
Abstract
Coral reefs are subject to coral bleaching manifested by the loss of endosymbiotic algae from coral host tissue. Besides algae, corals associate with bacteria. In particular, bacteria residing in the surface mucus layer are thought to mediate coral health, but their role in coral bleaching is unknown. We collected mucus from bleached and healthy Porites lobata colonies in the Persian/Arabian Gulf (PAG) and the Red Sea (RS) to investigate bacterial microbiome composition using 16S rRNA gene amplicon sequencing. We found that bacterial community structure was notably similar in bleached and healthy corals, and the most abundant bacterial taxa were identical. However, fine-scale differences in bacterial community composition between the PAG and RS were present and aligned with predicted differences in sulfur- and nitrogen-cycling processes. Based on our data, we argue that bleached corals benefit from the stable composition of mucus bacteria that resemble their healthy coral counterparts and presumably provide a conserved suite of protective functions, but monitoring of post-bleaching survival is needed to further confirm this assumption. Conversely, fine-scale site-specific differences highlight flexibility of the bacterial microbiome that may underlie adjustment to local environmental conditions and contribute to the widespread success of Porites lobata.
Similar content being viewed by others

Introduction
Corals live in an endosymbiotic relationship with photosynthetic algae of the genus Symbiodinium, along with other microorganisms, such as bacteria, archaea, fungi, and viruses1,2. This consortium constitutes a metaorganism commonly referred to as the coral holobiont3. While the algal symbionts provide a large part of the coral host’s metabolic requirements4, the role of coral-associated bacteria, although suggested to be functionally important, is less well understood5,6. Studies have shown that the bacterial community provides nutrients, confers protection from pathogen invasion through antimicrobial production, and is indicative of coral health states7,8,9,10,11,12,13,14.
Importantly, the coral host provides a range of habitats such as the coral tissue, skeleton, and surface mucus layer (SML) that harbor distinct and diverse bacteria, but only few studies characterized the differences in community composition and function15,16,17,18. In particular, the SML provides a protective barrier and constitutes a highly selected microbial environment critical for coral health8,19,20. Coral mucus is a complex mixture of carbohydrates, lipids, and proteins, and plays a fundamental role in structural support, heterotrophic feeding, sediment cleansing, defense against environmental stressors, besides a suite of other functions20,21,22. Coral mucus is produced in the ectodermal mucus gland cells and originates from photosynthates and compounds derived from heterotrophic feeding23,24. In many cases, the SML is a transparent coat that changes over days to mucus sheets. Aged mucus sheets are detached from the coral surface into the water column, after which new fluidic mucus is produced at the coral surface leading to a new cycle20,23. Detached coral mucus sheets are shown to play an important role in retaining and recycling nutrients and metabolites in coral reef ecosystems25.
Anthropogenic impacts in the form of local (e.g., overfishing and nutrient enrichment) and global (e.g., ocean warming and ocean acidification) stressors have resulted in a substantial loss of coral cover over the last decades, manifested by an increase in coral bleaching and coral disease26,27,28. Coral bleaching describes the physical whitening of the coral colony due to loss of its pigmented algal symbionts and can be induced by different stressors, of which climate changed-induced global warming is the most threatening29. Yet, the molecular mechanisms behind coral bleaching are still not completely understood. Previous studies have shown that bacteria can cause coral bleaching and demonstrated that coral bleaching is related to the presence of the bacterium Vibrio shilonii in the annual bleaching of the Mediterranean coral Oculina patagonica during warm summer months30, but not all studies found Vibrio shilonii in bleached corals31. More generally, studies have found differences in bacterial assemblages between bleached and healthy corals indicating that bacteria respond to coral bleaching, although the precise role of bacteria in coral health and coral bleaching is not well understood2,17,32.
Given the worldwide increase in coral bleaching and the projected increase in frequency of global mass bleaching events33, it is crucial to better understand the contribution of bacteria to coral bleaching. In particular, analysis of bacteria associated with the SML might provide further insight, given that the SML provides a first protective barrier of the coral host against invading microbes. However, only few studies have analyzed coral mucus-associated bacterial communities in coral bleaching. In addition, the effect of coral bleaching on bacterial communities of corals from different locations is virtually unknown. In particular, studies analyzing bacterial communities of corals from the PAG and RS might be highly informative in this regard, as both regions display high water temperatures and are considered unique environments where corals thrive under extreme environmental conditions. Studying these potential coral refugia34,35 may provide a model for understanding the effects of global environmental change36,37,38.
In this study, we analyzed bacterial communities of the SML from bleached and healthy coral colonies of Porites lobata that were collected in the PAG and the central RS using 16S rRNA gene amplicon sequencing. Our aim was to document bacterial community composition and potential bacterial shifts between bleached and healthy corals to further understand the role of SML-associated bacteria in coral bleaching. To our knowledge, this is the first comparison of mucus-associated bacterial communities of bleached and healthy corals from the PAG and RS.
