
Abstract
Parkinson’s disease (PD) is a progressive neurodegenerative disorder associated with both underlying genetic factors and neuroimaging findings. Existing neuroimaging studies related to the genome in PD have mostly focused on certain candidate genes. The aim of our study was to construct a linear regression model using both genetic and neuroimaging features to better predict clinical scores compared to conventional approaches. We obtained neuroimaging and DNA genotyping data from a research database. Connectivity analysis was applied to identify neuroimaging features that could differentiate between healthy control (HC) and PD groups. A joint analysis of genetic and imaging information known as imaging genetics was applied to investigate genetic variants. We then compared the utility of combining different genetic variants and neuroimaging features for predicting the Movement Disorder Society-sponsored unified Parkinson’s disease rating scale (MDS-UPDRS) in a regression framework. The associative cortex, motor cortex, thalamus, and pallidum showed significantly different connectivity between the HC and PD groups. Imaging genetics analysis identified PARK2, PARK7, HtrA2, GIGYRF2, and SNCA as genetic variants that are significantly associated with imaging phenotypes. A linear regression model combining genetic and neuroimaging features predicted the MDS-UPDRS with lower error and higher correlation with the actual MDS-UPDRS compared to other models using only genetic or neuroimaging information alone.
Similar content being viewed by others
Introduction
Parkinson’s disease (PD) is a progressive neurodegenerative disorder characterized by bradykinesia, resting tremors, rigidity, and difficulty with voluntary movement1. Typically, PD is diagnosed using criteria such as the United Kingdom’s Parkinson’s Disease Society Brain Bank2. The pathological hallmark of PD is the loss of dopaminergic neurons in the substantia nigra (SN) and those that project into the striatum. This neuronal loss leads to dopamine imbalance, which may result in inhibition of basal ganglia output and dysfunction within the cortico-basal ganglia-thalamocortical (CBGT) circuit3,4. Environmental toxins and genetic factors have been linked to the development of PD5. More than 16 PD-related genetic mutations have been recognized6.
Various neuroimaging approaches have been used to investigate how PD affects the brain4,7,8,9,10. These imaging approaches include positron emission tomography (PET), single-photon emission computed tomography (SPECT), and magnetic resonance imaging (MRI). Functional dopaminergic imaging such as dopamine transporter SPECT has been widely used to assess nigrostriatal degeneration11. White matter integrity could be assessed using diffusion MRI and tractography, which allows investigation of neuronal degeneration12. These various neuroimaging approaches allow us to better understand the neurobiological mechanism of PD13.
Existing neuroimaging studies related to the genome of PD have focused on certain candidate genes14,15. Recently, genome-wide association study (GWAS) has been used to identify genetic polymorphisms that influence disease phenotypes16. Common genetic variants that are associated with many diseases such as Alzheimer’s disease, PD, and diabetes have been successfully identified using GWAS16,17. Genes associated with PD have been identified using GWAS6,18. GWAS is frequently limited by the need for a large number of data samples to achieve statistical significance. Sample size requirements can be reduced using quantitative trait analysis, a form of genetic analysis that uses intermediate phenotypes of the disorder being studied19. Imaging genetics is a type of quantitative trait analysis that uses imaging measures (e.g., functional connectivity and brain activation) that are heritable and correlated with the disease in question.
Imaging genetics is an emerging tool that is capable of identifying the genetic risk of a disease based on both clinical factors and imaging techniques20. Imaging techniques provide complementary high dimensional information regarding the disease, and thus, GWAS could be improved by such additional information. Existing imaging genetic studies have been applied to Alzheimer’s disease and schizophrenia17,19,21,22,23. However, the application of imaging genetics for PD has been scarce. Prior imaging genetics studies have focused on specific gene candidates and have not used imaging measures for quantitative trait analysis14,15. One study investigating the cause of familial PD using functional PET imaging and genetic data reported that a mutation in the LRRK2 gene led to familial PD14. Another study investigated the early progression of PD in patients with mutant Parkin and PINK genes using functional MRI (fMRI)15. They showed that Parkin mutation carriers had abnormalities in the supplementary motor area and premotor cortex during a simple sequence task and that PINK mutation carriers were affected in the same regions during a finger sequence task.
