequency. (b) Evoked (that is, phase-locked) power is shown as function of time and frequency. (c) Raw and gamma-filtered traces are shown for wild type (WT) (red) and NR1neo−/− (black) mice. (d) Gamma-filtered responses demonstrate reduced stimulus-evoked activity for NR1 mice, consistent with the time-frequency plot. (e) NR1neo−/− mice demonstrated significantly elevated baseline local field potential (LFP) power, most significantly at gamma frequencies. (f) Auditory-evoked power was selectively reduced at gamma frequencies. Baseline gamma power inversely correlated with auditory-evoked synchronous gamma activity, yielding disrupted gamma signal-to-noise in NR1neo−/− mice. (g) Gamma power abnormalities predict deficits in social interactions and spatial memory. Figures show mean±s.e.m. (*P<0.05, **P<0.01, ***P<0.001).
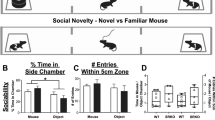
Gamma abnormalities predict social and working memory deficits
Behaviorally, gamma-band abnormalities in schizophrenia have been associated with treatment-resistant cognitive and negative symptoms, including working memory and social deficits, suggesting a common underlying mechanism.25, 32 To determine if gamma abnormalities mice were predictive of similar phenotypes in a preclinical setting, we measured spatial memory using a spontaneous alternation T-maze paradigm and sociability in an approach/avoidance paradigm. As expected,43, 59 NR1neo−/− mice demonstrated reduced social preference (see Figure 4; t18=7.76, P<0.0001) and impaired spatial memory (t22=4.58, P<0.0001). Intriguingly, baseline gamma power was highly correlated with both deficits (Figure 2g; Social: R=−0.72, P=0.002; T-maze: R=−0.61, P<0.025; Bonferroni corrected), suggesting that phenotypic and neural oscillatory abnormalities are related to a common circuit mechanism. Importantly, these results demonstrate that gamma synchrony is a translational biomarker with cross-species predictive utility.
Circuit hyperexcitability reduced by baclofen
Having linked changes in circuit excitability and neural synchrony to target social and cognitive phenotypes, we next investigated whether we could pharmacologically normalize circuit E/I balance. We chose the metabotropic GABAB-receptor agonist baclofen, which is known to increase inhibition via pre- and postsynaptic mechanisms. This would tonically reduce the likelihood of a cell firing, in contrast to the actions of a GABAA-receptor agonist, which would phasically modulate spike timing. Indeed, baclofen reduced the intrinsic hyperexcitability of pyramidal cells in NR1neo−/− mice (Figure 3a; F1,27=144.9, P<0.0001) such that spike-frequency approached levels in WT cells at baseline. Although group differences were not observed for the following measures at baseline, baclofen reduced evoked EPSC amplitudes (F1,28=13.4, P=0.001), spontaneous EPSC amplitudes (F1,28=8.1, P<0.01) and spontaneous EPSC frequency (F1,24=5.6, P<0.03). Therefore, baclofen improved E/I homeostasis via synaptic and intrinsic mechanisms.

Hyperexcitability of pyramidal cells following constitutive NMDAR hypofunction is normalized by baclofen. (a) NR1neo−/− cells demonstrated significant hyperexcitability, measured by the slope of the frequency–current relationship, which was reduced by baclofen (10 μM). Excitability of NR1neo−/− cells treated with baclofen approached levels seen in wild type (WT) cells at baseline. (b) Synaptic events (evoked excitatory postsynaptic currents (eEPSCs)) were evoked by local stimulation within 100 μm of the cell body. Although group differences were not observed, baclofen reduced eEPSC amplitudes. The bottom panel shows average traces of eEPSCs evoked with 100-μs pulses; stimulus artifacts are omitted. (c) Baclofen reduced spontaneous synaptic events (sEPSC), in terms of frequency (left) and amplitude (right). The bottom panel shows segments of raw sEPSC recordings and averaged sEPSC amplitudes. Figures show mean±s.e.m. (*P<0.05,**P<0.01,***P<0.001).
