
Abstract
Guanylyl cyclase-A (GC-A) signaling, a natriuretic peptide receptor, exerts renoprotective effects by stimulating natriuresis and reducing blood pressure. Previously we demonstrated massive albuminuria with hypertension in uninephrectomized, aldosterone-infused, and high salt-fed (ALDO) systemic GC-A KO mice with enhanced phosphorylation of p38 mitogen-activated protein kinase (MAPK) in podocytes. In the present study, we examined the interaction between p38 MAPK and GC-A signaling. The administration of FR167653, p38 MAPK inhibitor, reduced systolic blood pressure (SBP), urinary albumin excretion, segmental sclerosis, podocyte injury, and apoptosis. To further investigate the local action of natriuretic peptide and p38 MAPK in podocytes, we generated podocyte-specific (pod) GC-A conditional KO (cKO) mice. ALDO pod GC-A cKO mice demonstrated increased urinary albumin excretion with marked mesangial expansion, podocyte injury and apoptosis, but without blood pressure elevation. FR167653 also suppressed urinary albumin excretion without reducing SBP. Finally, we revealed that atrial natriuretic peptide increased phosphorylation of MAPK phosphatase-1 (MKP-1) concomitant with inhibited phosphorylation of p38 MAPK in response to MAPK kinase 3 activation, thereby resulting in decreased mRNA expression of the apoptosis-related gene, Bax, and Bax/Bcl2 ratio in cultured podocytes. These results indicate that natriuretic peptide exerts a renoprotective effect via inhibiting phosphorylation of p38 MAPK in podocytes.
Similar content being viewed by others

Introduction
Aldosterone has attracted much attention in the process of renal injury leading to proteinuria and chronic renal failure1. The mechanism underlying the contribution of aldosterone to worsening kidney function is thought to partly due to involve the activation of the mineralocorticoid receptor in podocytes, resulting in podocyte injury2, reactivation of renin-angiotensin-aldosterone system through positive feedback3, and the induction of reactive oxygen species (ROS) and activation of mitogen-activated protein kinases (MAPKs) in the kidney4. The site of action of aldosterone remains obscure, as aldosterone has been reported to exert its effect on podocytes5, mesangial cells6, endothelial cells7, and tubular cells8. In addition, aldosterone elevates blood pressure and regulates intraglomerular pressure8, which both contributes to the complex pathophysiology in aldosterone-induced renal injury.
Natriuretic peptides consist of atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), and C-type natriuretic peptide (CNP). ANP and BNP exert physiological effects such as diuresis and vasodilation through guanylyl cyclase-A (GC-A)/natriuretic peptide receptor-A (NPR-A)9. We previously demonstrated massive albuminuria, mesangial expansion, segmental sclerosis, and podocyte-injury with the elevation of systolic blood pressure in uninephrectomized, aldosterone-infused, and high salt diet-fed (ALDO) systemic GC-A knockout (KO) mice10. In the previous model, reduced blood pressure with the use of hydralazine fails to ameliorate renal injury; however, blockade of the renin angiotensin system by an angiotensin receptor blocker (ARB) or antioxidant tempol reduces glomerular injury10. Although our previous report may indicate that natriuretic peptides protect podocytes from aldosterone-induced renal injury, the precise site of action of natriuretic peptides/GC-A system, and the mechanism underlying amelioration of aldosterone-induced glomerular injury by natriuretic peptides have yet to be elucidated. In our previous study, we also demonstrated prominent phosphorylation of p38 MAPK in podocyte of ALDO GC-A KO mice10. Activation of p38 MAPK has been shown to be stimulated by UV light, heat, inflammation, and growth factors11. We and others have demonstrated that phosphorylation of p38 MAPK is activated in clinical glomerulonephritis, as well as in experimental podocyte injury glomerulopathy10,12,13. Blockade of p38 MAPK pathway reportedly ameliorates adriamycin nephropathy12 and crescentic glomerulonephritis14 in mice. However, the precise role of p38 MAPK in podocyte injury has yet to be elucidated. In addition, the mechanism underlying the renoprotective action of natriuretic peptides on podocytes remains to be clarified.
In the present study, we generated podocyte-specific (pod) GC-A conditional KO (cKO) mice and then examined the effects of p38 MAPK in systemic and pod GC-A cKO mice administered aldosterone, uninephrectomy, and a high salt diet.
Results
Inhibition of p38 MAPK ameliorates glomerular injury in ALDO systemic GC-A KO mice
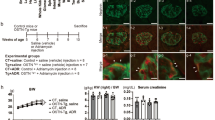
All ALDO mice were uninephrectomized and administered aldosterone and a 6% high salt diet for four weeks. First, we examined p38 MAPK phosphorylation in ALDO wild-type and systemic GC-A KO mice (Fig. 1a). Phosphorylation of p38 MAPK was predominantly detected in podocytes in ALDO wild-type mice (Fig. 1a). ALDO systemic GC-A KO mice demonstrated pronounced phosphorylation of p38 MAPK in podocytes (Fig. 1a), and had elevated systolic blood pressure (SBP) (Fig. 1b). Blockade of the p38 MAPK pathway using FR167653 (a p38 MAPK inhibitor) reduced SBP by 20 mmHg in ALDO systemic GC-A KO mice (Fig. 1b). Urinary albumin excretion in ALDO systemic GC-A KO mice was as high as 9,000 μg/mgCr; however, administration of FR167653 reduced urinary albumin excretion by 95% (Fig. 1c, Table 1). Body and kidney weights, and blood and urine parameters are presented in Table 1. ALDO systemic GC-A KO mice were found to have renal hypertrophy, and no effect of FR167653 on renal hypertrophy was observed. No significant differences in serum potassium, sodium, urinary potassium, or urinary sodium levels were observed (Table 1).

(a) Immunohistochemical study for phopho-p38 MAPK in glomeruli. Left panel shows a glomerulus in uninephrectomized, aldosterone-infused and high salt diet-fed (ALDO) wild-type mice. Right panel shows a glomerulus in ALDO systemic GC-A KO mice. Arrows, phospho-p38 MAPK-positive cells. Scale bar, 50 μm. (b) Time course of systolic blood pressure in ALDO wild-type mice, ALDO wild-type mice with FR167653, ALDO systemic GC-A KO mice, and ALDO systemic GC-A KO mice with FR167653. (c) Time course of albuminuria in these four groups. (d) Light microscopic analyses were performed at 4 weeks after aldosterone administration, stained with periodic acid-Schiff. In juxtamedullary glomeruli, ALDO systemic GC-A KO mice showed segmental sclerosis with glomerular hypertrophy. Treatment with FR167653 improved these changes. Scale bar, 50 μm. (e) Glomerular cross-sectional area and (f) mesangial area in juxtamedullary glomeruli at 4 weeks. (g) Histological examination of tubules and interstitial fibrosis by Masson’s trichrome-staining at 4 weeks. ALDO systemic GC-A KO mice exhibited tubular dilatation with protein cast deposition. These change were ameliorated by FR167653. Scale bar, 50 μm. n = 5, each. Mean ± SEM. ††p < 0.01 vs. ALDO wild-type mice. *p < 0.05, **p < 0.01 vs. ALDO systemic KO mice. WT, wild-type mice; KO, systemic GC-A knockout mice.
