
Abstract
Nine new bicyclic cembranoids, sarcophytrols M–U(1–9), were isolated from the South China Sea soft coral Sarcophyton trocheliophorum as minor components, along with one known related cembranoid 10. Their structures were elucidated by detailed spectroscopic analysis and chemical conversion. The chemical structures of these metabolites are characterized by the different patterns of the additional cyclization within the 14-member skeleton, which leading to the formation of furan, pyran, oxepane, and peroxyl rings, respectively. Among them, sarcophytrols R and S(6 and 7) share a rare decaryiol skeleton with an unusual C12/C15 cyclization. In addition, the absolute configurations of sarcophytrols M and T(1 and 8) were determined by the modified Mosher’s method. The research of these new secondary metabolites provided a further understanding of the diversity of cyclized cembranoids from the title species.
Similar content being viewed by others
Introduction
Cyclization is an extraordinary artistry that nature turn the simple cembranoids to a prodigious variety of structurally novel compounds, and it often links to a network of oxygenation process1, which lead to the formation of epoxyl2,3,4,5, furan4,6,7,8, pyran5,7,8, and oxepane5,9,10. Among them, the decaryiol-type cembranoids, characterized by a 6:14-fused ring system, are one of the most amazing examples. They are biogenetically derived from cembranoids by an uncommon transannular etherfication between C-12 and C-15 position and rarely discovered in nature. Actually, there are only four decaryiol-type cembranoids, decaryiols A–D, that have been reported from soft coral Sarcophyton decaryi11 and Lobophytum sp12. before.
It is widely recognized that the South China Sea soft coral S. trocheliophorum(phylum Cnidaria, class Anthozoa, subclass Octocorallia, order Alcyonacea, family Alcyoniidae) contains unusual cembranoids with a diversity of cyclizations13. Interestingly, these cembranoids are regarded as chemical defense compounds against predators such as other corals and fishes as well as against settlements of microorganisms14,15. In our ongoing studies of the chemistry and biology of the Hainan soft corals, we have considered the soft coral S. trocheliophorum as an important issue. A previous study we conducted on a collection of the title animal from Hainan had resulted in the isolation of a series of new cembranoids and cembranoid derivatives3,4,9,16. Many of these new secondary metabolites with different patterns of cyclizations showed significant inhibitory activity against human protein tyrosine phosphatase 1B(PTP1B) enzyme3,4,16, a promising drug target for the treatment of type 2 diabetes and obesity17. To accumulate these compounds for further biological study, we made a different collection of the same species from the same location(Yalong Bay, Hainan Province). Surprisingly, our chemical investigation on the crude acetone extract of the title animal showed the absence of aforementioned compounds existed in the former collection, while resulting to the discovery of three new capnosane diterpenoids18. We have now focused our attentions on the cembrane-type metabolites with diverse kinds of cyclizations from the latter collection to find more chemically interesting and biologically active compounds. This continuous investigation has now resulted in the isolation of nine new cembranoids(1–9), together with one known related cembranoid 10(Fig. 1). Among them, the characteristic chemical features of them are the diverse types of cyclized rings: furan rings possessed by sarcophytrols M–P(1–4), pyran rings formed at different positions in sarcophytrols Q–S(5–7), while oxepane and peroxyl rings appeared in sarcophytrols T(8) and U(9), respectively. In addition, sarcophytrols R and S(6 and 7) share a rare decaryiol skeleton with an unusual C12/C15 cyclization. We herein report the isolation and structure elucidation of these new cembranoids.

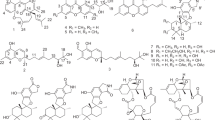
Structures of compounds 1–13.
Results and Discussion
Samples of S. trocheliophorum(dry weight 400 g) were extracted exhaustively with acetone, and the extract was partitioned between water and Et2O. The Et2O solvable fraction was subjected to repeated chromatography as usual work3,4,9,16, to afford ten pure metabolites, compounds 1–10(Fig. 1). A preliminary NMR analysis revealed that all the new molecules shared the same cembrane skeleton. Among them, the known compound was readily identified as sarglaucol(10)19 by comparison of its spectral data and [α]D values with those reported in the literatures.