Baclofen reverses gamma and behavioral deficits
Given that baclofen normalizes multiple aspects of circuit E/I balance, we investigated whether baclofen can reverse deficits in gamma SNR and target behavioral phenotypes (Figure 4, Supplementary Figure S2). To assess drug specificity, we also screened the antipsychotic risperidone and L-838,417, a subunit-selective GABAA-receptor α-2,3,5 (α-1 sparing) agonist similar to MK-0777, which was investigated on comparable measures in recent schizophrenia clinical trials.54, 60 L-838,417 represents a new class of benzodiazepines that lack the tolerance, withdrawal and adverse cognitive properties mediated by activity at the GABAA α-1 subunit. Both risperidone (dose × drug: F2,41=4.30, P=0.02) and L-838,417 (drug: F2,74=3.66, P=0.03) worsened gamma-SNR in NR1neo−/− mice. Conversely, baclofen dose-dependently improved gamma-SNR (drug: F2,49=8.39, P=0.0007).

Constitutive NMDAR circuit disruption causes electrophysiological and behavioral deficits that are reversed by the GABAB-receptor agonist baclofen. (a) Baclofen, but not risperidone or the selective GABAA-receptor agonist L-838,417, improved gamma signal-to-noise (SNR) in NR1neo−/− mice. (b) Social and (c) spatial memory deficits in NR1neo−/− mice were selectively reversed by baclofen. Baclofen reverses (d) deficits in prepulse inhibition and (e) elevated auditory startle responses in NR1-transgenic mice. (e) Locomotor hyperactivity is normalized by baclofen. Figures show mean±s.e.m., (*P<0.05, **P<0.01, ***P<0.001).
If gamma synchrony and target behaviors are truly linked via a common mechanism of E/I balance, we hypothesized that baclofen would rescue phenotypic deficits in mice with NMDAR-hypofunction. Indeed, baclofen reversed social deficits in a dose-dependent manner (Figure 4b; drug: F2,52=5.09, P<0.01) and significantly improved T-maze performance in NR1neo−/− mice (Figure 4c; drug: F1,22=6.31, P<0.02). Neither risperidone nor L-838,417 affected social or T-maze performance, consistent with negative clinical trials evaluating similar compounds (Supplementary Figure S2). Baclofen also reversed prepulse inhibition deficits (Figure 4d; drug: F2,28=4.3, P=0.02), elevated acoustic startle responses (Figure 4e; group × drug: F2,28=5.14, P=0.01) and locomotor hyperactivity (Figure 4f; drug: F2,42=9.0, P<0.001) in NR1neo−/− mice, which are all considered behavioral measures relevant to neuropsychiatric diseases that are less selective for treatment-resistant symptoms. Some of these behavioral deficits were also reversed by risperidone and/or L-838,418 (Supplementary Figure S2). These results fit with emerging evidence of reduced GABAB-signaling in schizophrenia, which predicts negative symptoms.61
Discussion
PV interneurons & gamma signal-to-noise ratio
In vivo electrophysiological studies demonstrated a pattern of reduced gamma frequency signal-to-noise (SNR) in mice with dysregulation of NMDA-receptor signaling. In particular, NR1neo−/− mice show a broad-spectrum increase in spontaneous LFP power (‘noise’) that was most significantly elevated at high (gamma) frequencies. In contrast, phase-locked auditory-evoked responses were significantly reduced, but only at gamma frequencies. This pattern is highly consistent with clinical studies, which demonstrate increased baseline activity,27, 62, 63 coupled with reduced sensory-evoked gamma synchrony,21, 23, 25, 27, 33 as we recently reviewed.22 Our data suggest that altered E/I balance, coincident with selective reduction of parvalbumin expression and pyramidal cell hyperexcitability, likely serves as a cellular mechanism underlying observed electrophysiological deficits.
Recent optogenetic work has shown that activation of PV+ interneurons is critical for generation of (stimulus-evoked) gamma synchrony in vivo.35, 39 Cardin et al.39 demonstrated that brief, rhythmic excitation of FSI at various frequencies selectively increases LFP gamma power, an effect that was not seen by driving pyramidal cells (even at gamma frequencies). Interestingly, this effect was blocked by NMDAR-antagonists. Sohal et al.35 investigated gamma synchrony in vivo by optogenetically stimulating pyramidal neurons while concomitantly inhibiting FSI. PV-cell silencing reduced LFP power and phase-locking in the gamma band (but not other frequencies), indicating that FSI activity is necessary to generate stimulus-evoked gamma synchrony. In the absence of pyramidal cell stimulation, however, PV-cell inhibition increased spontaneous LFP power, consistent with our findings of elevated background power in NR1neo−/− mice. Taken together, these findings indicate that precise PV-cell firing is critical for synchronizing stimulus-evoked gamma oscillations. However, in the absence of sensory (or optogenetic) stimulation, PV-cell silencing does not lead to a reduction in gamma synchrony, but rather causes an increase in LFP power. Behaviorally, targeted removal of FSI from hippocampal CA1 was recently shown to impair spatial working memory,64 in accordance with our findings. Recent work has demonstrated that NMDA-receptors on FSI are critical for the generation of gamma synchrony65, 66 (although pyramidal cell-specific perturbations also modulate gamma-band activity58).