We examined renal histology at four weeks after aldosterone infusion. A mild mesangial expansion in superficial glomeruli was observed in ALDO wild-type mice (Supplementary Fig. S1a). In contrast, ALDO systemic GC-A KO mice had marked mesangial expansion with glomerular hypertrophy (Supplementary Fig. S1a–c). Administration of FR167653 significantly reduced mesangial expansion and glomerular hypertrophy (Supplementary Fig. S1a–c). In juxtamedullary glomeruli, ALDO systemic GC-A KO mice exhibited severe segmental sclerosis with glomerular hypertrophy (Fig. 1d). Treatment with FR167653 inhibited segmental sclerosis and glomerular hypertrophy in ALDO systemic GC-A KO mice (Fig. 1d–f). We further examined renal fibrotic changes. ALDO systemic GC-A KO mice exhibited tubular dilatation with protein cast deposition, and these changes were ameliorated by FR167653 (Fig. 1g).
Next, we examined gene expression in glomeruli. TGF-β1 (Tgfb1) is a profibrotic growth factor and is closely associated with matrix accumulation and renal fibrosis. Analysis of glomerular expression revealed increased Tgfb1 mRNA levels in ALDO systemic GC-A KO mice, and this upregulation was significantly reduced by FR167653 (Supplementary Fig. S2a). Then, we examined the expression of genes associated with inflammation. We examined gene expression relevant to macrophages: Ccl2 (MCP1) is one of macrophage chemoattractant molecules, and Adgre1 (F4/80) is one of markers of macrophages. Expression of Ccl2 (MCP-1) mRNA was increased in ALDO systemic GC-A KO mice, and administration of FR167653 significantly reduced Ccl2 mRNA levels (Supplementary Fig. S2b). Consistent with this result, FR167653 also reduced Adgre1 (F4/80) mRNA expression in ALDO systemic GC-A KO mice (Supplementary Fig. S2c). A similar trend in Cybb (NOX-2) mRNA expression was observed (Supplementary Fig. S2d).
Inhibition of p38 MAPK reduces podocyte injury in ALDO systemic GC-A KO mice
We next evaluated podocyte injury. Electron microscopic analysis revealed that ALDO wild-type mice exhibited slightly widened podocytes foot processes (Fig. 2a,b). ALDO systemic GC-A KO mice demonstrated foot process effacement with irregular thickening of the glomerular basement membrane (GBM), and these changes were improved by FR167653 (Fig. 2a,b). We quantitated the width of foot process and revealed an improvement of foot process effacement by FR167653 (Fig. 2c). Immunofluorescent study demonstrated that reduced nephrin and podocin levels in ALDO systemic GC-A KO mice, and these changes were also ameliorated with administration of a p38 MAPK inhibitor (Fig. 2d,e), indicating that inhibition of the p38 MAPK pathway is closely associated with amelioration of podocyte injury.

(a,b) Electron microscopic photographs of lower (a) and higher (b) magnification show that treatment with FR167653 in ALDO systemic GC-A KO mice improved GBM thickening and widening foot processes in a superficial glomerulus. Scale bar, 500 nm. (c) The width of foot processes in electron microscopic photographs. n = 3. (d,e) Representative images of nephrin (d) and podocin (e) expression by immunofluorescent study in a juxtamedullary glomerulus. Scale bar, 50 μm. ††p < 0.01, vs. ALDO wild-type mice. **p < 0.01 vs. ALDO systemic GC-A KO mice.
We next examined glomerular apoptosis. ALDO wild-type mice demonstrated almost no apoptotic cells within the glomeruli (Fig. 3a). In contrast, ALDO systemic GC-A KO mice had a large number of TUNEL-positive cells within the glomeruli and tubules, and this increase in the number of apoptotic cells was significantly inhibited by treatment with FR167653 (Fig. 3a). We also examined glomerular p53 expression, as it has been shown to be closely associated with apoptosis15. Expression of p53 in glomeruli was not detected in ALDO wild-type mice but was observed in ALDO systemic GC-A KO mice (Fig. 3b). Inhibition of p38 MAPK reduced glomerular expression of p53 in ALDO systemic GC-A KO mice (Fig. 3b). We next counted the number of podocytes by immunohistochemical analysis for Wilms’ tumor 1 (WT1). Administration of FR167653 did not change podocyte numbers in ALDO wild-type mice (Fig. 3c,d). ALDO systemic GC-A KO mice had a significant reduction in the number of podocytes (Fig. 3c,d), and this reduction was reversed by administration of FR167653 (Fig. 3c,d). Phosphorylation of p38 MAPK in glomeruli was augmented in ALDO systemic GC-A KO mice, and this phosphorylation was ameliorated in ALDO systemic GC-A KO mice administered FR167653 (Supplementary Fig. S3). MAPK kinase 3 (MKK3) is an upstream kinase of p38 MAPK16. We examined the phosphorylation of MKK3 in these mice by immunohistochemical study. ALDO systemic GC-A KO mice showed enhanced phosphorylation of MKK3 in glomeruli, and this phosphorylation was reduced with the treatment of FR167653 (Supplementary Fig. S4). These results indicate that inhibition of p38 MAPK ameliorates podocyte injury and apoptosis.

(a) TUNEL staining shows that TUNEL-positive cells were increased in glomeruli of ALDO systemic GC-A KO mice. Treatment with FR167653 in ALDO systemic GC-A KO mice reduced the number of TUNEL-positive cells. (b) Immunohistochemical study for p53. ALDO systemic GC-A KO mice showed increased number of p53-positive cells in glomeruli. Administration with FR167653 reduced the number of p53-positive cells. (c) Immunohistochemical study for WT1 in juxtamedullary glomeruli. (d) The number of WT1-positive cells in juxtamedullary glomeruli. ALDO systemic GC-A mice showed reduced number of WT1-positive cells, and its decrease was ameliorated with FR167653. Scale bar, 50 μm. Mean ± SEM. ††p < 0.01, vs. ALDO wild-type mice. *p < 0.05 vs. ALDO systemic GC-A KO mice.