The HRESIMS of sarcophytrol M(1) established the molecular formula C20H34O4. 1H and 13C NMR spectra of 1(Tables 1 and 2) were reminiscent of a known cembranoid,(2E,7E)-4,11-dihydroxy-1,12-oxidocembra-2,7-diene(11), previously isolated from Sinularia ovispiculata20. The distinct difference between them was the presence of a hydroxyl at C-15(δC 72.4) in 1, which was further confirmed by the observation of HMBC correlations from H3-16(δH 1.12)/H3-17(δH 1.06) to C-15(Fig. 2). Similar to 11, a trans-disubstituted olefin at C-2/C-3 was recognized by the doublet coupling constant(15.4 Hz) of H-2(δH 5.62) and H-3(δH 5.92), while the chemical shift of Me-19(δC < 20 ppm) indicated the 7E assignment in 120,21. The relative configurations of the stereogenic centers were determined by NOE relationships(Fig. 2), which exhibited similar key cross-peaks to those in the cembranoids sinulariols D and F7. The NOE correlation between H-3 and H-11(δH 3.54) implied that the trans-disubstituted olefin and the hydroxyl group at C-11 were hind the same face toward the five-membered ring. Thus, Me-20 was oriented to the same face with the isopropyl group, and tentatively assigned to be α-oriented. In addition, the significant NOE correlations of H-2/H3-16 and H-2/H-14b(δH 1.67) suggested that H-2 and the isopropyl group to be on the same side of 1. The trans-disubstituted olefin group favoring ‘up’ or ‘down’-orientation toward the 14-membered ring, and thus H-3 was oriented to ‘up’ face. The configuration of H-11 was assigned as β mainly on the basis of the aforementioned strong NOE correlation between H-11 and H-3 and the very weak NOE correlation between H-11 and H3-20(δH 1.11). According to the NOE correlation from H-3 to H3-18(δH 1.27) and the lack of correlation from H-2 to H3-18, we tentatively assigned the configuration of H3-18 to β face. To obtain its absolute configuration, two aliquots of compound 1 were treated with(R)- and(S)-α-methoxy-α-trifluoromethylphenyl acetyl(MTPA) chlorides to obtain the(S)- and(R)-esters, respectively. Analysis of ΔδSR values(δS - δR) observed for the signals of the protons close to 11-OH(Fig. 3) indicated the S configuration at this carbon. Consequently, the absolute configuration of compound 1 was determined as(1S, 4S, 11S, 12R).

Selected key COSY, HMBC and ROESY correlations for compound 1.

The ΔδSR [Δ(δS-δR)] data for the MTPA esters of compounds 1 and 8.
Sarcophytrol N(2) has a molecular formula of C21H36O4, as established by HRESIMS and NMR data, 14 mass units more than that of 1. The 1H NMR data of 2(Tables 1 and 2) were closely reminiscent to those of 1, except for the newly appeared signal at δH 3.23(3H, s), which suggested the presence of an additional methoxyl group in 2, consistent with one carbon resonance at δC 50.6(q) in its 13C NMR spectrum. The introduction of the methoxyl group in 2 resulted in the significant downfield shift of C-15 from δC 72.4(s) in 1 to 78.4(s) in 2 and the upfield shifts of C-16/C-17. Furthermore, the methoxyl group was secured at C-15 by a diagnostic HMBC correlation from methoxyl group to C-15. Furthermore, ROESY correlations of compound 2 were similar with those of 1. On the basis of above evidences, compound 2 was identified as 15-methoxyl derivative of 1.
Sarcophytrol P(3) possesses a molecular formula of C22H36O5 as determined by HRESIMS data, 42 mass units more than that of 1. The NMR spectroscopic features of 3(Tables 1 and 2) mostly resembled those of 1. In fact, the only difference was at C-11 position, where the hydroxyl group(δH 3.54; δC 76.4) in 1 was replaced by an acetyl(δH 5.09, 2.04; δC 77.3, 21.3, 171.0) in 3. A detailed 2D NMR analysis further confirmed the planar structure of compound 3. As the patterns of ROESY correlations of 3 were similar with those of 1, accordingly the structure of 3 was established as the 11-acetyl derivative of 1. The chemical conversion of 1 into 3 by simple acetylation in Ac2O/pyridine further indicated the absolute configuration of 3 is the same as 1.
Sarcophytrol P(4) was isolated as an optically active colorless oil with molecular formula C20H34O4, the same as 1. The NMR spectra of 4(Tables 1 and 2) were strongly reminiscent of those of 1 and a careful 2D NMR analysis suggested that they share the same gross structure. In fact, the only difference was found in the segment from C-2 to C-5, where 13C NMR chemical shifts of C-3, C-4 and C-5 in 4 were all upfield shifted while that of γ-carbon C-2 was downfield shifted with respect of 1. This evidence clearly suggested that the relative configuration of the hydroxyl group at C-4 of 4 was different from that of 1. The NOE interactions between H-2(δH 5.55) and H3-18(δH 1.33) in 1 rather than H-3(δH 6.07) and H3-18 in 4 confirmed this assignment. Herein, 4 was 4-epimer of 1.