Elevated microcircuit noise
These results indicate that appropriate PV cell function is integral for the generation of stimulus-evoked gamma-band synchrony, which is disrupted in NR1neo−/− mice. In addition, these findings indicate that PV cell firing is necessary to reduce background (that is, spontaneous) LFP power, which is elevated following NMDAR-hypofunction. Elevated background LFP power is likely caused by an increase in spontaneous pyramidal cell firing, as indicated by our data demonstrating hyperexcitability of pyramidal cells from NR1neo−/− mice. Elevated spontaneous cell firing reduces the ability of a neural system to synchronize its activity in a temporally precise manner in response to a salient stimulus, such as an auditory tone. As such, enhanced microcircuit ‘noise’ could, itself, be a pathologic disease state contributing to reduced stimulus-evoked synchrony and associated phenotypic deficits,67 as supported by our data demonstrating a significant, negative relationship between pre- and post-stimulus oscillatory activity, as well as the significant relationship between gamma abnormalities and phenotypic deficits. Indeed, a recent study demonstrated that induction of pyramidal cell hyperexcitability (for example, elevated microcircuit noise) caused an increase in baseline gamma power, as well as cognitive and social deficits in wild-type mice in vivo.58 We extend these findings in transgenic mice relevant to multiple neurodevelopmental disorders, demonstrate that such deficits are pharmacologically reversible by baclofen and show that gamma synchrony can be used as a preclinical, translational biomarker for novel therapeutic development.
Baclofen mechanism of action
Based on these results, the efficacy of baclofen may relate to its ability to dampen hyperexcitability via pre- and postsynaptic mechanisms. Baclofen stimulates metabotropic GABAB-receptors, which function as presynaptic autoreceptors to inhibit vesicular release and activate postsynaptic, inward-rectifying potassium channels. Together, these mechanisms serve to tonically hyperpolarize neurons, decrease resting membrane potential and reduce cell firing. This hyperpolarization likely affects gamma SNR in two ways. First, by decreasing intrinsic pyramidal cell excitability, as well as spontaneous and evoked synaptic input into these cells (Figure 3), we would expect that spontaneous firing of these cells (‘noise’) would be reduced, leading to a reduction in spontaneous LFP power. Likewise, hyperpolarization of pyramidal cells by baclofen would be expected to increase stimulus-evoked gamma-band signal, given the significant negative correlation between pre- and post-stimulus gamma power. Given the dysfunction of FSI, we speculate that such a reduction in membrane potential of postsynaptic neurons may seek to augment the downstream effects of phasic GABA release by these interneurons. Instead of attempting to directly mimic the phasic output of FSI (and other interneurons), as GABAA-receptor agonists such as L-838,417 would, baclofen places the system in a state where GABA that is released is amplified due to a lower resting membrane potential. As temporally precise GABA release by FSI onto pyramidal cells is crucial for the emergence of oscillatory activity, this amplification of the output from FSI would be expected to facilitate gamma synchrony. As such, the efficacy of tonic GABAB-agonists but not phasic GABAA-agonists suggests that elevated microcircuit noise is the primary insult following NMDAR-hypofunction. Baclofen broadly reduces the likelihood of a cell firing, in contrast to the actions of a GABAA-receptor agonist that would phasically modulate spike timing.