In our study, we applied imaging genetics combining genetic polymorphisms and brain connectivity measurements as intermediate phenotypes to identify clinically relevant genetic variants in PD. Another aim of our study was to determine if the use of genetic variants along with their associated neuroimaging features could better predict clinical scores of PD progression compared to using conventional approaches with GWAS or neuroimaging alone. We obtained diffusion MRI and DNA genotyping data from the Parkinson’s disease Progression Marker Initiative (PPMI) database24. Connectivity analysis was used to identify neuroimaging features within the CBCT circuit that could differentiate between healthy control (HC) and PD groups. These neuroimaging features and associated clinical scores were used as intermediate phenotypes in imaging genetics analysis to identify clinically relevant genetic variants. We then analyzed the utility of combining different genetic variants and neuroimaging features for predicting the Movement Disorder Society-sponsored unified Parkinson’s disease rating scale (MDS-UPDRS), a well-established measure reflecting motor symptoms and clinimetric properties of PD, using a regression framework25.
Results
Subject selection
First, quality control of genotype data was performed based on the Enhancing Neuro Imaging Genetics through Meta Analysis (ENIGMA) protocol. In total, single-nucleotide polymorphisms (SNPs) with a minor allele frequency <1% (n = 53,482), Hardy-Weinberg equilibrium <10−6 (n = 683), and a genotype missing rate >5% (n = 14,378) were excluded from the study. Following quality control, 136,350 SNPs remained for further analysis. Second, the population stratification of the genetic data was adjusted using multidimensional scaling. After population stratification, only unrelated Caucasian subjects (n = 418) remained (Supplementary Figure S1). Of these unrelated Caucasian subjects, 146 (HC = 40, PD = 106) had both neuroimaging and genetic data available in the database. Forty of these PD cases were then randomly chosen to achieve equal numbers of samples in each group.
Connectivity differences
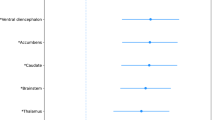
Differences between the HC and PD groups were quantified in terms of degree centrality (DC) values using t-tests with Bonferroni correction. Four regions, namely the associative cortex, motor cortex, thalamus, and pallidum, had significantly different DC values between the HC and PD groups (Table 1). The associative cortex, motor cortex, and thalamus showed increased DC values in PD patients compared with HC subjects, while the pallidum showed reduced DC values in PD patients compared with HC subjects.
Genetic variants in imaging genetics analysis
The results of imaging genetics analysis using the DC values of the associative cortex, motor cortex, thalamus, and pallidum and MDS-UPDRS are shown in Table 2. Three SNPs were associated with the DC values of these four regions, and four SNPs were associated with the MDS-UPDRS. PARK2 (which contained three SNPs) was associated with the DC value of the associative cortex and MDS-UPDRS. PARK7 was associated with the DC value of the motor cortex, and SNCA was associated with the DC value of the thalamus. HtrA2 and GIGYF2 were associated with the MDS-UPDRS.
Linear model analysis
We compared three multiple linear regression models to determine which model best explained the MDS-UPDRS. Stepwise regression was applied to construct three linear regression models, and the process led to different regression models depending on the initial model. Model C (the one with both genetic variants and neuroimaging features) (3) showed the best performance (adjusted R-squared (adj-R2) = 0.714, p < 0.001) compared to model A (the one with neuroimaging features) (1) and model B (the one with genetic variants) (2), which yielded adj-R2 values of 0.508 (p < 0.001) and 0.442 (p < 0.001), respectively (Tables 3,4 and 5). Details regarding the three equations are found in the Methods section. Specifically, the DC value of the pallidum singly explained the greatest (9.70%) variance (F = 21.52, p < 0.001) in model C (3). Model C (3) explained 72.51% of the variance in the MDS-UPDRS, while model A (1) and model B (2) explained 53.25% and 42.74% of the variance in the MDS-UPDRS (Tables 3,4 and 5). Thus, combining genetic variants with neuroimaging features resulted in an improved adj-R2 value, indicating that they provide complementary information. Each model included a distinct set of independent variables (IVs), and thus, the number of included IVs varied from model to model. A given variable might appear in two different models, but its contribution in terms of percentage of explained variance could be different as there was a distinct set of IVs in each model. For example, the DC of the thalamus explained 10.75% of the variance in conjunction with 5 other IVs in model A, while the DC of the same region explained 6.87% in conjunction with 9 other IVs in model C.
Prediction of MDS-UPDRS
The model combining genetic and neuroimaging features, model C (3), predicted the MDS-UPDRS with root mean squared error (RMSE) of 7.82 and showed a high correlation with the actual MDS-UPDRS (r = 0.788, p < 0.001) compared with the models using genetic and neuroimaging features alone (model A (1) and model B (2)). The first model with only neuroimaging features, model A (1), showed a high RMSE of 10.24 and a correlation of 0.616 (p < 0.001). The second model with only genetic variants, model B (2), yielded a RMSE of 11.71 and a correlation of 0.475 (p < 0.001). Plots of the actual and predicted MDS-UPDRS scores for these three models are given in Fig. 1.