Sarcophytrol Q(5) possesses the same molecular formula C22H36O5 as 4, established by HRESIMS, whereas its NMR data(Tables 1 and 2) were almost identical to those of sinulariol Z(12)22. The distinction was attributed to the NMR data of 5 presenting an extra hydroxyl group at C-15, which was further confirmed by the observation of HMBC correlations from H3-16(δH 1.12)/H3-17(δH 1.07) to C-15(δC 74.6). Due to the presence of the 15-OH, 13C NMR chemical shifts of C-15, C-16, C-17, and C-1 were all downfield shifted reasonably while those of γ-carbons 2 and 14 were upfield shifted with respect of those of 12. Furthermore, the significant NOE interactions between H-3(δH 5.98) and H-11(δH 3.34) and the lack of NOE correlations of H-11/H3-16(δH 1.12), H-11/H3-17(δH 1.07), H-11/H3-20(δH 1.16) confirmed the isopropyl group and H3-20 were in opposite face against H-11 in the hexatomic ring. Additional strong NOE interaction between H-3 and H3-18 indicated H3-18 at the same side as H-3 in the case of 5. Thus, 5 had the same relative configuration relationships as those of 12, which were further confirmed by the closely identical spectral data for the segment C-3–C-13.
Sarcophytrol R(6) was obtained as a colorless, optically active oil {[α − 13.3(c 0.06, MeOH)}. HRESIMS and 13C NMR spectra analysis established the molecular formula of 6 as C20H34O4. Thus, four degrees of unsaturation were determined for 6. The NMR data(Tables 1 and 2) revealed the presence of two trisubstituted double bonds(δH 5.36, δC 146.33, 121.54; δH 5.30, δC 126.73, 135.48), which accounted for two degrees of unsaturation. The remaining two degrees of unsaturation strongly indicated that 6 has a bicyclic structure with a carbocyclic ring bridged by oxygen. The 13C NMR spectrum of 6 contained five oxygenated carbon signals at δC 74.04(CH), 76.80(C), 69.49(CH), 75.32(C) and 74.44(C), which were assigned by 2D NMR spectrum to C-3, C-4, C-11, C-12 and C-15, respectively. In order to define the carbon atoms linked to these functionalities, 2D NMR experiments(in DMSO-d6) of 6 were measured. The formation of an ether bridge across C-12 and C-15 was deduced from the observation of OH-3(δH 4.31), OH-4(δH 3.95) and OH-11(δH 4.10). The location of these three –OH groups are determined by the 1H-1H COSY correlations of H-3(δH 3.90) with OH-3 and H-11(δH 3.40) with OH-11, and the HMBC correlations from OH-4 to C-3(δC 72.9), C-4(δC 75.1), C-5(δC 38.1).
− 13.3(c 0.06, MeOH)}. HRESIMS and 13C NMR spectra analysis established the molecular formula of 6 as C20H34O4. Thus, four degrees of unsaturation were determined for 6. The NMR data(Tables 1 and 2) revealed the presence of two trisubstituted double bonds(δH 5.36, δC 146.33, 121.54; δH 5.30, δC 126.73, 135.48), which accounted for two degrees of unsaturation. The remaining two degrees of unsaturation strongly indicated that 6 has a bicyclic structure with a carbocyclic ring bridged by oxygen. The 13C NMR spectrum of 6 contained five oxygenated carbon signals at δC 74.04(CH), 76.80(C), 69.49(CH), 75.32(C) and 74.44(C), which were assigned by 2D NMR spectrum to C-3, C-4, C-11, C-12 and C-15, respectively. In order to define the carbon atoms linked to these functionalities, 2D NMR experiments(in DMSO-d6) of 6 were measured. The formation of an ether bridge across C-12 and C-15 was deduced from the observation of OH-3(δH 4.31), OH-4(δH 3.95) and OH-11(δH 4.10). The location of these three –OH groups are determined by the 1H-1H COSY correlations of H-3(δH 3.90) with OH-3 and H-11(δH 3.40) with OH-11, and the HMBC correlations from OH-4 to C-3(δC 72.9), C-4(δC 75.1), C-5(δC 38.1).
The E geometry of the Δ1 and Δ9 double bonds was deduced by the ROESY correlations of H-2(δH 5.36)/H3-17(δH 1.31) and of H-7(δH 5.30)/H2-9(δH 2.12)(Fig. 4). Moreover, H-14a(δH 2.85) correlated with H-3(δH 4.19) and H-11(δH 3.57), suggesting H-3 and H-11 to be on the same side of 6. When H-3 was assigned tentatively as β-orientation, H-11 was accordingly oriented in β-face. The presence of the cross-peak between H-2 and H3-18(δH 1.27) in combination with the absence of the correlations of H-3/H-2 and H-3/H3-18 in the ROESY spectrum, clarified H3-18 and H-2 to be oriented in opposite to H-3. Finally, the α-oriented H3-20 was tentatively deduced by the absence of correlations between H3-20 and H-11.

Selected key ROESY correlations for compounds 6 and 7.