Baclofen clinical trials
In the 1970s, several small clinical trials and/or case studies were conducted with baclofen in schizophrenia patients.68, 69, 70, 71, 72, 73, 74, 75 Although the general impression was that baclofen was unsuccessful, none of these studies explicitly investigated the efficacy of baclofen on negative symptoms or cognitive measures. Instead, outcomes consisted of subjective clinical impression or measures of psychosis (i.e., positive symptoms). In addition, only three of these studies assessed the effect of baclofen with concomitant, stable neuroleptic treatment.68, 73, 74 Of these three studies, one found no change,73 whereas two reported improvement, especially in ‘symptoms of autism’.68, 74 Many of the remaining studies did not report clinical efficacy, but are confounded by the concurrent withdrawal or lack of antipsychotic treatment. Importantly, all studies reported that baclofen was generally well tolerated in schizophrenia patients. Indeed, our results suggest that baclofen would be most effective in cognitive and negative symptom domains, given that this is the strength of the NR1 model and that gamma synchrony seems most related to these symptoms. Finally, arbaclofen, the R enantiomer of baclofen, is currently in phase 2 and phase 3 clinical trials for social withdrawal in autism and Fragile X Syndrome, respectively (http://clinicaltrials.gov/ct2/show/NCT01288716; http://clinicaltrials.gov/ct2/show/NCT01282268).
Model validity
One potential limitation of the current study centers on the construct validity of NR1neo−/− mice for the clinical populations from a risk gene perspective. Rather than focusing on a single polymorphism that is likely present in a small proportion of patients, this work employed a pathway approach using model that reflects the myriad of mechanisms that can lead to disruptions in NMDAR signaling, likely increasing its translational potential and generalizability relative to single, vulnerability gene approaches. It also seems unlikely that patients with schizophrenia or autism have the same extent of reduced NMDAR activity as that in NR1neo−/− mice. However, a deleterious GRIN1 mutation associated with ID nearly completely abolishes receptor function,19 suggesting that the model has physiological relevance to a continuum of human conditions. The main challenge in forward translation of novel therapeutics from preclinical settings, a major focus of this study, has been the preponderance of false-positive results in animal studies. For example, more subtle pharmacologic NMDAR disruption often employed as a preclinical disease model can be reversed with GABAA-receptor agonists,76, 77, 78 despite the fact that these drugs do not improve, and often worsen, symptoms of schizophrenia.60, 79 In addition, recent studies have investigated selective, interneuron-specific knockout of NMDA receptors as a preclinical disease model, given the GABAergic neuropathology of schizophrenia.51, 65, 66 However, these models have either failed to recapitulate core clinical phenotypes, including social deficits,66 spatial memory deficits,65 and working memory deficits at high memory loads,65 or have demonstrated complete phenotypic reversal of treatment-resistant symptoms (i.e., working memory deficit) with risperidone,51 despite the drug's lack of clinical efficacy on this domain.80, 81 Thus, although these studies have added important mechanistic insights, we employed a model with clear phenotypic deficits that would favor false-negative predictive validity as a conservative approach to advance a treatment target. The observation that baclofen dose-dependently reversed multiple neural deficits in this model provides more confidence for successful forward translation. In addition, neither risperidone nor the subunit-selective GABAA-receptor agonist affected target phenotypes in NR1-transgenic mice, consistent with clinical studies, confirming the appropriate predictive validity of this model and highlighting the specificity of the GABAB findings. We propose that future studies should investigate the effects of baclofen on electrophysiological and behavioral phenotypes in addition to transgenic disease models.
In conclusion, this study demonstrated a relationship between constitutive NMDAR-hypofunction, disrupted excitatory/inhibitory balance, reduced gamma signal-to-noise and the treatment-resistant symptoms of schizophrenia, autism and ID. The GABAB-receptor agonist baclofen, but not risperidone or the subunit-selective GABAA-receptor agonist L-838,417, improved gamma-SNR, social function and spatial memory deficits in NR1neo−/− mice. As such, we provide a novel, clinically translatable paradigm based on gamma-band activity for preclinical and clinical therapeutic screening for neurodevelopmental, neuropsychiatric disorders. Finally, we demonstrate that GABAB-receptors represent an appropriate therapeutic target for restoring circuit and phenotypic abnormalities in diseases characterized by constitutive NMDAR-hypofunction.
References
Begni S, Moraschi S, Bignotti S, Fumagalli F, Rillosi L, Perez J et al. Association between the G1001C polymorphism in the GRIN1 gene promoter region and schizophrenia. Biol Psychiatry 2003; 53: 617–619.
Galehdari H, Pooryasin A, Foroughmand A, Daneshmand S, Saadat M . Association between the G1001C polymorphism in the GRIN1 gene promoter and schizophrenia in the Iranian population. J Mol Neurosci 2009; 38: 178–181.
Zhao X, Li H, Shi Y, Tang R, Chen W, Liu J et al. Significant association between the genetic variations in the 5′ end of the N-methyl-D-aspartate receptor subunit gene GRIN1 and schizophrenia. Biol Psychiatry 2006; 59: 747–753.