Sub-figures (a,b and c) show actual and predicted MDS-UPDRS from Models A (1), B (2), and C (3), respectively. The dashed line indicates the identity line (i.e., actual score = predicted score). (d) shows error plots of the three models.
Discussion
Our main finding showed that the linear prediction model combining neuroimaging and genetics could explain 72.51% of the variance in the MDS-UPDRS, and the model resulted in a high correlation (r = 0.788) between the predicted and actual MDS-UPDRS. Our connectivity analysis revealed that the associative cortex, motor cortex, thalamus, and pallidum had significantly different DC values between the HC and PD groups. Imaging genetics analysis identified PARK2, PARK7, HtrA2, GIGYRF2, and SNCA as genetic variants that are significantly associated with intermediate phenotypes. These findings illustrate that the identified PD-related genes might yield structural connectivity changes that could be recognized using diffusion MRI within the CBGT circuit. The linear regression model combining genetic and neuroimaging features predicted the MDS-UPDRS with a lower RMSE and higher correlation with the actual MDS-UPDRS compared to the other models using only genetic or neuroimaging information alone.
Many recent neuroimaging studies have investigated PD using brain connectivity4,8,10,26,27. Connectivity analysis assumes that the brain is a complex interconnected network, and it is suitable for exploring large-scale circuits that include the cortical-thalamic and cerebro-cerebellar circuits28. Connectivity could be assessed using centrality measures including betweenness, eigenvector, degree, and page-rank centrality to assess network properties in the brain29. Each centrality characterizes the importance of a given node in terms of global or local contribution to the network. There is no single centrality measure that works well in all network assessments, and the optimal centrality measure is different depending on the imaging modality and the disease of concern30. DC has a straightforward neurobiological interpretation in structural connectivity networks, and thus, we utilized DC in this study. Our connectivity analysis revealed that the associative cortex, motor cortex, thalamus, and pallidum had significantly different DC values between the HC and PD groups. Some previous studies also showed that these regions were important in PD patients based on fMRI, diffusion MRI, and SPECT8,27,31,32,33.
Conventional genetic analyses have identified genetic variants linked with PD5,18. Imaging genetics combines imaging information with genetic information, allowing for identification of additional genetic variants22. Thus, the analysis of PD with imaging genetics might identify additional information to better investigate the neurobiological mechanisms of PD. In this study, an effect of PARK2 (Parkin) was observed on the DC value of the associative cortex and clinical scores. PARK2 plays an important role in presynaptic and postsynaptic neuronal processes34. Mutations in the Parkin protein are responsible for disrupting E3 ubiquitin ligase activity in PD35. The associative cortex plays a secondary role in processing motor information in the classic model of the CBGT motor circuit31. Previous work showed that a mutation of PARK2 was associated with activity in the left lateral orbitofrontal cortex, which is part of the associative cortex36. Another study reported that the DC value of the dorsolateral prefrontal sub-region of the associative cortex was increased in PD patients compared to HC using resting state-fMRI8. These findings suggest that PARK2 may be linked with connectivity in PD patients and in turn may affect the MDS-UPDRS. An effect of PARK7 (DJ-1) was observed on DC values in the motor cortex. PARK7 is involved in transcriptional regulation and response to oxidative stress37. The motor cortex is a key region involved in motor processing, serving as a major input into the basal ganglia1,13. Our findings suggest that PARK7 may affect neurons in the motor cortex and in turn may affect PD symptoms. Previous reports showed that characteristic PD findings, such as dopaminergic neuronal death in the SN and motor abnormalities, are affected by mutations in PARK737. One resting state-fMRI study showed that connectivity between the putamen and motor cortex increased in PD patients27. Another study reported that the DC value of the parietal cortex, a sub-region of the motor cortex, was increased in PD patients compared with HC subjects8. Moreover, one diffusion MRI study reported that track density in the primary somatosensory cortex was increased in PD patients compared with HC subjects32. Thus, the physiologic association of PARK7 with neurons in the motor cortex suggested by our results could be feasible. The interaction term between rs9346876 (PARK2) and rs363611 (HtrA2) led to 15.28% and 9.11% of explained variance for models B and C, respectively, in the linear regression analysis. This finding is consistent with previous studies reporting that HtrA2 specifically binds to and directly cleaves the E3 ubiquitin ligase PARK238,39.