Sarcophytrol S(7) was isolated as an optically active, colorless oil {[α − 39.3(c 0.07, MeOH)}. Its molecular formula, C20H32O3, was established by HRESIMS, 18 units less than that of 6. Careful comparison of NMR data of 7 and 6(Tables 1 and 2) revealed that the former differs from the latter only by the presence of a terminal methylene group(δH 5.13, 4.93; δC 111.37, 147.92) in 7 instead of a methyl(δH 1.27; δC 22.91) and a oxygenated quarternary carbon(δC 76.8) in 6, in agreement with 18 mass units difference between them, while the rest of the molecules was the same. Base on the HMBC correlations between H2-18(δH 5.13, 4.93) and C-3(δC 75.24) and C-5(δC 28.32), the terminal methylene group was located at C-4. Moreover, 1H-1H COSY, HSQC, and HMBC experiments allowed the unambiguous definition of the structure of 7. Analogously to 6, the relative stereochemistry of three chiral centers at C-3, C-11 and C-12 was elucidated to be the same as those of 6 by the ROESY experiments(Fig. 4).
− 39.3(c 0.07, MeOH)}. Its molecular formula, C20H32O3, was established by HRESIMS, 18 units less than that of 6. Careful comparison of NMR data of 7 and 6(Tables 1 and 2) revealed that the former differs from the latter only by the presence of a terminal methylene group(δH 5.13, 4.93; δC 111.37, 147.92) in 7 instead of a methyl(δH 1.27; δC 22.91) and a oxygenated quarternary carbon(δC 76.8) in 6, in agreement with 18 mass units difference between them, while the rest of the molecules was the same. Base on the HMBC correlations between H2-18(δH 5.13, 4.93) and C-3(δC 75.24) and C-5(δC 28.32), the terminal methylene group was located at C-4. Moreover, 1H-1H COSY, HSQC, and HMBC experiments allowed the unambiguous definition of the structure of 7. Analogously to 6, the relative stereochemistry of three chiral centers at C-3, C-11 and C-12 was elucidated to be the same as those of 6 by the ROESY experiments(Fig. 4).
The HRESIMS of sarcophytrol T(8) established the molecular formula C20H30O2, indicating six degrees of unsaturation. The presence of four double bonds(δH 6.09, δC 140.2, 123.5; δH 5.66, δC 137.2, 123.4; δH 5.05, δC 125.5, 133.5; δH 5.07, δC 124.8, 145.9) accounted for four degrees of unsaturation. The remaining two degrees of unsaturation strongly indicated that 8 has a bicyclic structure. A HMBC relationship between H2-20(δH 4.25 and 4.07) and C-15(δC 78.9) ascertained that an ether bridge formed across C-20 and C-15. Key NOE correlations from H-3(δH 5.66) to H-5b(δH 1.98) and H-14a(δH 2.60) and from H-2(δH 6.09) to H3-18(δH 1.75) and H3-17(δH 1.35) suggested the olefinic geometries were assigned to 1E and 3E. The chemical shift of Me-19(δC < 20 ppm) indicated the 7E assignment21. The additional hydroxylation at C-10 was supported by the 1H-1H COSY correlations of H-10(δH 4.26)/H-11(δH 5.07) and H-10/H-9(δH 2.40 and 2.16). Since compound 8 contained a secondary alcohol at C-10, its absolute configuration of C-10 was determined to be S, by applying the modified Mosher’s method following the same protocol as used for 1(Fig. 3).
Sarcophytrol U(9), a colorless oil, had a molecular formula of C20H32O4 established by HRESIMS(m/z 359.2199 [M + Na]+) and NMR data, indicating five degrees of unsaturation. The presence of a secondary and a tertiary hydroxyl group was clearly deduced from NMR signals at δC 76.9(d), 74.4(s) and δH 3.44(1H, dd, J = 10.2, 2.2 Hz, H-11). One additional terminal double bond was inferred by 13C NMR signals at δC 145.9(C) and 119.1(CH2). The conjugated olefinic group was also evident by four sp2 carbon signals in the 13C NMR spectrum at δC 130.3(s), 129.0(d), 132.8(d), and 123.0(s), and two olefinic doublets in the 1H NMR spectrum at δH 6.33(1H, d, J = 16.6 Hz, H-2) and 5.97(1H, d, J = 16.6 Hz, H-3). Detailed analysis of the 2D spectra(Fig. 5), allowed assigning all the chemical shifts in the NMR spectra, which led to the cembrane skeleton of 9. An exo-CH2, the conjugated olefinic groups and the 14-member ring of cembrane skeleton accounted for four degrees of unsaturation. As a consequence, there must be two oxygen atoms unassigned had to be ascribed to a peroxide bridge that linked at C-4 and C-7, respectively, to complete the required unsaturation degrees of five. In addition, the analysis of the NMR data of 9, in comparison with that of a known cembranoid,(1S,2E,4S,6R,7S,8R,11S)-8,11-epidioxy-2,12(20)-cembradiene-4,6,7-triol(13), previously reported from Greek tobacco23, confirmed the partial structure from C-4-C-9. Thus, the gross planar structure of 9 was determined as a 4,7-epidioxy-15(1),2,8(19)-cembratriene-11,12-diol.