Greenwood TA, Lazzeroni LC, Murray SS, Cadenhead KS, Calkins ME, Dobie DJ et al. Analysis of 94 candidate genes and 12 endophenotypes for schizophrenia from the Consortium on the Genetics of Schizophrenia. Am J Psychiatry 2011; 168: 930–946.
Iwayama-Shigeno Y, Yamada K, Itokawa M, Toyota T, Meerabux JM, Minabe Y et al. Extended analyses support the association of a functional (GT)n polymorphism in the GRIN2A promoter with Japanese schizophrenia. Neurosci Lett 2005; 378: 102–105.
Li D, He L . Association study between the NMDA receptor 2B subunit gene (GRIN2B) and schizophrenia: a HuGE review and meta-analysis. Genet Med 2007; 9: 4–8.
Barnby G, Abbott A, Sykes N, Morris A, Weeks DE, Mott R et al. Candidate-gene screening and association analysis at the autism-susceptibility locus on chromosome 16p: evidence of association at GRIN2A and ABAT. Am J Hum Genet 2005; 76: 950–966.
O’Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet 2011; 43: 585–589.
Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011; 474: 380–384.
Gai X, Xie HM, Perin JC, Takahashi N, Murphy K, Wenocur AS et al. Rare structural variation of synapse and neurotransmission genes in autism. Mol Psychiatry 2012; 17: 402–411.
Bangash MA, Park JM, Melnikova T, Wang D, Jeon SK, Lee D et al. Enhanced polyubiquitination of Shank3 and NMDA receptor in a mouse model of autism. Cell 2011; 145: 758–772.
Blundell J, Blaiss CA, Etherton MR, Espinosa F, Tabuchi K, Walz C et al. Neuroligin-1 deletion results in impaired spatial memory and increased repetitive behavior. J Neurosci 2010; 30: 2115–2129.
Yun SH, Trommer BL . Fragile X mice: reduced long-term potentiation and N-Methyl-D-Aspartate receptor-mediated neurotransmission in dentate gyrus. J Neurosci Res 2011; 89: 176–182.
Maliszewska-Cyna E, Bawa D, Eubanks JH . Diminished prevalence but preserved synaptic distribution of N-methyl-D-aspartate receptor subunits in the methyl CpG binding protein 2(MeCP2)-null mouse brain. Neuroscience 2010; 168: 624–632.
Hayashi-Takagi A, Takaki M, Graziane N, Seshadri S, Murdoch H, Dunlop AJ et al. Disrupted-in-Schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat Neurosci 2010; 13: 327–332.
Kim J, Jung SY, Lee YK, Park S, Choi JS, Lee CJ et al. Neuroligin-1 is required for normal expression of LTP and associative fear memory in the amygdala of adult animals. Proc Natl Acad Sci USA 2008; 105: 9087–9092.
Ventruti A, Kazdoba TM, Niu S, D’Arcangelo G . Reelin deficiency causes specific defects in the molecular composition of the synapses in the adult brain. Neuroscience 2011; 189: 32–42.
Escobar M, Crouzin N, Cavalier M, Quentin J, Roussel J, Lante F et al. Early, time-dependent disturbances of hippocampal synaptic transmission and plasticity after in utero immune challenge. Biol Psychiatry 2011; 70: 992–999.
Hamdan FF, Gauthier J, Araki Y, Lin DT, Yoshizawa Y, Higashi K et al. Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am J Hum Genet 2011; 88: 306–316.
Endele S, Rosenberger G, Geider K, Popp B, Tamer C, Stefanova I et al. Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat Genet 2010; 42: 1021–1026.
Gandal MJ, Edgar JC, Ehrlichman RS, Mehta M, Roberts TP, Siegel SJ . Validating gamma oscillations and delayed auditory responses as translational biomarkers of autism. Biol Psychiatry 2010; 68: 1100–1106.
Gandal MJ, Edgar JC, Klook K, Siegel SJ . Gamma synchrony: towards a translational biomarker for the treatment-resistant symptoms of schizophrenia. Neuropharmacology 2012; 62: 1504–1518.
Kwon JS, O’Donnell BF, Wallenstein GV, Greene RW, Hirayasu Y, Nestor PG et al. Gamma frequency-range abnormalities to auditory stimulation in schizophrenia. Arch Gen Psychiatry 1999; 56: 1001–1005.