We performed additional analysis with 14 right-sided and 14 left-sided onset cases in the PD group using contralateral ROIs, and detailed results are shown in the supplemental material. We observed better results for the reduced 28 patient group compared to using the previous 40 patients in terms of adj-R2 values. Model C’s adj-R2 improved from 0.714 to 0.768, model A’s adj-R2 improved from 0.508 to 0.650, and model B’s adj-R2 improved from 0.422 to 0.485. Similar to the main results with 40 patients, model C (adj-R2 = 0.768) outperformed models A (adj-R2 = 0.650) and B (adj-R2 = 0.485) implying that combining genetic variants with neuroimaging features indeed was the best model. Similar to the main results, for the reduced 28 patients, model C showed the best prediction performance with low RMSE (RMSE = 3.35) and a high correlation with the actual MDS-UPDRS (r = 0.847, p < 0.001).
Our study was limited by the small number of available samples. This issue was partly mitigated by analyzing SNPs and genes previously linked with PD6. Our work focused on known genetic variants, thereby establishing a foundation for future research. The neuroimaging data in our study came from nine different clinical sites, but all nine sites adopted the same image acquisition protocol using the same MRI scanner model (Siemens 3T scanner) to reduce confounding effects. In addition, we added multi-center information as a nuisance co-variate to account for multi-center confounding effects. We restricted the connectivity analysis to eight known regions within the CBGT circuit, which did not cover the entire brain. We used a single imaging modality (i.e., diffusion MRI) to compute intermediate phenotypes of PD. A multi-modal imaging study, potentially including PET and SPECT, could provide complementary information and thus has the potential to provide further insight into the pathophysiology of PD. Finally, we retrieved data from a research database that lacked follow-up longitudinal data. Future work should consider longitudinal data to test the validity of our models.
Methods
Subjects and imaging acquisition
This study was a retrospective analysis of anonymized data, and institutional review board (IRB) approval was obtained at Sungkyunwkan University. Our study was performed in full accordance with the local IRB guidelines. Informed consent was obtained from all subjects. The baseline neuroimaging (T1-, T2-, and diffusion MR) and DNA genotyping data were obtained from the PPMI research database24. In detail, diffusion MRI scans were acquired with the following acquisition parameters: TR = 900 ms, TE = 88 ms, b = 1000 s/mm2, 64 diffusion gradient directions with one b0 image, image matrix = 116 × 116 × 72, and voxel resolution = 1.98 × 1.98 × 2 mm3. T1-weighted MRI scans were acquired using the following parameters: TR = 2300 ms, TE = 2.98 ms, TI = 900 ms, image matrix = 240 × 256 × 176, and voxel resolution = 1 × 1 × 1 mm3. T2-weighted MRI scans were acquired using the following parameters: TR = 3000 ms, TE = 101 ms, image matrix = 228 × 256 × 48, and voxel resolution = 0.9375 × 0.9375 × 3 mm3. The PPMI database included 423 PD and 196 HC cases as of July 2015. PD cases were classified using the criteria established by the PPMI consortium24. Subjects who underwent both MRI imaging and genetic testing were considered for this study. We used off-medication subjects so that the effects of levodopa therapy were excluded. Additional patient data including MDS-UPDRS, Hohn and Yahn, Geriatric Depression Scale (GDS), Montreal Cognitive Assessment (MoCA), handedness and family history were collected for each case at the baseline visit (Supplementary Table S1). Population stratification of genetic data was adjusted using multidimensional scaling. We used multidimensional scaling analysis based on the ENIGMA protocol to reduce the population stratification and to ensure genetic homogeneity for all subjects40. All quality controlled SNPs were compared to the HapMap3 reference population using Plink v1.09 software41. Identity by state distance was calculated for all autosomal SNPs so that only unrelated Caucasian subjects remained. Of these unrelated Caucasian subjects, we chose 40 HC and 40 PD cases as shown in the Results section. The age, sex ratio, handedness, and cognitive assessments of the HC and PD groups were matched (Supplementary Table S1). Among 40 PD patients, 14 were right-sided, 14 were left-sided, and 12 were bilateral onset cases.
Tractography
Diffusion MRI data were processed with tractography, an algorithm that can extract in vivo neuronal fiber information, as described in Fig. 2. In brief, distortions due to eddy currents and head motion were first corrected using FSL. Second, T1-, T2-, and diffusion-weighted MRI scans were aligned to the Montreal Neurological Institute space with non-linear registration using FSL42. Then, white matter, gray matter, and cerebrospinal fluid were segmented from the registered T1-weighted image using Freesurfer43. The segmented white matter was used to guide the tractography algorithm. Fiber information was computed using the deterministic tractography algorithm implemented in the Diffusion Toolkit44. For connectivity analysis (performed with MATLAB), structural connectivity matrices were constructed using a deterministic tractography algorithm.