Selected key COSY, HMBC and ROESY correlations for compound 9.
The relative stereochemistry of the chiral centers at C-4, C-7, C-11 and C-12 was established by a ROESY experiment running on 9. In MM2 energy-minimized conformation(Fig. 5), H-7 was suggested to be axial position toward the hexatomic ring(β-orientation). The diagnostic correlations between H-7(δH 4.71) and H-5a(δH 1.87) as well as H3-18(δH 1.28) and H-5a suggested H-5a and H3-18 were determined as axial and equatorial position, respectively(β-orientation). The ROESY correlation of H-11(δH 3.44) with H3-20(δH 1.28) suggested the H-11 and H3-20 are in the same face, which was compatible with that of the co-exist cembranoid sarglaucol(10)19. However, the relationship of H-7 and H-11 cannot be determined from the ROESY spectrum.
In conclusion, nine new cembranoids, sarcophytrols M–U(1–9), were isolated from the South China Sea soft coral Sarcophyton trocheliophorum, along with one known related cembranoid 10. Among them, the characteristic chemical features of them are the diverse types of cyclized rings: furan rings possessed by sarcophytrols M-P(1–4), pyran rings formed in sarcophytrols Q-S(5–7), while oxepane and peroxyl rings appeared in sarcophytrols T(8) and U(9), respectively. In addition, sarcophytrols R and S(6 and 7) share a rare bicyclic skeleton of the decaryiol-type. These group of diterpenes were first reported from the same genus of soft coral S. decaryi11, and described so far only from the species of soft corals Lobophytum sp.12. The co-isolation of these diterpenes is an example of the productivity of the title animal.
In light of a wide range of biological activities and pharmacological properties of cembranoids13, we performed in vitro investigation of inhibitory activity against human protein tyrosine phosphatase 1B(PTP1B) enzyme, a promising drug target for the treatment of type 2 diabetes and obesity17, for compounds 1–10, since the previously reported cembranoids from the title animals displayed significant PTP1B inhibitory activity3,4,16. Unfortunately, the bioassay result showed that none of the tested compounds exhibited interesting PTP1B inhibitory activities. In addition, the antitumor and antibacterial activities were also tested for compounds 1–10. However, they exhibited neither cytotoxicities against the human tumor cell lines HL-60 and K-562, nor antibacterial activity against Pseudomonas aeruginosa.
Methods
General experimental procedures
Optical rotations were measured on a Perkin-Elmer 341 polarimeter. IR spectra were recorded on a Perkin-Elmer 577 spectrometer with KBr disks. HRESIMS spectra were recorded on a Waters-Micromass Q-TOF Ultima Global electrospray mass spectrometer. NMR spectra were measured on a Bruker Avance III 500 and Varian INOVA 600 spectrometers with the residual CHCl3(δH 7.26 ppm, δC 77.0 ppm) as internal standard. Chemical shifts are expressed in δ(ppm) and coupling constants(J) in Hz. 1H and 13C NMR assignments were supported by 1H-1H COSY, HSQC, and HMBC experiments. Commercial Silica gel(Qing Dao Hai Yang Chemical Group Co., 200–300 and 400–600 mesh), C18 reversed-phase silica gel(150–200 mesh, Merck) and Sephadex LH-20(Amersham Biosciences) were used for column chromatography. Reversed phase HPLC(Agilent 1100 series liquid chromatography using a VWDG1314A detector at 210 nm and a semi-preparative ODS-HG-5 [5 μm, 10 mm(i.d.) × 25 cm] column was also employed. Pre-coated silica gel GF254 plates(Qing Dao Hai Yang Chemical Group Co. Ltd. Qingdao, People’s Republic of China) were used for analytical thin-layer chromatography(TLC). All solvents used were of analytical grade(Shanghai Chemical Reagents Company, Ltd.).
Animal materials
The soft corals S. trocheliophorum was collected by scuba at Yalong Bay, Hainan Province, China, in February 26, 2006, at a depth of −15 to −20 m, and identified by Professor R.-L. Zhou of South China Sea Institute of Oceanology, Chinese Academy of Sciences. The voucher sample is deposited at the Shanghai Institute of Materia Medica, CAS, under registration No. YAL-4.
Extraction and isolation
The lyophilized bodies of S. trocheliophorum(400 g, dry weight) were minced into pieces and exhaustively extracted with Me2CO at room temperature(3 × 1 L). The solvent-free Me2CO extract was partitioned between Et2O and H2O. The organic phase was evaporated under reduced pressure to give a dark brown residue(16 g), which was subjected to Si gel column chromatography(CC) and eluted with petroleum ether(PE) in acetone(0–100%, gradient) to yield 14 fractions(A–M). These fractions were subjected to repeated chromatography as usual work3,4,9,16, to afford ten pure metabolites: 1(5.7 mg), 2(4.8 mg), 3(4.2 mg), 4(3.6 mg), 5(6.7 mg), 6(3.1 mg), 7(2.0 mg), 8(5.6 mg), 9(1.0 mg), 10(2.1 mg).