Deletion of GC-A in podocytes aggravates glomerular injury
Although inhibition of p38 MAPK in ALDO systemic GC-A KO mice ameliorated podocyte and glomerular injury, the relevance of p38 MAPK inhibition with natriuretic peptides in podocyte injury remains obscure. To reveal the role of p38 MAPK signaling in podocytes of ALDO systemic GC-A KO mice, we generated podocyte-specific (pod) GC-A conditional KO (cKO) mice by crossing Nephrin-Cre mice with GC-A floxed mice, and then administering FR167653. Vehicle (VEH)-treated mice were also uninephrectomized, and administered 6% high salt diet and ethanol alone instead of ethanol-containing aldosterone. GC-A floxed mice without Cre recombinase were used as control mice. VEH control and pod GC-A cKO mice had normal SBP (Fig. 4a). ALDO control mice exhibited an increase in SBP of 10 mmHg at four weeks, and ALDO pod GC-A cKO mice had comparable SBP to ALDO control mice (123.2 ± 2.2 vs. 119.4 ± 2.3 mmHg; Fig. 4a). FR167653 was administered to pod GC-A cKO mice as the activation of p38 MAPK is reportedly involved in glomerular injury in ALDO systemic GC-A KO mice. No change in SBP was observed following the administration of FR167653 in ALDO pod GC-A cKO mice (119.0 ± 2.4 mmHg; Fig. 4a). Body weights, kidney weights and serum and urinary parameters are presented in Table 2. Pod GC-A cKO mice exhibited normal kidney weights comparable with control mice (Table 2). ALDO control and pod GC-A cKO mice exhibited renal hypertrophy, and administration of FR167653 had no effect on renal hypertrophy in pod GC-A cKO mice (Table 2). Pod GC-A cKO mice had similar serum and urinary potassium levels to control mice, and aldosterone infusion increased urinary potassium levels and decreased serum potassium levels to the same extent in both control and pod GC-A cKO mice (Table 2). There was no significant difference in serum creatinine and urea nitrogen (UN) levels between ALDO control and pod GC-A cKO mice.

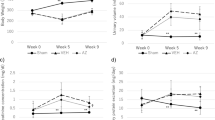
(a) Time course of systolic blood pressure of VEH control mice, ALDO control mice, VEH pod GC-A cKO mice, ALDO pod GC-A cKO mice, and ALDO pod GC-A cKO mice with FR167653 is shown. Administration of aldosterone increased systemic blood pressure at comparable levels in both control mice and pod GC-A cKO mice. Administration with FR167653 did not change blood pressure. (b) Time course of urinary albumin excretion in these five groups. (c) PAS staining of renal section were performed at 4 weeks after aldosterone administration. In juxtamedullary glomeruli, ALDO pod GC-A cKO mice showed mesangial expansion with glomerular hypertrophy. Treatment with FR167653 improved these changes. Scale bar, 50 μm. (d) Glomerular cross-sectional area and (e) mesangial area in juxtamedullary glomeruli at 4 weeks were quantified in five groups. (f) Glomerular mRNA expression of Npr1 in VEH control and pod GC-A cKO mice.n = 7, each. Mean ± SEM. †p < 0.05, ††p < 0.01 vs. VEH control mice, **p < 0.01 vs. VEH pod GC-A cKO mice, ¶¶p < 0.01 vs. ALDO pod GC-A cKO mice.
At basal levels, urinary albumin excretion was not different between VEH control mice and pod GC-A cKO mice (18.5 ± 3.3 and 14.2 ± 2.6 μg/mgCr, respectively; Fig. 4b). ALDO control mice had a threefold increase in urinary albumin excretion at four weeks (84.0 ± 6.5 vs. 31.3 ± 3.6 μg/mgCr; Fig. 4b). Of note, ALDO pod GC-A cKO mice demonstrated a significantly increased urinary albumin excretion by 13-fold compared to ALDO control mice at four weeks (84.0 ± 6.5 vs. 1,188 ± 152 μg/mgCr; Fig. 4b). These results indicate that deletion of GC-A in podocytes aggravates aldosterone-induced renal injury. Treatment of ALDO pod GC-A cKO mice with FR167653 suppressed urinary albumin excretion by 95% compared with ALDO pod-GC-A cKO mice (80.5 ± 10.5 μg/mgCr; Fig. 4b), indicating the importance of p38 MAPK pathway in aldosterone-induced podocyte injury.
Pod GC-A cKO mice had normal glomerular histology at baseline. In superficial glomeruli, there was no significant difference in glomerular cross-sectional or mesangial areas between ALDO control and ALDO pod GC-A cKO mice (Supplementary Fig. S5a,b). In juxtamedullary glomeruli, ALDO control mice had mild glomerular hypertrophy and mesangial expansion (Fig. 4c–e). ALDO pod GC-A cKO mice were observed to have marked mesangial expansion and glomerular hypertrophy (Fig. 4c–e). Administration of FR167653 ameliorated glomerular hypertrophy and mesangial expansion (Fig. 4c–e). We examined Npr1 mRNA to examine the deletion efficacy of Npr1 gene. Glomerular mRNA expression of Npr1 (GC-A) in VEH pod GC-A cKO mice was decreased by 40% compared with that in VEH control mice (Fig. 4f).
Deletion of GC-A in podocytes aggravates podocyte injury and its injury is reversed by inhibition of p38 MAPK
To evaluate podocyte injury, we performed immunofluorescent analyses of juxtamedullary glomeruli for nephrin and podocin. ALDO control mice exhibited no difference in nephrin and podocin expression compared with VEH control mice (Fig. 5a,b). Although VEH pod GC-A cKO mice had normal nephrin and podocin staining, ALDO pod GC-A cKO mice demonstrated decreased staining intensity for nephrin and podocin (Fig. 5a,b). ALDO pod GC-A cKO mice with FR167653 maintained similar nephrin and podocin staining intensities as VEH pod GC-A cKO mice (Fig. 5a,b). We examined podocyte injury in juxtamedullary glomeruli by electron microscopic analysis. VEH pod GC-A cKO mice demonstrated normal foot processes and GBM (Fig. 5c). ALDO pod GC-A cKO mice exhibited mild thickening of the GBM and foot process effacement, and these changes were improved with the administration of FR167653 (Fig. 5c). Next, we examined glomerular p38 MAPK phosphorylation. No glomerular p38 MAPK phosphorylation was observed in VEH pod GC-A cKO mice (Fig. 5d). Administration of aldosterone much increased phosphorylation of p38 MAPK, particularly in podocytes in pod GC-A cKO mice compared to control mice (Fig. 5d). Phosphorylation of p38 MAPK in glomeruli was reduced by administration of FR167653 (Fig. 5d).

(a,b) Representative images of nephrin (a) and podocin (b) expression by immunofluorescent study in a juxtamedullary glomerulus. Scale bars, 50 μm. (c) Transmission electron microscopic microphotographs of a juxtamedullary glomerulus. ALDO pod GC-A cKO mice exhibited mild thickening GBM and foot process effacement and its injury was ameliorated with the administration of FR167653. (d) Immunohistochemical staining for phosphorylated p38 MAPK. Arrows, phospho-p38 MAPK positive cells. Scale bars, 50 μm.