Overview of neuroimaging and image genetics processing steps.
Region of interest specification
Connectivity analysis requires nodes and edges in a graph structure. Eight ROIs associated with PD were adopted from the existing literature and used as connectivity nodes4. The eight ROIs were the caudate, putamen, pallidum, thalamus, sensorimotor cortex, associative cortex, limbic cortex (specified by the Desikan-Killiany atlas) and SN (specified by transferring ROI information from a pre-defined atlas via image co-registration) (Fig. 3)45,46. These regions are members of the CBGT circuit and are known to be associated with motor, executive, and movement functions1,4,47,48.

Eight ROIs associated with PD in the CBGT circuit were defined based on MR images. The ROIs include the associative cortex, limbic cortex, sensorimotor cortex, caudate, putamen, pallidum, thalamus, and substantia nigra.
Structural connectivity analysis
Structural connectivity was computed using eight regions as nodes and the fiber density between two regions as edges. The fiber density between two regions was defined as follows:  where f is the fiber connection between regions i and j, l(f) is the length of the fiber connection, Si is the surface area of region i, and mean FA(i, j) is the mean fractional anisotropy value along all fiber connections between regions i and j. In other words, fiber density was calculated as the connection efficiency between two regions, which was further defined as the number of fibers normalized by length and surface area49,50,51,52,53. The edge values were entered as elements of the matrix, referred to as the connectivity matrix. A simple network model that considered undirected and weighted edges was used to construct the brain network. We adopted DC, which is a simple and sensitive measure of structural connectivity, to measure the properties of the brain networks30. DC was computed as the sum of all edge weights connected in a given node54. DC represents the importance of a given node, where a high DC value signifies high information flow in the node. The microstructure of white matter is under strong genetic control, and thus, the white matter structural connectivity might be affected by genes55.
where f is the fiber connection between regions i and j, l(f) is the length of the fiber connection, Si is the surface area of region i, and mean FA(i, j) is the mean fractional anisotropy value along all fiber connections between regions i and j. In other words, fiber density was calculated as the connection efficiency between two regions, which was further defined as the number of fibers normalized by length and surface area49,50,51,52,53. The edge values were entered as elements of the matrix, referred to as the connectivity matrix. A simple network model that considered undirected and weighted edges was used to construct the brain network. We adopted DC, which is a simple and sensitive measure of structural connectivity, to measure the properties of the brain networks30. DC was computed as the sum of all edge weights connected in a given node54. DC represents the importance of a given node, where a high DC value signifies high information flow in the node. The microstructure of white matter is under strong genetic control, and thus, the white matter structural connectivity might be affected by genes55.
Group-wise differences in neuroimaging
Group-wise differences in neuroimaging were assessed with t-tests and Bonferroni correction between the HC and PD groups. For each group, the DC values of the eight nodes were stacked into three-dimensional matrices. Each group had a single three-dimensional matrix. Each element in the stacked connectivity matrix contained 40 observations. We performed t-tests to compare between the HC and PD groups to assess group-wise differences. Multiple comparison issues were corrected using the Bonferroni correction (p < 0.05, corrected)56,57. DC values of regions with significant differences were used as intermediate phenotypes for imaging genetics.
Genetic quality control
We quality controlled DNA genotyping data based on the ENIGMA protocol40. Quality control of genotype data was performed based on minor allele frequency, genotype missing rate, Hardy-Weinberg equilibrium, and genotyping rate. SNPs that did not satisfy the criteria were excluded, and subjects with a low genotyping rate were also excluded. In total, SNPs with a minor allele frequency <1%, Hardy-Weinberg equilibrium <10−6, and a genotype missing rate >5% were excluded from the study. Following quality control, the remaining SNPs were used for further analysis.
Imaging genetics
Imaging genetics analysis was conducted using intermediate phenotypes to identify genetic variants associated with PD. In the first analysis, neuroimaging features (i.e., DC values of regions with significant differences between HC and PD) were used as intermediate phenotypes. In the second analysis, the clinical score (i.e., MDS-UPDRS) was used as an intermediate phenotype. The genetic variance was described with an additive model58. Imaging genetics was performed using Plink software41. A linear regression analysis was performed at each SNP using the number of minor alleles as the independent variable and various types of intermediate phenotype information as the dependent variable. Age and sex were included as covariates in the linear model. Significant genetic variants were determined using a permutation test by performing random group assignments 10,000 times23,59. We focused on SNPs that were previously linked with PD.