Chemical structure data
All investigated compounds were ≥95% pure(HPLC, wavelength = 210 nm).
The NMR spectra of the compounds are provided in the Supporting Information.
Sarcophytrol M(1): colorless oil;  − 60.8(c 0.17, MeOH). IR(KBr) νmax 3438, 2936, 1645, 1098, 950, 754 cm−1; for 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 361.2356 [M + Na]+(calcd for 361.2355, C20H34O4Na).
− 60.8(c 0.17, MeOH). IR(KBr) νmax 3438, 2936, 1645, 1098, 950, 754 cm−1; for 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 361.2356 [M + Na]+(calcd for 361.2355, C20H34O4Na).
Sarcophytrol N(2): colorless oil;  − 120.0(c 0.15, MeOH). IR(KBr) νmax 3435, 2922, 1648, 1116, 954 cm−1; for 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 375.2504 [M + Na]+(calcd for 375.2511, C21H36O4Na).
− 120.0(c 0.15, MeOH). IR(KBr) νmax 3435, 2922, 1648, 1116, 954 cm−1; for 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 375.2504 [M + Na]+(calcd for 375.2511, C21H36O4Na).
Sarcophytrol O(3): colorless oil;  − 30.0(c 0.10, MeOH). IR(KBr) νmax 3433, 2932, 1733, 1638, 1262, 1024 cm−1; for 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 403.2458 [M + Na]+(calcd for 403.2455, C22H36O5Na).
− 30.0(c 0.10, MeOH). IR(KBr) νmax 3433, 2932, 1733, 1638, 1262, 1024 cm−1; for 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 403.2458 [M + Na]+(calcd for 403.2455, C22H36O5Na).
Sarcophytrol P(4): colorless oil;  + 24.4(c 0.10, MeOH). IR(KBr) νmax 3441, 2942, 1648, 1262, 1098, 802 cm-1; for 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 361.2345 [M + Na]+(calcd for 361.2355, C20H34O4Na).
+ 24.4(c 0.10, MeOH). IR(KBr) νmax 3441, 2942, 1648, 1262, 1098, 802 cm-1; for 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 361.2345 [M + Na]+(calcd for 361.2355, C20H34O4Na).
Sarcophytrol Q(5): colorless oil;  + 45.9(c 0.22, MeOH). IR(KBr) νmax 3458, 2950, 1650, 1232, 1028, 756 cm−1; for 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 361.2354 [M + Na]+(calcd for 361.2355, C20H34O4Na).
+ 45.9(c 0.22, MeOH). IR(KBr) νmax 3458, 2950, 1650, 1232, 1028, 756 cm−1; for 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 361.2354 [M + Na]+(calcd for 361.2355, C20H34O4Na).
Sarcophytrol R(6): colorless oil;  − 13.3(c 0.06, MeOH). IR(KBr) νmax 3455, 2932, 1653, 1255, 768 cm−1; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 361.2346 [M + Na]+(calcd for 361.2355, C20H34O4Na).
− 13.3(c 0.06, MeOH). IR(KBr) νmax 3455, 2932, 1653, 1255, 768 cm−1; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 361.2346 [M + Na]+(calcd for 361.2355, C20H34O4Na).
Sarcophytrol S(7): colorless oil;  − 39.3(c 0.07, MeOH). IR(KBr) νmax 3437, 2947, 1633, 1198, 879 cm−1; for 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 343.2252 [M + Na]+(calcd for 343.2249, C20H32O3Na).
− 39.3(c 0.07, MeOH). IR(KBr) νmax 3437, 2947, 1633, 1198, 879 cm−1; for 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 343.2252 [M + Na]+(calcd for 343.2249, C20H32O3Na).
Sarcophytrol T(8): colorless oil;  − 35.4(c 0.08, MeOH); UV(MeOH) λmax 242 nm. IR(KBr) νmax 3418, 2962, 1568, 1262, 782 cm−1; for 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 325.2141 [M + Na]+(calcd for 325.2144, C20H30O2Na).
− 35.4(c 0.08, MeOH); UV(MeOH) λmax 242 nm. IR(KBr) νmax 3418, 2962, 1568, 1262, 782 cm−1; for 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 325.2141 [M + Na]+(calcd for 325.2144, C20H30O2Na).
Sarcophytrol U(9): colorless oil;  + 29.0(c 0.10, MeOH); UV(MeOH) λmax 238 nm. IR(KBr) νmax 3425, 2955, 1753, 1662, 778 cm−1; for 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 359.2199 [M + Na]+(calcd for 359.2198, C20H32O4Na).