We next analyzed glomerular mRNA expression levels. As a previous report showed that natriuretic peptide counteracts aldosterone-induced renal injury10, we examined the expression level of Npr1 in this model. Administration of aldosterone upregulated Npr1 expression in both control mice and pod GC-A cKO mice, with no significant difference between the two groups (Fig. 6a). FR167653 treatment reduced Npr1 mRNA expression by 60% (Fig. 6a). We next examined the expression of Tgfb1 and Fn1, because fibronectin (Fn1) is one of main extracellular matrix in the kidney and these gene expression is relevant to extracellular matrix accumulation. The expression of extracellular matrix-related gene, such as Tgfb1 and Fn1, was enhanced with administration of aldosterone both in control mice and pod GC-A cKO mice, and was ameliorated by treatment with FR167653 (Fig. 6b,c). We examined gene expression relevant to macrophages: Ccl2 (MCP1) is one of macrophage chemoattractant molecules, and Adgre1 (F4/80) is one of markers of macrophages. Expression patterns of Ccl2 and Adgre1 were similar to expression levels of Tgfb1 and Fn1 (Fig. 6d,e). We next examined the expression of oxidative stress-related genes and downstream target genes of p38 MAPK. Aldosterone also upregulated Cybb and Trp53 (p53) mRNA levels in both control mice and pod GC-A cKO mice, and FR167653 ameliorated these changes (Fig. 6f,g).

Real-time RT-PCR analysis of Npr1 (a; GC-A), Tgfb1 (b; TGF-β1), Fn1 (c; Fibronectin), Ccl2 (d; MCP-1), Adgre1 (e; F4/80), Cybb (f; Cybb), Trp53 (g; p53) are shown. Mean ± SEM. ††p < 0.01, vs. VEH control mice. ‡p < 0.05, vs. VEH control mice. ‡‡p < 0.01 vs. VEH pod GC-A cKO mice. ¶¶p < 0.01 vs. ALDO pod GC-A cKO mice.
Next, we performed TUNEL assays to evaluate podocyte apoptosis. The number of TUNEL-positive cells in the juxtamedullary glomeruli was increased in ALDO pod GC-A cKO mice (Fig. 7a). Treatment with FR167653 completely abolished TUNEL-positive cells in the glomeruli of ALDO pod GC-A cKO mice (Fig. 7a). The number of WT1-positive cells in superficial glomeruli was not different among groups (Supplementary Fig S6). The number of WT1-positive cells in the juxtamedullary glomeruli of VEH pod GC-A cKO mice did not differ from VEH control mice (Fig. 7b,c). Administration of aldosterone decreased the number of WT1-positive cells in pod GC-A cKO mice compared with pod GC-A cKO mice that were not treated with aldosterone, and this decrease tended to be ameliorated by FR167653 (Fig. 7b,c).

(a) The number of TUNEL-positive cells was increased in ALDO pod GC-A cKO mice. Administration of FR167653 decreased the number of TUNEL-positive cells in ALDO pod GC-A cKO mice. (b,c) Administration of aldosterone decreased the number of WT1-positive cells in ALDO pod GC-A cKO mice. Inhibition of p38 MAPK maintained the number of WT1-positive cells. Mean ± SEM. ‡p < 0.05 vs. VEH pod GC-A cKO mice. Scale bar, 50 μm.
Natriuretic peptides inhibit phosphorylation of p38 MAPK via activating MKP-1
We next investigated whether ANP can inhibit expression of apoptosis-related proteins induced by activation of p38 MAPK in cultured human podocytes. ANP was administered to the human podocytes transfected with plasmids expressing wild-type MKK3 (MKK3) or constitutive active MKK3 (CA) (Fig. 8a). Phosphorylation of p38 MAPK was increased with following transfection of MKK3 or CA plasmids, and this increase was abolished not by treatment with ANP in MKK3-transfected podocytes, but by treatment with ANP in CA-transfected podocytes (Fig. 8b,c). Expression of Bax was not significantly different in MKK3-transfected podocytes, but significantly upregulated in cultured podocytes transfected with CA, and this increase was ameliorated only with high dose ANP (Fig. 8d). Consistent with this result, Bcl2 mRNA expression was reduced by transfection with CA, and ANP ameliorated this effect in cultured podocytes (Fig. 8e). As a result, the Bax/Bcl2 ratio was increased following transfection with MKK3 or CA plasmids, and this increase was ameliorated with ANP (Fig. 8f). To confirm the mechanism of ANP-mediated inhibition of p38 MAPK phosphorylation, we examined the phosphorylation status of MAPK phosphatase-1 (MKP-1) which dephosphorylates p38 MAPK, in cultured podocytes (Fig. 8g). Administration of ANP increased phosphorylation of MKP-1 in cultured human podocytes (Fig. 8h,i), indicating that ANP dephosphorylates p38 MAPK via activation of MKP-1.

(a) Experimental protocol. We used (1) empty plasmids, (2) plasmids expressing MKK3 (MKK3; pRc/RSV Flag MKK3), or (3) plasmids expressing constitutive active MKK3 (CA; pRc/RSV Flag MKK3 (glu)). MKK3 is an upstream kinase of p38 MAPK. ANP was exchanged at every 24 h. (b,c) Western blotting of phosphorylation of p38 MAPK and total p38 MAPK in human podocytes by transfection with empty, MKK3 or CA plasmids with or without ANP at 48 h after transfection. (n = 4, each group). Mean ± SEM. **p < 0.01 vs. human podocytes transfected with empty plasmids without ANP, ¶p < 0.05 vs. CA plasmids without ANP. (d–f) Real-time RT-PCR analysis for Bax (d), Bcl2 (e) were examined (n = 6, each group), and Bax/Bcl2 ratio are shown (f). Mean ± SEM. **p < 0.01, *p < 0.05 vs. human podocytes transfected with empty plasmids without ANP, ††p < 0.01 vs. MKK3 plasmids without ANP, ¶¶p < 0.01 vs. CA plasmids without ANP. (g) Phosphorylation of MKP-1 in human cultured podocytes stimulated with ANP. (h,i) ANP increased phosphorylation of MKP-1 (arrow) in human podocytes. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as control. Mean ± SEM. **p < 0.01 vs. human podocytes with vehicle. M, MagicMark XP Western Protein standard, a molecular weight marker.