Results
Algal symbionts associated with Porites lobata
To reveal potential correlations between bacterial communities and algal symbionts associated with mucus from P. lobata from the PAG and RS, we conducted ITS2 DGGE-fingerprinting and subsequent sequencing of prominent bands. Our analysis revealed that P. lobata from the PAG were exclusively associated with Symbiodinium type C3 (Supplementary Table S1). In comparison, colonies from the RS were associated with Symbiodinium type C15 and C15 variants (C15h, C15n, C97), but we also found Symbiodinium types from clade D (D1, D1a, D6) in a considerable number of coral colonies (41%; Supplementary Table S1). In addition, one sample harbored Symbiodinium type A1. Overall, we did not detect differences in symbiont types between health states, but symbiont assemblages were different between the PAG and RS and were more variable among corals from the RS. It should be noted, however, that coral colonies were sampled based on visual inspection, hence, we cannot positively exclude that some healthy samples were affected by temperature stress, but did not yet show visual signs of paling. Further, due to difficulties in amplifying Symbiodinium DNA from some mucus samples (primarily bleached samples), ITS2 types could not be determined for all samples (i.e., for about a third of all coral samples).
Bacterial community composition of coral mucus and seawater
We produced 55 16S rRNA gene libraries totaling 4,421,127 sequences from 50 P. lobata mucus samples (five bleached and five healthy colonies from each of two reefs of the PAG and three reefs of the RS) and five water samples (one water sample from each reef) (Table 1). After quality filtering, 2,840,780 sequences with an average length of 292 bp were retained.
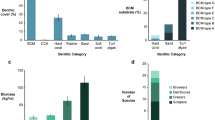
To assess overall differences in bacterial community composition, sequences were classified to the family level (Fig. 1; Supplementary Fig. S1). There was no apparent difference between bleached and healthy coral colonies from either the PAG or RS. Rather, mucus samples from the PAG and RS appeared to be composed of the same bacterial families, but with varying degrees of abundance. For instance, corals from the PAG and RS were both dominated by bacteria from the family Pseudomonadaceae (~5% to 47%), Dermabacteraceae (~5% to 18%), and Flavobacteraceae (~3% to 19%). The former two families were particularly abundant in samples from the RS, whereas the latter one in samples from the PAG. Water samples from the PAG and RS appeared highly similar and were dominated by Flavobacteriaceae (~12% to 16%), Halomonadaceae (~10% to 18%), and Pelagibacteraceae (~9% to 16%), and markedly different from mucus samples (Fig. 1; Supplementary Fig. S1).

Depicted is a taxonomy stacked column plot on the phylogenetic level of family. Each color represents one of the 27 most abundant families. Remaining taxa are grouped under category ‘others’. Samples are ordered by site, reef, and health-state. Values displayed as means of n = 5 for corals and n = 1 for water samples, B: Bleached; H: Healthy, PAG reef1: Saadiyat; PAG reef 2: Ras Ghanada; RS reef1: Shib Nazar; RS reef 2: Al Fahal; RS reef3: Inner Fsar.
Bacterial community differences of coral mucus across regions
Besides the overall similarity of bacterial community composition of mucus from P. lobata colonies, we were interested to assess fine-scale differences in bacterial community composition across health states (i.e., bleached and healthy) and across regions (i.e., PAG and RS). For this purpose, sequences were clustered in to OTUs after subsampling to 2,827 sequences per sample (Supplementary Dataset S1).
Species richness (Chao1) and bacterial diversity (Inverse Simpson) were highest in seawater (Table 1), but bacterial communities were not significantly different between PAG and RS (Fs = 5.09, PAMOVA ≥ 0.05), although a separation by region was noticeable (Supplementary Fig. S2). Conversely, water samples from the PAG and RS were significantly different from all coral mucus samples (Fs = 18.42, PAMOVA < 0.001). Seawater samples were excluded from subsequent analyses to emphasize on fine-scale differences of coral mucus-associated bacterial communities.
Species richness as well as Simpson evenness and Inverse Simpson Indices (diversity measures) in mucus samples were on average higher for coral bacterial communities from the PAG in comparison to the RS (all Pt-test ≤ 0.001) pointing to a more diverse and heterogeneous bacterial community in coral mucus from the PAG (Table 1, Supplementary Table S2). Importantly, we did not find significant differences between bleached and healthy corals, irrespective of region (Fig. 2, Fs = 0.92, PAMOVA = 0.35 PAG; Fs = 0.61, PAMOVA = 0.72 RS). Following this, we assessed differences between all samples from the PAG and all samples from the RS and found significant differences between mucus-associated bacteria (Fig. 2, Fs = 26.03, PAMOVA < 0.001).

Principal coordinate analysis based on Operational Taxonomic Unit (OTU) abundance (sequence counts) shows a partitioning of mucus microbiomes by region, but not by health state. Ellipses denote 95% confidence intervals per group.
To further analyze this, we tested for indicator bacterial taxa that are significantly associated with coral mucus from either region. We found 70 bacterial OTUs that were significantly associated with mucus samples from the PAG and 24 OTUs that were significantly associated with mucus samples from the RS regardless of health state (both P ≤ 0.01) (Supplementary Table S3). Notably, these regional indicator taxa were mostly low abundant in the mucus samples (Supplementary Table S3) in comparison to core microbiome members (see below, Table 2).