+ 29.0(c 0.10, MeOH); UV(MeOH) λmax 238 nm. IR(KBr) νmax 3425, 2955, 1753, 1662, 778 cm−1; for 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 359.2199 [M + Na]+(calcd for 359.2198, C20H32O4Na).
Preparation of( S )- and( R )-MTPA Esters of 1. The 1 S derivative was obtained by treating 1(2.0 mg) with(R)-MTPA-Cl in dry pyridine for ca. 16 h under stirring at room temperature. The reaction mixture was purified by CC(silica gel) to afford pure 1 S(1.4 mg). In a similar manner, 1 R(1.2 mg) was prepared from(S)-MTPA-Cl.
1 S: Selected 1H NMR(CDCl3, 400 MHz) δ 5.997(1H, d, J = 15.4 Hz, H-3), 5.657(1H, d, J = 15.4 Hz, H-2), 5.504(1H, dd, J = 10.4, 4.3 Hz, H-7), 5.273(1H, d, J = 9.3 Hz, H-11), 1.673(3H, s, Me-19), 1.284(3H, s, Me-18), 1.122(3H, s, Me-16), 1.100(3H, s, Me-20), 1.072(3H, s, Me-17).
1 R: Selected 1H NMR(CDCl3, 400 MHz) δ 6.000(1H, d, J = 15.4 Hz, H-3), 5.656(1H, d, J = 15.4 Hz, H-2), 5.515(1H, dd, J = 11.1, 4.3 Hz, H-7), 5.288(1H, d, J = 9.9 Hz, H-11), 1.684(3H, s, Me-19), 1.285(3H, s, Me-18), 1.117(3H, s, Me-16), 1.072(3H, s, Me-20), 1.072(3H, s, Me-17).
Preparation of( S )- and( R )-MTPA Esters of 8. The 8 S derivative was obtained by treating 8(2.0 mg) with(R)-MTPA-Cl in dry pyridine for ca. 16 h under stirring at room temperature. The reaction mixture was purified by CC(silica gel) to afford pure 8 S(1.4 mg). In a similar manner, 8 R(1.2 mg) was prepared from(S)-MTPA-Cl.
8 S: Selected 1H NMR(CDCl3, 400 MHz) δ 6.111(1H, d, J = 10.9 Hz, H-2), 5.642(1H, d, J = 10.9 Hz, H-3), 5.591(1H, m, H-10), 5.098(1H, dd, J = 9.9, 5.2 Hz, H-7), 4.969(1H, d, J = 9.4 Hz, H-11), 4.206(1H, dd, J = 16.8, 1.4 Hz, Ha-20), 4.008(1H, dd, J = 16.8, 2.2 Hz, Hb-20), 1.767(3H, s, Me-18), 1.458(3H, s, Me-19), 1.379(3H, s, Me-16), 1.357(3H, s, Me-17).
8 R: Selected 1H NMR(CDCl3, 400 MHz) δ 6.128(1H, d, J = 10.9 Hz, H-2), 5.659(1H, d, J = 10.9 Hz, H-3), 5.626(1H, dt, J = 10.2, 3.8 Hz, H-10), 5.070(1H, dd, J = 9.9, 5.2Hz, H-7), 5.070(1H, d, J = 9.4 Hz, H-11), 4.255(1H, dd, J = 16.8, 1.1 Hz, Ha-20), 4.055(1H, dd, J = 16.8, 1.9 Hz, Hb-20), 1.769(3H, s, Me-18), 1.449(3H, s, Me-19), 1.386(3H, s, Me-16), 1.357(3H, s, Me-17).
Additional Information
How to cite this article: Liang, L.-F. et al. New Bicyclic Cembranoids from the South China Sea Soft Coral Sarcophyton trocheliophorum. Sci. Rep. 7, 46584; doi: 10.1038/srep46584(2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Li, Y. & G. Pattenden Perspectives on the structural and biosynthetic interrelationships between oxygenated furanocembranoids and their polycyclic congeners found in corals. Nat. Prod. Rep. 28, 1269–1310(2011).
Chen, W.-T. et al. Further new highly oxidative cembranoids from the Hainan soft coral Sarcophyton trocheliophorum . Nat. Prod. Bioprospect. 6, 97–102(2016).
Liang, L.-F. et al. Unprecedented diterpenoids as a PTP1B inhibitor from the Hainan soft coral Sarcophyton trocheliophorum Marenzeller. Org. Lett. 15, 274–277(2013).
Liang, L.-F. et al. Cembrane diterpenoids from the soft coral Sarcophyton trocheliophorum Marenzeller as a new class of PTP1B inhibitors. Bioorg. Med. Chem. 21, 5076–5080(2013).
Chen, W.-T. et al. Structural diversity of terpenoids in the soft coral Sinularia flexibilis, evidenced by a collection from the South China Sea. RSC Adv. 5, 23973–23980(2015).