Discussion
The present study demonstrated that blockade of p38 MAPK ameliorates glomerular injury in ALDO systemic GC-A KO mice. As we previously reported, ALDO wild-type mice show mild albuminuria, a slight increase in mesangial area, and mild podocyte injury; however, ALDO systemic GC-A KO mice show massive albuminuria with prominent mesangial expansion and podocyte injury10. One of the differences between ALDO wild-type and ALDO systemic GC-A KO mice was the phosphorylation status of p38 MAPK, because these two types of mice were found to have similar mRNA expression of inflammation, profibrotic, and oxidative stress genes10. The present study clearly demonstrated that the blockade of p38 MAPK reduced albuminuria, mesangial expansion, podocyte injury and apoptosis of glomerular cells in ALDO systemic GC-A mice. Previous studies have reported that blockade of p38 MAPK reduces glomerular injury in several experimental glomerulopathy models, including adriamycin nephropathy12, complement-mediated glomerular injury17, anti-GBM glomerulonephritis13, lupus nephritis18, thrombotic microangiopathy19, and diabetic nephropathy20. However, the role of p38 MAPK activation in response to aldosterone has yet to be fully elucidated, especially in the glomeruli. The present study has unveiled the novel pathway of aldosterone-induced glomerular injury. Aldosterone has been shown to promote apoptosis in many cell types, including podocytes through phosphoinositide-3/Akt, p38 MAPK and p53 pathways in vitro21. However, the role of p38 MAPK in aldosterone-induced podocyte apoptosis remains elusive. The present study demonstrates that blockade of p38 MAPK ameliorates apoptosis in podocytes, potentially indicating the mechanism by which MAPK signaling reduces podocyte injury.
Interestingly, administration of FR167653 reduced systolic blood pressure in ALDO systemic GC-A KO mice by 20 mmHg, although it had no effect on systolic blood pressure in ALDO wild-type mice. ALDO systemic GC-A KO mice had similar levels of serum aldosterone, and daily urinary sodium excretion, suggesting that the reduction in systolic blood pressure was not due to increased sodium excretion. Other possible explanations are changes in blood vessel tone and cardiac output. Tojo et al. reported that administration of FR167653 to Dahl salt-sensitive rats reduces expression of interleukin-1β and tumor necrosis factor-α in the whole kidney without altering blood pressure22. However, Potthoff et al. recently reported that chronic p38 MAPK inhibition improves vascular function and reduces systolic blood pressure in angiotensin II-stimulated hypertension in mice23. Interestingly, they also reported that non-treated mice do not exhibit blood pressure reduction23, consistent with our results.
Kuhn et al. reported that BNP activates phosphorylation of p38 MAPK in microvascular endothelial cells24, a finding contradictory to our results. However, phosphorylation of p38 MAPK is context-dependent rather than ligand-dependent. Phosphorylation of p38 MAPK is prominent in podocytes in ALDO systemic GC-A KO mice; therefore, decreased p38 MAPK activity in response to FR167653 appears to function in the protection of podocytes. However, we were unable to exclude the effects of blood pressure reduction and the role of mesangial cells as well as endothelial cells in ALDO systemic GC-A KO mice administered FR167653. To overcome these limitations, we generated podocyte-specific GC-A cKO mice and administered aldosterone and high salt following uninephrectomy. Recently, Staffel et al. reported that uninephrectomized pod GC-A cKO mice with deoxycorticosterone acetate and high salt diet exhibit similar blood pressure and increased albuminuria compared with control mice, confirming the protective role of natriuretic peptides/GC-A pathway in podocytes25. We also constructed pod GC-A cKO mice and demonstrated increased urinary albumin and glomerular injury without blood pressure elevation in ALDO pod GC-A cKO mice. Especially, juxtamedullary glomeruli rather than superficial glomeruli exhibited prominent mesangial expansion and podocyte injury in ALDO pod GC-A cKO mice. We speculate that hemodynamic changes will be the important factor on glomerular injury in ALDO pod GC-A cKO mice, because aldosterone concentration should be same between superficial and juxtamedullary glomeruli. Administration of FR167653 to ALDO pod GC-A cKO mice ameliorated albuminuria, mesangial expansion, loss of nephrin and podocin, and podocyte foot process effacement, suggesting that podocytes are one of the main targets of FR167653. Several studies showed genetic deletion of a specific molecule in podocytes affects mesangial expansion26,27, probably because of paracrine effects between podocytes and mesangial cells. However, GC-A was localized in mesangial cells28 and endothelial cells29 as well as in podocytes. In addition, urinary albumin excretion of ALDO pod GC-A cKO mice was much less than that of ALDO systemic GC-A KO mice, although ALDO pod GC-A cKO mice showed lower SBP. These finding may indicate the importance of mesangial or endothelial cells in the signaling of natriuretic peptides. This is the limitation of this study.
Aldosterone has been shown to phosphorylate p38 MAPK in many cell types, including vascular smooth muscle cells30, cardiomyocytes31, neutrophils32, peritoneal mesothelial cells33, and podocytes21. However, the relationship between natriuretic peptides and phosphorylation of p38 MAPK is cell-type and context-dependent. Previous studies have reported that natriuretic peptides activates p38 MAPK in hypoxia-exposed hepatocytes34, lung endothelial cells24 and brown adipocytes35. In contrast, other reports, including our previous study, have demonstrated that natriuretic peptides abolish p38 MAPK activation induced by inflammatory stimuli in human umbilical endothelial cells36, bovine pulmonary microvascular endothelial cells37, fibroblasts38 and podocytes10. The mechanism by which natriuretic peptides reduce phosphorylation of p38 MAPK has yet to be fully evaluated; however, a study has reported that natriuretic peptides augment MKP-1, which dephosphorylates p38 MAPK, in endothelial cells39. Consistent with this study, the present study also demonstrated the MKP-1 phosphorylation following treatment of podocytes with ANP. These results indicate that deletion of GC-A enhances phosphorylation of p38 MAPK in the kidney following stimulation with aldosterone.
Previous studies have reported that aldosterone induces apoptosis in mesangial cells40, endothelial cells41, renal tubular cells42, and podocytes43 in the kidney. Natriuretic peptides have been documented to have activity against apoptosis in cells in renal cortex and medulla in SHR rats44, macrophages45, cardiomyocytes in db/db mice46, and hepatocytes47. Conversely, other reports have shown that ANP induces apoptosis48,49. However, the contribution of natriuretic peptides to apoptosis in podocytes remains unclear. In the present study, we demonstrate that natriuretic peptides inhibit Bax expression, leading to decreased levels of apoptosis.
In conclusion, inhibition of p38 MAPK pathway ameliorates glomerular injury in uninephrectomized, high-salt fed, and aldosterone-infused mice by protecting against podocyte injury. These results indicate that p38 MAPK inhibitors may represent promising therapeutic targets against renal injury.