Construction of multiple linear regression models
We constructed linear models using a stepwise approach to explain the clinical score (i.e., MDS-UPDRS) for all subjects (i.e., for both the HC and PD groups). Multiple linear regression models were constructed using imaging features, genetic features, and two-way interactions between them. The multiple linear regression also considered family history information, MoCA, GDS, sex, age, and multi-center information as co-variants to control for confounding effects. Stepwise regression was applied to construct the regression models explaining the MDS-UPDRS scores60. An initial model with all possible independent variables along with interaction terms among them was constructed. Then, a candidate independent variable was removed if the removal of the variable led to a smaller p-value for the overall regression model. The process of variable removal was repeated until there was no improvement of the p-value of the regression model. For instance, the initial model considered DC values of four significant ROIs (from Table 1) and numerical SNP values of significant genetic variants (from Table 2) as independent variables. Then, each independent variable was tested for possible removal as described above. Subsequently, three linear regression models were constructed to explain the MDS-UPDRS score. The first model considered only neuroimaging features, the second model considered only genetic variants, and the third model considered both genetic variants and neuroimaging features:



where Y is the clinical score; IMG is the imaging features; SNP is the genetic features; COV is the co-variants; INTER are the two-way interactions of predictors; α, β, γ and δ are estimated coefficients; and ε is a residual error term. The quality of multiple linear regression was assessed with adj-R2 values.
Prediction and validation of the model
To validate the models, a leave-one-out cross validation method was applied by assigning each case as the test set and the remaining cases as the training set. The process was repeated 80 times, each time assigning a different test set. The performance of prediction was assessed with Pearson’s correlation coefficients between actual and predicted MDS-UPDRS scores. The RMSE was computed to quantify differences between the predicted and actual MDS-UPDRS.
Additional Information
How to cite this article: Kim, M. et al. Imaging genetics approach to Parkinson’s disease and its correlation with clinical score. Sci. Rep. 7, 46700; doi: 10.1038/srep46700 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Stoessl, A. J. Neuroimaging in Parkinson’s disease. Neurotherapeutics 8, 72–81 (2011).
National Collaborating Centre for Chronic Conditions (Great Britain). Parkinson’s disease: national clinical guideline for diagnosis and management in primary and secondary care. Royal College of Physicians, (2006).
Obeso, J. A. et al. Functional organization of the basal ganglia: therapeutic implications for Parkinson’s disease. Movement disorders: official journal of the Movement Disorder Society 23 Suppl 3, S548–559, doi: 10.1002/mds.22062 (2008).
Sharman, M. et al. Parkinson’s disease patients show reduced cortical‐subcortical sensorimotor connectivity. Movement Disorders 28, 447–454 (2013).
Dauer, W. & Przedborski, S. Parkinson’s disease: mechanisms and models. Neuron 39, 889–909 (2003).
Rodriguez-Oroz, M. C. et al. Initial clinical manifestations of Parkinson’s disease: features and pathophysiological mechanisms. The Lancet Neurology 8, 1128–1139 (2009).
Ravina, B. et al. The role of radiotracer imaging in Parkinson disease. Neurology 64, 208–215 (2005).
Wu, T. et al. Changes of functional connectivity of the motor network in the resting state in Parkinson’s disease. Neuroscience letters 460, 6–10 (2009).
Zhang, K. et al. Voxel-based analysis of diffusion tensor indices in the brain in patients with Parkinson’s disease. European journal of radiology 77, 269–273 (2011).
Zhang, Yu et al. Diffusion tensor imaging of the nigrostriatal fibers in Parkinson’s disease. Movement Disorders 30, 1229–1236 (2015).
Pavese, N. & Brooks, D. J. Imaging neurodegeneration in Parkinson’s disease. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 1792, 722–729 (2009).
Le Bihan, D. et al. Diffusion tensor imaging: concepts and applications. Journal of magnetic resonance imaging 13, 534–546 (2001).
Weingarten, C. P., Sundman, M. H., Hickey, P. & Chen, N.-k. Neuroimaging of Parkinson’s disease: Expanding views. Neuroscience & Biobehavioral Reviews 59, 16–52 (2015).
Khan, N. L. et al. Mutations in the gene LRRK2 encoding dardarin (PARK8) cause familial Parkinson’s disease: clinical, pathological, olfactory and functional imaging and genetic data. Brain 128, 2786–2796 (2005).
van Nuenen, B. F. et al. Mapping preclinical compensation in Parkinson’s disease: an imaging genomics approach. Movement Disorders 24, S703–S710 (2009).
Burton, P. R. et al. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447, 661–678 (2007).