Jia, R. et al. Sarcophytonolides E−H, cembranolides from the Hainan soft coral Sarcophyton latum . J. Nat. Prod. 69, 819–822(2006).
Lai, D. et al. Sinulariols A-S, 19-oxygenated cembranoids from the Chinese soft coral Sinularia rigida . Tetrahedron 67, 6018–6029(2011).
Lu, Y. et al. Anti-inflammatory cembranoids from the soft corals Sinularia querciformis and Sinularia granosa . J. Nat. Prod. 71, 1754–1759(2008).
Liang, L.-F. et al. Sartrolides A-G and bissartrolide, new cembranolides from the South China Sea soft coral Sarcophyton trocheliophorum Marenzeller. Tetrahedron 69, 7381–7386(2013).
Cheng, S.-Y. et al. Cembranoids from the octocoral Sarcophyton ehrenbergi . J. Nat. Prod. 73, 197–203(2010).
Carmely, S., A. Groweiss & Y. Kashman Decaryiol, a new cembrane diterpene from the marine soft coral Sarcophyton decaryi . J. Org. Chem. 46, 4279–4284(1981).
Fattorusso, E. et al. Oxygenated cembranoids of the decaryiol type from the Indonesian soft coral Lobophytum sp. Tetrahedron, 65, 2898–2904(2009).
Liang, L.-F. & Y.-W. Guo Terpenes from the soft corals of the genus Sarcophyton: chemistry and biological activities. Chem. Biodivers. 10, 2161–2196(2013).
Coll, J. C. The chemistry and chemical ecology of octocorals(Coelenterata, Anthozoa, Octocorallia). Chem. Rev. 92, 613–631(1992).
Coll, J. C. et al. Algal overgrowth of alcyonacean soft corals. Mar. Biol. 96, 129–135(1987).
Liang, L.-F. et al. Sarsolenane and capnosane diterpenes from the Hainan soft coral Sarcophyton trocheliophorum Marenzeller as PTP1B inhibitors. Eur. J. Org. Chem. 2014, 1841–1847(2014).
Jiang, C.-S., L.-F. Liang & Y.-W. Guo Natural products possessing protein tyrosine phosphatase 1B(PTP1B) inhibitory activity found in the last decades. Acta Pharmacol. Sin. 33, 1217–1245(2012).
Chen, W.-T. et al. Sarcophytrols A–C, new capnosane diterpenoids from the South China Sea soft coral Sarcophyton trocheliophorum . Tetrahedron Lett. 56, 1348–1352(2015).
Zhang, C.-X. et al. New precursor of tetraterpenoids from the soft coral Sarcophyton glaucum . Nat. Prod. Res. 27, 782–786(2013).
Rao, C. B. et al. Metabolites of the soft coral Sinularia ovispiculata from the Indian Ocean. J. Nat. Prod., 56, 2003–2007(1993).
Bowden, B. F. et al. Studies of Australian soft corals. X. The isolation of epoxyisoneocembrene-A from Sinularia grayi and isoneocembrene-A from Sarcophyton ehrenbergi . Aust. J. Chem. 31, 2707–2712(1978).
Lai, D. et al. Cembranoids from the soft coral Sinularia rigida with antifouling activities. J. Agric. Food Chem. 61, 4585–4592(2013).
Arndt, R. et al. Tobacco chemistry. 68. Structure determination and biomimetic syntheses of two new tobacco cembranoids. Acta Chem. Scand. 42B, 294–302(1988).
Acknowledgements
This research was financially supported by the National Natural Science Foundation of China(Nos. 81520108028, 41506187, 21672230, 41676073, and 81603022), the SCTSM Project(Nos. 15431901000 and 14431901100), the Hunan Provincial Natural Science Foundation of China(No. 2015JJ3176), the SKLDR/SIMM Projects(Nos. SIMM1501ZZ-03). Lin-Fu Liang thanks to the financial support of the China Postdoctoral Science Foundation(No. 2016M601677). The authors are grateful to Prof. Margherita Gavagnin and Mr. Ernesto Mollo of Institute of Biomolecular Chemistry(ICB)-CNR for their valuable work on cooperation including collection of the sample and suggestion for the experiments.
Author information
Authors and Affiliations
Contributions
L.-F.L., H.-Y.W. and W.-T.C. conducted the main experiments; L.-F.L. and X.-W.L. did the data analyzes, and wrote the manuscript; Y.-W.G. designed the experiments, revised and polished the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Liang, LF., Chen, WT., Li, XW. et al. New Bicyclic Cembranoids from the South China Sea Soft Coral Sarcophyton trocheliophorum. Sci Rep 7, 46584 (2017). https://doi.org/10.1038/srep46584
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep46584
This article is cited by
-
Chemistry and bioactivity of secondary metabolites from South China Sea marine fauna and flora: recent research advances and perspective
Acta Pharmacologica Sinica (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