Methods
Reagents and antibodies
Aldosterone was obtained from Sigma Aldrich (St. Louis, MO). FR167653, p38α MAP Kinase inhibitor, was kindly provided by Astellas Pharma Inc. (Tokyo, Japan). Primary antibodies used for immunohistochemical studies and Western blotting were goat anti-nephrin (R&D Systems, Minneapolis, MN), rabbit anti-podocin (Sigma Aldrich), rabbit anti-phospho-p38 MAPK (Cell Signaling Technology, Boston, MA), rabbit total p38 MAPK (Cell Signaling Technology), rabbit anti-WT1 (Santa Cruz, Dallas, TX), and rabbit anti-p53 (Vector Laboratories, Burlingame, CA), rabbit anti-phospho-MKP-1 (Cell Signaling Technology), rabbit anti-phospho-MKK3 (Cell Signaling Technology), and mouse anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Santa Cruz) antibodies. pRc/RSV Flag MKK350 and pRc/RSV Flag MKK3 (glu)51 were gifts from Professor Roger Davis (Addgene plasmid #14671 and #14670, respectively).
Animal experiments
All animal experiments were performed in accordance with Fundamental Guidelines for Proper Conduct of Animal Experiment and Related Activities in Academic Research Institutions and were approved by the Animal Experimentation Committee of Kyoto University Graduate School of Medicine. Male systemic GC-A KO mice52 or their wild-type littermates received left uninephrectomy under intraperitoneal pentobarbital anesthesia and osmotic minipump (ALZET 2004, Cupertino, CA) implantation subcutaneously to infuse aldosterone (0.2 μg/kg body weight per minute) or vehicle. All mice were fed a diet containing 6% NaCl10. FR167653 was administered by dissolving it in drinking water12. Blood pressure was measured indirectly by using the tail-cuff method (MK-2000ST; Muromachi Kikai, Tokyo, Japan) at −2, 0, 1, 2, and 4 weeks. Urine samples were collected from metabolic cages (Shinano manufacturing, Tokyo, Japan) for 24 h at −2, 0, 1, 2, and 4 weeks and urine volumes were measured. Blood and kidney samples were harvested at 4 weeks. Urinary albumin levels were measured with albumin ELISA kits (Exocell, Philadelphia, PA). Serum and urinary Cr, sodium, and potassium levels were measured by enzymatic methods (SRL, Tokyo, Japan).
Construction of GC-A floxed mice and nephrin-promotor driven Cre recombinase transgenic mice were as described previously53,54. By crossing GC-A floxed mice (C57BL/6 J background) with Nephrin Cre mice (C57BL/6 J background), we generated pod GC-A cKO mice (Npr1fl/fl, Nephrin Cre/+mice) and control littermates (Npr1fl/fl,+/+). Male pod GC-A cKO mice or control mice at 12 weeks of age were uninephrectomized, and administered with 6% NaCl diet and aldosterone or vehicle using the same protocol for systemic GC-A KO mice.
Renal histology and electron microscopy
Histological and electron microscopic examinations were performed as described previously55. Periodic acid-Schiff (PAS) stained kidney samples were examined by light microscopy. Mesangial areas in juxtamedullary glomeruli were measured using MetaMorph Software (Molecular Devices, Sunnyvale, CA). Electron microscopic examination was performed using an electron microscope (H-7600, Hitachi, Tokyo, Japan).
Immunohistochemistry
Immunofluorescent studies for nephrin and podocin were performed as descried previously10. Briefly, cryostat sections were incubated with goat anti-nephrin antibody or rabbit anti-podocin antibody, and then incubated with fluorescein isothiocyanate (FITC)-labeled secondary antibody. Immunohistochemical analysis for phospho-p38 MAPK, WT1, p53, phospho-MKK3 were as previously described with some modifications10. Staining for the presence of apoptotic cells was performed using in situ cell death detection kits (Roche, Mannheim, Germany). Paraffin-embedded renal sections were reacted with fluorescent dyed mixed solutions.
Cell culture and transfection
Conditionally immortalized human podocytes were provided by Dr. Moin Saleem, and cultured and differentiated as described previously56,57. Briefly, cells were cultured with RPMI 1640 medium (Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS; Biowest, Nuaille, France) and insulin-transferrin-selenite supplement (LifeTechnologies, Carlsbad CA). Differentiated podocytes were transfected with pRc/RSV Flag MKK3, pRc/RSV Flag MKK3 (glu) or empty plasmid using Nucleofector Kit for primary Mammalian Epithelia cells (Lonza, Basel, Switzerland) as described previously57. Transfected cells were incubated with vehicle or ANP, with the exchange of the medium containing ANP at every 24 h, and then harvested for RNA and protein analysis at 48 h. To confirm phosphorylation of MKP-1, differentiated podocytes were treated with ANP for 10 min and then harvested.
Glomerular RNA, protein extraction and real-time RT-PCR
Glomeruli were isolated by the graded sieving methods as described previously10. RNA and protein extraction was performed using AllPrep DNA/RNA/protein kits (QIAGEN, Hilden, Germany). Quantitative real-time PCR was performed using StepOnePlus System (Thermo Fischer Scientific, Waltham, MA), as descried previously58. Npr1, Tgfb1, Ccl2, Fn1, Cybb, Adgre1, Trp53, Bax, and Bcl2 mRNA expression levels were evaluated. Some primer and probe sets were described previously10. Adgre1 forward primer, 5′-tggtggtcataatctctgcttctg-3′; Adgre1 reverse primer, 5′-agacaggccccaggaaactc-3′; Adgre1 probe, 5′-FAM-cccgtctctgtattcaaccagcagcgatt-TAMRA-3′; Trp53 forward primer, 5′-gcttctccgaagactggatgac-3′; Trp53 reverse primer, 5′-gcagtgaggtgatggcagg-3′; Trp53 probe primer, 5′-FAM-tcacagtcggatatcagcctcgagctcc-TAMRA-3′; Bax forward primer, 5′-aactggtgctcaaggccct-3′; Bax reverse primer, 5′-cccggaggaagtccaatgtc-3′; Bax probe primer, 5′-tgcaccaaggtgccggaactgatca-TAMRA-3′; Bcl2 forward primer, 5′-gatgggatcgttgccttatgca-3′; Bcl2 reverse primer, 5′-cagtctacttcctctgtgatgttgta-3′; 5′-cagcatgatcctctgtcaagtttcctt-TAMRA-3′, Bcl2 probe primer, 5′-FAM-cagcatgatcctctgtcaagtttcctt -TAMRA-3′.
Western blotting
Western blotting was performed as previously described10. Filters on isolated cell extracts were incubated with rabbit anti-phospho p38 MAPK antibody, rabbit anti-p38 MAPK antibody, rabbit anti-phospho MKP-1 antibody, or mouse anti-GAPDH antibody. Immunoblot were developed using horseradish peroxidase-linked donkey anti-rabbit or anti-mouse antibody and a chemiluminescent kit.
Statistical Analysis
Data are expressed as the mean ± SE. Statistical analysis was performed using ANOVA or Student’s t test as appropriate.
Additional Information
How to cite this article: Kato, Y. et al. Natriuretic peptide receptor guanylyl cyclase-A pathway counteracts glomerular injury evoked by aldosterone through p38 mitogen-activated protein kinase inhibition. Sci. Rep. 7, 46624; doi: 10.1038/srep46624 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Brown, J. N. Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nat. Rev. Nephrol. 9, 459–469 (2013).