Lewis DA, Hashimoto T, Volk DW . Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci 2005; 6: 312–324.
Light GA, Hsu JL, Hsieh MH, Meyer-Gomes K, Sprock J, Swerdlow NR et al. Gamma band oscillations reveal neural network cortical coherence dysfunction in schizophrenia patients. Biol Psychiatry 2006; 60: 1231–1240.
Virji-Babul N, Cheung T, Weeks D, Herdman AT, Cheyne D . Magnetoencephalographic analysis of cortical activity in adults with and without Down syndrome. J Intellect Disabil Res 2007; 51 (Pt 12): 982–987.
Politoff AL, Stadter RP, Monson N, Hass P . Cognition-related EEG abnormalities in nondemented Down syndrome subjects. Dementia 1996; 7: 69–75.
Howard MW, Rizzuto DS, Caplan JB, Madsen JR, Lisman J, Aschenbrenner-Scheibe R et al. Gamma oscillations correlate with working memory load in humans. Cereb Cortex 2003; 13: 1369–1374.
Engel AK, Singer W . Temporal binding and the neural correlates of sensory awareness. Trends Cogn Sci 2001; 5: 16–25.
Gray CM, Konig P, Engel AK, Singer W . Oscillatory responses in cat visual cortex exhibit inter-columnar synchronization which reflects global stimulus properties. Nature 1989; 338: 334–337.
Lee KH, Williams LM, Breakspear M, Gordon E . Synchronous gamma activity: a review and contribution to an integrative neuroscience model of schizophrenia. Brain res 2003; 41: 57–78.
Williams LM, Whitford TJ, Nagy M, Flynn G, Harris AW, Silverstein SM et al. Emotion-elicited gamma synchrony in patients with first-episode schizophrenia: a neural correlate of social cognition outcomes. J Psychiatry Neurosci 2009; 34: 303–313.
Kirihara K, Rissling AJ, Swerdlow NR, Braff DL, Light GA . Hierarchical organization of gamma and theta oscillatory dynamics in schizophrenia. Biol Psychiatry 2012; 71: 873–880.
Brock J, Brown CC, Boucher J, Rippon G . The temporal binding deficit hypothesis of autism. Dev Psychopathol 2002; 14: 209–224.
Sohal VS, Zhang F, Yizhar O, Deisseroth K . Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature 2009; 459: 698–702.
Kehrer C, Maziashvili N, Dugladze T, Gloveli T . Altered excitatory-inhibitory balance in the NMDA-hypofunction model of schizophrenia. Front Mol Neurosci 2008; 1: 6.
Sun Y, Farzan F, Barr MS, Kirihara K, Fitzgerald PB, Light GA et al. Gamma oscillations in schizophrenia: mechanisms and clinical significance. Brain Res 2011; 1413: 98–114.
Rubenstein JL, Merzenich MM . Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav 2003; 2: 255–267.
Cardin JA, Carlen M, Meletis K, Knoblich U, Zhang F, Deisseroth K et al. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature 2009; 459: 663–667.
Lewis DA, Gonzalez-Burgos G . Pathophysiologically based treatment interventions in schizophrenia. Nat Med 2006; 12: 1016–1022.
Javitt DC, Spencer KM, Thaker GK, Winterer G, Hajos M . Neurophysiological biomarkers for drug development in schizophrenia. Nat Rev 2008; 7: 68–83.
Mohn AR, Gainetdinov RR, Caron MG, Koller BH . Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell 1999; 98: 427–436.
Halene TB, Ehrlichman RS, Liang Y, Christian EP, Jonak GJ, Gur TL et al. Assessment of NMDA receptor NR1 subunit hypofunction in mice as a model for schizophrenia. Genes Brain Behav 2009; 8: 661–675.
Ehrlichman RS, Gandal MJ, Maxwell CR, Lazarewicz MT, Finkel LH, Contreras D et al. N-methyl-d-aspartic acid receptor antagonist-induced frequency oscillations in mice recreate pattern of electrophysiological deficits in schizophrenia. Neuroscience 2009; 158: 705–712.
Ehrlichman RS, Luminais SN, White SL, Rudnick ND, Ma N, Dow HC et al. Neuregulin 1 transgenic mice display reduced mismatch negativity, contextual fear conditioning and social interactions. Brain Res 2009; 1294: 116–127.