Hirschhorn, J. N. & Daly, M. J. Genome-wide association studies for common diseases and complex traits. Nature Reviews Genetics 6, 95–108 (2005).
Klein, C. & Schlossmacher, M. G. The genetics of Parkinson disease: implications for neurological care. Nature clinical practice Neurology 2, 136–146 (2006).
Potkin, S. G. et al. A genome-wide association study of schizophrenia using brain activation as a quantitative phenotype. Schizophrenia bulletin 35, 96–108 (2009).
Thompson, P. M., Martin, N. G. & Wright, M. J. Imaging genomics. Current opinion in neurology 23, 368 (2010).
Stein, J. L. et al. Voxelwise genome-wide association study (vGWAS). neuroimage 53, 1160–1174 (2010).
Tost, H., Bilek, E. & Meyer-Lindenberg, A. Brain connectivity in psychiatric imaging genetics. Neuroimage 62, 2250–2260 (2012).
Wang, Q. et al. Genome-wide association analysis with gray matter volume as a quantitative phenotype in first-episode treatment-naive patients with schizophrenia. PloS one 8, e75083 (2013).
Marek, K. et al. The parkinson progression marker initiative (PPMI). Progress in neurobiology 95, 629–635 (2011).
Disease, M. D. S. T. F. o. R. S. f. P. s. The Unified Parkinson’s Disease Rating Scale (UPDRS): status and recommendations. Movement disorders: official journal of the Movement Disorder Society 18, 738 (2003).
Kim, H. J. et al. Alterations of mean diffusivity in brain white matter and deep gray matter in Parkinson’s disease. Neuroscience letters 550, 64–68 (2013).
Yu, R., Liu, B., Wang, L., Chen, J. & Liu, X. Enhanced functional connectivity between putamen and supplementary motor area in Parkinson’s disease patients. PloS one 8, e59717 (2013).
Van Dijk, K. R. et al. Intrinsic functional connectivity as a tool for human connectomics: theory, properties, and optimization. Journal of neurophysiology 103, 297–321 (2010).
Zuo, X.-N. et al. Network centrality in the human functional connectome. Cerebral cortex 22, 1862–1875 (2012).
Rubinov, M. & Sporns, O. Complex network measures of brain connectivity: uses and interpretations. Neuroimage 52, 1059–1069 (2010).
Purves, D., Augustine, G. J., Fitzpatrick, D. et al. Neuroscience. 2nd edition. Sunderland (MA): Sinauer Associates(2001).
Ziegler, E. et al. Mapping track density changes in nigrostriatal and extranigral pathways in Parkinson’s disease. Neuroimage 99, 498–508 (2014).
Tolosa, E., Wenning, G. & Poewe, W. The diagnosis of Parkinson’s disease. The Lancet Neurology 5, 75–86 (2006).
Schlossmacher, M. G. et al. Parkin localizes to the Lewy bodies of Parkinson disease and dementia with Lewy bodies. The American journal of pathology 160, 1655–1667 (2002).
Sriram, S. R. et al. Familial-associated mutations differentially disrupt the solubility, localization, binding and ubiquitination properties of parkin. Human molecular genetics 14, 2571–2586 (2005).
Anders, S. et al. Compensatory premotor activity during affective face processing in subclinical carriers of a single mutant Parkin allele. Brain 135, 1128–1140 (2012).
Nuytemans, K., Theuns, J., Cruts, M. & Van Broeckhoven, C. Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: a mutation update. Human mutation 31, 763–780 (2010).
Park, H.-M. et al. The serine protease HtrA2/Omi cleaves Parkin and irreversibly inactivates its E3 ubiquitin ligase activity. Biochemical and biophysical research communications 387, 537–542 (2009).
Cilenti, L. et al. Inactivation of Omi/HtrA2 protease leads to the deregulation of mitochondrial Mulan E3 ubiquitin ligase and increased mitophagy. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research 1843, 1295–1307 (2014).
ENIGMA2 Genetics Support Team. ENIGMA2 1KGP Cookbook (v3) [Online]. The Enhancing Neuroimaging Genetics through MetaAnalysis (ENIGMA) Consortium. http://enigma.ini.usc.edu/wpcontent/uploads/2012/07/ENIGMA2_1KGP_cookbook_v3.pdf (2013).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. The American Journal of Human Genetics 81, 559–575 (2007).
Smith, S. M. et al. Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage 23 Suppl 1, S208–219, doi: 10.1016/j.neuroimage.2004.07.051 (2004).
Fischl, B. FreeSurfer. Neuroimage 62, 774–781 (2012).
Wang, R. et al. Diffusion toolkit: a software package for diffusion imaging data processing and tractography. Proc Intl Soc Mag Reson Med. 5, 3720 (2007).