Shibata, S. et al. Podocyte as the target for aldosterone: roles of oxidative stress and Sgk1. Hypertension 49, 355–364 (2007).
Harada, E. et al. Aldosterone induces angiotensin-converting-enzyme gene expression in cultured neonatal rat cardiocytes. Circulation 104, 137–139 (2001).
Nishiyama, A. et al. Possible contributions of reactive oxygen species and mitogen-activated protein kinase to renal injury in aldosterone/salt-induced hypertensive rats. Hypertension 43, 841–848 (2004).
Nagase, M. & Fujita, T. Aldosterone and glomerular podocyte injury. Clin. Exp. Nephrol. 12, 233–242 (2008).
Terada, Y. et al. Aldosterone stimulates proliferation of mesangial cells by activating mitogen-activated protein kinase 1/2, cyclin D1, and cyclin A. J. Am. Soc. Nephrol. 16, 2296–2305 (2005).
Oberleithner, H. Aldosterone makes human endothelium stiff and vulnerable. Kidney Int. 67, 1680–1682 (2005).
Remuzzi, G., Cattaneo, D. & Perico, N. The aggravating mechanisms of aldosterone on kidney fibrosis. J. Am. Soc. Nephrol. 19, 1459–1462 (2008).
Nakao, K., Ogawa, Y., Suga, S. & Imura, H. Molecular biology and biochemistry of the natriuretic peptide system. I: Natriuretic peptides. J. Hypertens. 10, 907–912 (1992).
Ogawa, Y. et al. Natriuretic peptide receptor guanylyl cyclase-A protects podocytes from aldosterone-induced glomerular injury. J. Am. Soc. Nephrol. 23, 1198–1209 (2012).
Kumar, S., Boehm, J. & Lee, C. J. p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat. Rev. Drug. Discov. 2, 717–726 (2003).
Koshikawa, M. et al. Role of p38 mitogen-activated protein kinase activation in podocyte injury and proteinuria in experimental nephrotic syndrome. J. Am. Soc. Nephrol. 16, 2690–2701 (2005).
Stambe, C. et al. Blockade of p38α MAPK ameliorates acute inflammatory renal injury in rat anti-GBM glomerulonephritis. J. Am. Soc. Nephrol. 14, 338–351 (2003).
Sheryanna, A. et al. Inhibition of p38 mitogen-activated protein kinase is effective in the treatment of experimental crescentic glomerulonephritis and suppresses monocyte chemoattractant protein-1 but not IL-1β or IL-6. J. Am. Soc. Nephrol. 18, 1167–1179 (2007).
Pippin, W. J. et al. DNA damage is a novel response to sublytic complement C5b-9-induced injury in podocytes. J. Clin. Invest. 111, 877–885 (2003).
Derijard, B. et al. Independent human MAP-kinase signal transduction pathways defined by MEK and MKK isoforms. Science 267, 682–685 (1995).
Aoudjit, L., Stanciu, M., Li, H., Lemay, S. & Takano, T. p38 mitogen-activated protein kinase protects glomerular epithelial cells from complement-mediated cell injury. Am. J. Physiol. Renal Physiol. 285, F765–774 (2003).
Iwata, Y. et al. p38 mitogen-activated protein kinase contributes to autoimmune renal injury in MRL-Faslpr mice. J. Am. Soc. Nephrol. 14, 57–67 (2003).
Fu, J. X. et al. Role of p38 MAP kinase pathway in a toxin-induced model of hemolytic uremic syndrome. Pediatr. Nephrol. 19, 844–852 (2004).
Peng, L. et al. The protective effect of beraprost sodium on diabetic nephropathy by inhibiting inflammation and p38 MAPK signaling pathway in high-fat diet/streptozotocin-induced diabetic rats. Int. J. Endocrinol. 2016, 1690474 (2016).
Chen, C. et al. Aldosterone induces apoptosis in rat podocytes: role of PI3-K/Akt and p38MAPK signaling pathways. Nephron Exp. Nephrol. 113, e26–34 (2009).
Tojo, A. et al. Antioxidative effect of p38 mitogen-activated protein kinase inhibitor in the kidney of hypertensive rat. J. Hypertens. 23, 165–174 (2005).
Potthoff, A. S. et al. Chronic p38 mitogen-activated protein kinase inhibition improves vascular function and remodeling in angiotensin II-dependent hypertension. J. Renin Angiotensin. Aldosterone Syst. 17, 1470320316653284 (2016).
Kuhn, M. et al. The natriuretic peptide/guanylyl cyclase-A system functions as a stress-responsive regulator of angiogenesis in mice. J. Clin. Invest. 119, 2019–2030 (2009).
Staffel, J. et al. Natriuretic peptide receptor guanylyl cyclase-A in podocytes is renoprotective but dispensable for physiologic renal function. J. Am. Soc. Nephrol. 28, 260–277 (2017).
Lenoir, O. et al. Direct action of endothelin-1 on podocytes promotes diabetic glomerulosclerosis. J. Am. Soc. Nephrol. 25, 1050–1062 (2014).
Mariya, T. et al. Notch1 and Notch2 in podocytes play differential roles during diabetic nephropathy development. Diabetes 64, 4099–4111 (2015).
Tripathi, S. & Pandey, K. N. Guanylyl cyclase/natriuretic peptide receptor-A signaling antagonizes the vascular endothelial grwoth factor-stimulated MAPKs and downstream effectors AP-1 and CREB in mouse mesangial cells. Mol. Cell. Biochem. 368, 47–59 (2012).
Chen, W. et al. Atrial natriuretic peptide enhances microvascular albumin permeability by the caveolae-mediated transcellular pathway. Cardiovasc Res 93, 141–151 (2012).
Callera, E. G. et al. Aldosterone activates vascular p38MAP kinase and NADPH oxidase via c-Src. Hypertension 45, 773–779 (2005).
Lopez-Andres, N., Inigo, C., Gallego, I., Diez, J. & Fortuno, A. M. Aldosterone induces cardiotrophin-1 expression in HL-1 adult cardiomyocytes. Endocrinology 149, 4970–4978 (2008).
Walczak, C. et al. Aldosterone increases VEGF-A production in human neutrophils through PI3K, ERK1/2 and p38 pathways. Biochim. Biophys. Acta. 1813, 2125–2132 (2011).
Yu, M. et al. Effect of aldosterone on epithelial-to-mesenchymal transition of human peritoneal mesothelial cells. Kidney Res. Clin. Pract. 34, 83–92 (2015).
Carini, R. et al. Mechanisms of hepatocyte protection against hypoxic injury by atrial natriuretic peptide. Hepatology 37, 277–285 (2003).
Bordicchia, M. et al. Cardiac natriuretic peptides act via p38 MAPK to induce the brown fat thermogenic program in mouse and human adipocytes. J. Clin. Invest. 122, 1022–1036 (2012).