Connolly PM, Maxwell C, Liang Y, Kahn JB, Kanes SJ, Abel T et al. The effects of ketamine vary among inbred mouse strains and mimic schizophrenia for the P80, but not P20 or N40 auditory ERP components. Neurochem Res 2004; 29: 1179–1188.
Connolly PM, Maxwell CR, Kanes SJ, Abel T, Liang Y, Tokarczyk J et al. Inhibition of auditory evoked potentials and prepulse inhibition of startle in DBA/2J and DBA/2Hsd inbred mouse substrains. Brain Res 2003; 992: 85–95.
Siegel SJ, Connolly P, Liang Y, Lenox RH, Gur RE, Bilker WB et al. Effects of strain, novelty, and NMDA blockade on auditory-evoked potentials in mice. Neuropsychopharmacology 2003; 28: 675–682.
Gandal MJ, Ehrlichman RS, Rudnick ND, Siegel SJ . A novel electrophysiological model of chemotherapy-induced cognitive impairments in mice. Neuroscience 2008; 157: 95–104.
Delorme A, Makeig S . EEGLAB: an open source toolbox for analysis of single-trial EEG dynamics including independent component analysis. Neurosci Methods 2004; 134: 9–21.
Belforte JE, Zsiros V, Sklar ER, Jiang Z, Yu G, Li Y et al. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat Neurosci 2010; 13: 76–83.
Deacon RM, Rawlins JN . T-maze alternation in the rodent. Nat Protoc 2006; 1: 7–12.
Levitt P, Eagleson KL, Powell EM . Regulation of neocortical interneuron development and the implications for neurodevelopmental disorders. Trends Neurosci 2004; 27: 400–406.
Lewis DA, Cho RY, Carter CS, Eklund K, Forster S, Kelly MA et al. Subunit-selective modulation of GABA type A receptor neurotransmission and cognition in schizophrenia. Am J Psychiatry 2008; 165: 1585–1593.
Volk DW, Pierri JN, Fritschy JM, Auh S, Sampson AR, Lewis DA . Reciprocal alterations in pre- and postsynaptic inhibitory markers at chandelier cell inputs to pyramidal neurons in schizophrenia. Cereb Cortex 2002; 12: 1063–1070.
Medoff DR, Holcomb HH, Lahti AC, Tamminga CA . Probing the human hippocampus using rCBF: contrasts in schizophrenia. Hippocampus 2001; 11: 543–550.
Pagani M, Manouilenko I, Stone-Elander S, Odh R, Salmaso D, Hatherly R et al. Brief report: alterations in cerebral blood flow as assessed by PET/CT in adults with autism spectrum disorder with normal IQ. J Autism Dev Disord 2012; 42: 313–318.
Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O’Shea DJ et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 2011; 477: 171–178.
Dzirasa K, Ramsey AJ, Takahashi DY, Stapleton J, Potes JM, Williams JK et al. Hyperdopaminergia and NMDA receptor hypofunction disrupt neural phase signaling. J Neurosci 2009; 29: 8215–8224.
Buchanan RW, Keefe RS, Lieberman JA, Barch DM, Csernansky JG, Goff DC et al. A randomized clinical trial of MK-0777 for the treatment of cognitive impairments in people with schizophrenia. Biol Psychiatry 2011; 69: 442–449.
Liu SK, Fitzgerald PB, Daigle M, Chen R, Daskalakis ZJ . The relationship between cortical inhibition, antipsychotic treatment, and the symptoms of schizophrenia. Biol Psychiatry 2009; 65: 503–509.
Itil TM, Saletu B, Davis S . EEG findings in chronic schizophrenics based on digital computer period analysis and analog power spectra. Biol Psychiatry 1972; 5: 1–13.
Winterer G, Coppola R, Goldberg TE, Egan MF, Jones DW, Sanchez CE et al. Prefrontal broadband noise, working memory, and genetic risk for schizophrenia. Am J Psychiatry 2004; 161: 490–500.
Murray AJ, Sauer JF, Riedel G, McClure C, Ansel L, Cheyne L et al. Parvalbumin-positive CA1 interneurons are required for spatial working but not for reference memory. Nat Neurosci 2011; 14: 297–299.
Carlen M, Meletis K, Siegle JH, Cardin JA, Futai K, Vierling-Claassen D et al. A critical role for NMDA receptors in parvalbumin interneurons for gamma rhythm induction and behavior. Mol Psychiatry 2012; 17: 537–548.