Keuken, M. C. et al. Quantifying inter-individual anatomical variability in the subcortex using 7T structural MRI. NeuroImage 94, 40–46 (2014).
Desikan, R. S. et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage 31, 968–980 (2006).
Parent, A. & Hazrati, L.-N. Functional anatomy of the basal ganglia. I. The cortico-basal ganglia-thalamo-cortical loop. Brain Research Reviews 20, 91–127 (1995).
Braak, H. & Del Tredici, K. Cortico-basal ganglia-cortical circuitry in Parkinson’s disease reconsidered. Experimental neurology 212, 226–229 (2008).
Kim, M. & Park, H. Structural connectivity profile of scans without evidence of dopaminergic deficit (SWEDD) patients compared to normal controls and Parkinson’s disease patients. SpringerPlus 5, 1421 (2016).
Tuch, D. S., Wedeen, V. J., Dale, A. M., George, J. S. & Belliveau, J. W. Conductivity tensor mapping of the human brain using diffusion tensor MRI. Proceedings of the National Academy of Sciences 98, 11697–11701 (2001).
Hagmann, P. et al. White matter maturation reshapes structural connectivity in the late developing human brain. Proceedings of the National Academy of Sciences 107, 19067–19072 (2010).
Fischi-Gómez, E. et al. Structural brain connectivity in school-age preterm infants provides evidence for impaired networks relevant for higher order cognitive skills and social cognition. Cerebral cortex 25, 2793–2805 (2015).
Feldman, H. M., Yeatman, J. D., Lee, E. S., Barde, L. H. & Gaman-Bean, S. Diffusion tensor imaging: a review for pediatric researchers and clinicians. Journal of developmental and behavioral pediatrics: JDBP 31, 346 (2010).
Schwarz, A. J. & McGonigle, J. Negative edges and soft thresholding in complex network analysis of resting state functional connectivity data. Neuroimage 55, 1132–1146 (2011).
Thompson, P. M., Ge, T., Glahn, D. C., Jahanshad, N. & Nichols, T. E. Genetics of the connectome. Neuroimage 80, 475–488 (2013).
Nichols, T. E. & Holmes, A. P. Nonparametric permutation tests for functional neuroimaging: a primer with examples. Human brain mapping 15, 1–25 (2002).
Bullmore, E. T. et al. Global, voxel, and cluster tests, by theory and permutation, for a difference between two groups of structural MR images of the brain. Medical Imaging, IEEE Transactions on 18, 32–42 (1999).
Brown, G. G. et al. Voxel-based morphometry of patients with schizophrenia or bipolar I disorder: a matched control study. Psychiatry Research: Neuroimaging 194, 149–156 (2011).
Smith, S. M. et al. Functional connectomics from resting-state fMRI. Trends in cognitive sciences 17, 666–682 (2013).
Draper, N. R. & Smith, H. Applied regression analysis. (John Wiley & Sons, 2014).
Acknowledgements
This study was supported by the Institute for Basic Science (grant number IBS-R015-D1). This work was also supported by National Research Foundation of Korea (grant number NRF-2016R1A2B4008545). Imaging data were obtained from the PPMI. PPMI is a public-private partnership funded by The Michael J. Fox Foundation for Parkinson’s Research, Abbott, Avid Pharmaceuticals, Biogen Idec, Bristol-Myers Squibb, Covance, Elan, GE Healthcare, Genentech, GlaxoSmithKline, Lilly, Merck, Meso Scale Discovery, Pfizer, Roche, and UCB (www. ppmi-info.org/fundingpartners).
Author information
Authors and Affiliations
Contributions
H.P. and M.K. conceptualized the design of the study. M.K., J.K., and S.L. performed the experiments and analyses. H.P. oversaw the entire study. H.P. and M.K. co-wrote the draft. All authors reviewed and provided feedback for the draft.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Kim, M., Kim, J., Lee, SH. et al. Imaging genetics approach to Parkinson’s disease and its correlation with clinical score. Sci Rep 7, 46700 (2017). https://doi.org/10.1038/srep46700
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep46700
This article is cited by
-
Predictive modelling of Parkinson’s disease progression based on RNA-Sequence with densely connected deep recurrent neural networks
Scientific Reports (2022)
-
A three-stage, deep learning, ensemble approach for prognosis in patients with Parkinson’s disease
EJNMMI Research (2021)
-
An Efficient WRF Framework for Discovering Risk Genes and Abnormal Brain Regions in Parkinson’s Disease Based on Imaging Genetics Data
Journal of Computer Science and Technology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