Kiemer, K. A. et al. Inhibition of p38 MAPK activation via induction of MKP-1: atrial natriuretic peptide reduces TNF-α-induced actin polymerization and endothelial permeability. Circ. Res., 90, 874–881 (2002).
Irwin, C. D., Patot Tv, C. M, Tucker, A. & Bowen, R. Direct ANP inhibition of hypoxia-induced inflammatory pathways in pulmonary microvascular and macrovascular endothelial monolayers. Am. J. Physiol. Lung Cell Mol. Physiol. 288, L849–L859 (2005).
Song, Z. et al. Recombinant human brain natriuretic peptide attenuates LPS-induced cellular injury in human fetal lung fibroblasts via inhibiting MAPK and NF-κB pathway activation. Mol. Med. Rep. 14, 1785–1790 (2016).
Furst, R. et al. Atrial natriuretic peptide induces mitogen-activated protein kinase phosphatase-1 in human endothelial cells via Rac1 and NAD(P)H oxidase/Nox2-activation. Circ. Res. 96, 43–53 (2005).
Shi, H., Zhang, A., He, Y., Yang, M. & Gan, W. Effects of p53 on aldosterone-induced mesangial cell apoptosis in vivo and in vitro . Mol. Med. Rep. 13, 5102–5108 (2016).
Qiao, W., Zhang, W., Shao, S., Gai, Y. & Zhang, M. Effect and mechanism of poly (ADP-ribose) polymerase-1 in aldosterone-induced apoptosis. Mol. Med. Rep. 12, 1631–1638 (2015).
Kitada, K. et al. Aldosterone induces p21-regulated apoptosis via increased synthesis and secretion of tumour necrosis factor-α in human proximal tubular cells. Clin. Exp. Pharmacol. Physiol. 39, 858–863 (2012).
Yuan, Y. et al. The roles of oxidative stress, endoplasmic reticulum stress, and autophagy in aldosterone/mineralocorticoid receptor-induced podocyte injury. Lab. Invest. 95, 1374–1386 (2015).
Romero, M. et al. Chronic treatment with atrial natriuretic peptide in spontaneously hypertensive rats: beneficial renal effects and sex differences. PLoS One 10, e0120362 (2015).
Chang, C. et al. The inhibition of oxidised low-density lipoprotein-induced apoptosis of macrophages by recombinant human brain natriuretic peptide and the underlying mechanism. Cardiology 132, 137–146 (2015).
Plante, E. et al. Treatment with brain natriuretic peptide prevents the development of cardiac dysfunction in obese diabetic db/db mice. Diabetologia 57, 1257–1267 (2014).
Yamada, T., Kotake, Y., Nagata, H. & Takeda, J. Atrial natriuretic peptide reduces hepatic ischemia-reperfusion injury in rabbits. J. Anesth. 27, 901–908 (2013).
Wu, F. C., Bishopric, H. N. & Pratt, E. R. Atrial natriuretic peptide induces apoptosis in neonatal rat cardiac myocytes. J. Biol. Chem. 272, 14860–14866 (1997).
Suenobu, N., Shichiri, M., Iwashina, M., Marumo, F. & Hirata, Y. Natriuretic peptides and nitric oxide induce endothelial apoptosis via a cGMP-dependent mechanism. Arterioscler. Thromb. Vasc. Biol. 19, 140–146 (1999).
Derijard B. et al. Independent human MAP-kinase signal transduction pathways defined by MEK and MKK isoforms. Science 267, 682–685 (1995).
Raingeaud, J. et al. MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol. Cell Biol. 16, 1247–1255 (1996).
Lopez, J. M. et al. Salt-resistant hypertension in mice lacking the guanylyl cyclase-A receptor for atrial natriuretic peptide. Nature 378, 65–68 (1995).
Nojiri, T. et al. Atrial natriuretic peptide prevents cancer metastasis through vascular endothelial cells. Proc. Natl. Acad. Sci. USA 112, 4086–4091 (2015).
Asano, T. et al. Permanent genetic tagging of podocytes: fate of injured podocytes in a mouse model of glomerular sclerosis. J. Am. Soc. Nephrol. 16, 2257–2262 (2005).
Yokoi, H. et al. Overexpression of connective tissue growth factor in podocytes worsens diabetic nephropathy in mice. Kidney Int. 73, 446–455 (2008).
Saleem, A. M. et al. A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J. Am. Soc. Nephrol. 13, 630–638 (2002).
Koga, K. et al. MicroRNA-26a inhibits TGF-β -induced extracellular matrix protein expression in podocytes by targeting CTGF and is downregulated in diabetic nephropathy. Diabetologia 58, 2169–2180 (2015).
Yokoi, H. et al. Pleiotrophin triggers inflammation and increased peritoneal permeability leading to peritoneal fibrosis. Kidney Int. 81, 160–169 (2012).
Acknowledgements
This work was supported in part by research grants from JSPS KAKENHI (Grant Numbers 21790806, 25461246, 26461225), the Integration Research for Agriculture and Interdisciplinary Fields from the National Agriculture and Food Research Organization, and Japan Agency for Medical Research and Development (AMED; Grant Number 15652238). We gratefully acknowledge M. Fujimoto, Y. Sakashita, C. Kimura, N. Igarashi, E. Nishimura, and T. Nagase in Department of Nephrology, Kyoto University Graduate School of Medicine and other lab members for technical assistance, and A. Yamamoto, S. Ogino and Y. Mizukami in the Department of Nephrology, Kyoto University Graduate School of Medicine for secretarial assistance.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: Y.K., K.M., M.K., A.I., K.P.M., N.T., S.O., T.K., K.N., M.M., M.Y., H.Y. Performed the experiments and interpretation in results: Y.K., K.O., T.T., I.K., M.A.S., T.M., H.Y. Wrote a paper: Y.K., H.Y. Y.K. and H.Y. are the guarantors of this work and had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Kato, Y., Mori, K., Kasahara, M. et al. Natriuretic peptide receptor guanylyl cyclase-A pathway counteracts glomerular injury evoked by aldosterone through p38 mitogen-activated protein kinase inhibition. Sci Rep 7, 46624 (2017). https://doi.org/10.1038/srep46624
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep46624
This article is cited by
-
Current and Emerging Classes of Pharmacological Agents for the Management of Hypertension
American Journal of Cardiovascular Drugs (2022)
-
Osteocrin ameliorates adriamycin nephropathy via p38 mitogen-activated protein kinase inhibition
Scientific Reports (2021)
-
Novel therapeutics for the treatment of hypertension and its associated complications: peptide- and nonpeptide-based strategies
Hypertension Research (2021)
-
Low-dose atrial natriuretic peptide for prevention or treatment of acute kidney injury: a systematic review and meta-analysis
Critical Care (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