Korotkova T, Fuchs EC, Ponomarenko A, von Engelhardt J, Monyer H . NMDA receptor ablation on parvalbumin-positive interneurons impairs hippocampal synchrony, spatial representations, and working memory. Neuron 2010; 68: 557–569.
Winterer G, Ziller M, Dorn H, Frick K, Mulert C, Wuebben Y et al. Schizophrenia: reduced signal-to-noise ratio and impaired phase-locking during information processing. Clin Neurophysiol 2000; 111: 837–849.
Frederiksen PK . Letter: Baclofen in the treatment of schizophrenia. Lancet 1975; 1: 702.
Davis KL, Hollister LE, Berger PA . Letter: Bacloffen in schizophrenia. Lancet 1976; 1: 1245.
Gulmann NC, Bahr B, Andersen B, Eliassen HM . A double-blind trial of baclofen against placebo in the treatment of schizophrenia. Acta Psychiatr Scand 1976; 54: 287–293.
Simpson GM, Branchey MH, Shrivastava RK . Letter: Baclofen in schizophrenia. Lancet 1976; 1: 966–967.
Beckmann H, Frische M, Ruther E, Zimmer R . Baclofen (para-chlorphenyl-GABA) in schizophrenia. Pharmakopsychiatr Neuropsychopharmakol 1977; 10: 26–31.
Bigelow LB, Nasrallah H, Carman J, Gillin JC, Wyatt RJ . Baclofen treatment in chronic schizophrenia: a clinical trial. Am J Psychiatry 1977; 134: 318–320.
Schopf J, Hucker H . Baclofen in the treatment of schizophrenia: a pilot study. Pharmakopsychiatr Neuropsychopharmakol 1977; 10: 89–91.
Simpson GM, Lee JH, Shrivastava RK, Branchey MH . Baclofen in the treatment of tardive dyskinesia and schizophrenia. Psychopharmacol Bull 1978; 14: 16–18.
Behrens MM, Ali SS, Dao DN, Lucero J, Shekhtman G, Quick KL et al. Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science 2007; 318: 1645–1647.
Castner SA, Arriza JL, Roberts JC, Mrzljak L, Christian EP, Williams GV . Reversal of ketamine-induced working memory impairments by the GABAAalpha2/3 agonist TPA023. Biol Psychiatry 2010; 67: 998–1001.
Ma J, Leung LS . The supramammillo-septal-hippocampal pathway mediates sensorimotor gating impairment and hyperlocomotion induced by MK-801 and ketamine in rats. Psychopharmacology (Berl) 2007; 191: 961–974.
Menzies L, Ooi C, Kamath S, Suckling J, McKenna P, Fletcher P et al. Effects of gamma-aminobutyric acid-modulating drugs on working memory and brain function in patients with schizophrenia. Arch Gen Psychiatry 2007; 64: 156–167.
Goldberg TE, Goldman RS, Burdick KE, Malhotra AK, Lencz T, Patel RC et al. Cognitive improvement after treatment with second-generation antipsychotic medications in first-episode schizophrenia: is it a practice effect? Arch Gen Psychiatry 2007; 64: 1115–1122.
Reilly JL, Harris MS, Keshavan MS, Sweeney JA . Adverse effects of risperidone on spatial working memory in first-episode schizophrenia. Arch Gen Psychiatry 2006; 63: 1189–1197.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Translational Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Gandal, M., Sisti, J., Klook, K. et al. GABAB-mediated rescue of altered excitatory–inhibitory balance, gamma synchrony and behavioral deficits following constitutive NMDAR-hypofunction. Transl Psychiatry 2, e142 (2012). https://doi.org/10.1038/tp.2012.69
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2012.69
Keywords
This article is cited by
-
Ketamine in neuropsychiatric disorders: an update
Neuropsychopharmacology (2024)
-
Synergistic, long-term effects of glutamate dehydrogenase 1 deficiency and mild stress on cognitive function and mPFC gene and miRNA expression
Translational Psychiatry (2023)
-
Mouse mutants in schizophrenia risk genes GRIN2A and AKAP11 show EEG abnormalities in common with schizophrenia patients
Translational Psychiatry (2023)
-
Auditory processing in rodent models of autism: a systematic review
Journal of Neurodevelopmental Disorders (2022)
-
Valproic acid-exposed astrocytes impair inhibitory synapse formation and function
Scientific Reports (2021)