We also tested for bacterial taxa significantly associated with health state for each region and across regions. We found 3 and 5 bacterial taxa that were significantly associated with bleached and healthy mucus samples from PAG, respectively (Supplementary Table S4). In comparison, only 1 bacterial taxon was significantly associated with healthy samples from RS, and we did not find any bacteria significantly associated with bleached samples in the RS.
Core microbiome of coral mucus
Despite the site-specific differences in bacterial communities from the PAG and RS, we were interested in determining how many and which OTUs were consistently associated with coral mucus of P. lobata, as indicated by the bacterial community composition analysis (Fig. 1; Supplementary Fig. S1). To do this, we determined all OTUs that were present in at least 75% of all mucus samples and considered them members of the core microbiome. Following these criteria, we identified 42 core OTUs in mucus from P. lobata colonies, 6 of which were only present in mucus samples, while another 10 were primarily found in mucus. The remaining 26 core OTUs were consistently associated with coral mucus, although more abundant in seawater (Table 2). Importantly, 5 of the 42 core OTUs constituted the most abundant bacterial taxa. This suggests that despite differences in mucus-associated bacterial communities of corals from the PAG and RS, the core microbiome consistently encompasses the most abundant bacteria shared between bleached and healthy coral mucus samples, irrespective of site. Notably, Pseudomonas veronii (OTU0001), Brachybacterium sp. (OTU0002), and Dietzia sp. (OTU0003) that constituted the most abundant taxa are commonly associated with saline environments39,40,41 and corals41. Interestingly, and in line with Ainsworth et al.42, the 6 core taxa exclusive to mucus, i.e. Herbaspirillum sp. (OTU0012), Brevibacterium aureum (OTU0018), Sphingomonas echinoides (OTU0029), Delftia sp. (OTU0063), Sphingobium yanoikuyae (OTU0071), and Staphylococcus epidermidis (OTU0068), were present at comparatively low abundance.
Taxonomy-based functional profiling of bacterial communities
To gain insight into putative functional differences associated with bacterial community differences, we applied taxonomy-based functional profiling (Fig. 3). The majority of samples grouped by region, although some samples from the PAG clustered with samples from the RS (Fig. 3). Within clustered samples from the PAG, the functions ‘Sulfate reducer’, ‘Ammonia oxidizer’, ‘Chitin degradation’, ‘Nitrite reducer’, ‘Xylan degrader’, ‘Sugar fermentor’, and ‘Dinitrogen-fixing’ were all less abundant in comparison to the RS where these processes were more abundant. In contrast, ‘Nitrogen fixation’ and ‘Sulfide oxidizer’ were abundant in the PAG and either unchanged or scarce in the RS (Fig. 3). To confirm an enrichment of ‘Dinitrogen-fixing’ in the RS, we assessed the abundance of diazotroph communities via qPCR of the nifH gene and could confirm an on average higher abundance of diazotrophs in samples from the RS in comparison to the PAG (27% increase; Supplementary Fig. S3), but this difference was not statistically significant (PT-test ≥ 0.05).

The heatmap displays putative changes in bacterial community function. Changes are displayed on a relative scale with enrichment in red and depletion in blue. Rightmost column indicates sample names and health status (B: Bleached, H: Healthy).
Discussion
In this study, we compared bacterial community composition of the SML from bleached and healthy coral colonies of P. lobata from the PAG and RS in order to determine their structure, stability, and putative functional profiles. Our results show that bacterial community composition of coral mucus is highly similar between bleached and healthy corals. Importantly, core bacterial microbiome members are comprised of abundant and rare bacterial associates, whereas site-specific and health state differences exist for less abundant bacteria. Further, algal symbiont association shows a similar pattern to bacterial community patterns, as we did not find differences between health states, but regional differences. For instance, we could confirm the presence of Symbiodinium type C3 in P. lobata from the PAG43,44,45, whereas P. lobata from the RS were associated with Symbiodinium types C15 (and variants thereof) as well as types from clade D43,46. However, we did not find apparent differences between bleached and healthy colonies. Hence, it remains to be seen whether a causal relationship between Symbiodinium and bacterial community patterns exists. Given that corals from both the PAG and RS are able to survive seasonal temperature maxima exceeding those form other regions, at least in part due to harboring algal thermal tolerant symbionts that are commonly associated with high temperature environments44,45,47, it would be intriguing to find bacterial associates that co-occur with symbiont types. In a recent study with the coral model Aiptasia, microbial community patterns were distinct between symbiotic and aposymbiotic anemones, arguing for a connection between bacterial community composition and the cnidarian-algal symbiosis48. At large, the presence of photosymbionts distinguishes the microbiomes of hosts from those without photosymbionts49, but microbiomes of juvenile corals hosting different Symbiodinium clades were indistinguishable50.
To our knowledge, this is the first study that compares mucus-associated bacteria from bleached and healthy P. lobata colonies. Porites spp. have high production rates of mucus that cover coral colonies in the form of mucus sheets that exhibit a distinct ageing cycle making it an ideal model system to study dynamics of the mucus-associated microbiota20,23. Commonly, a new fluid mucus layer is produced about every four weeks20,23. The microbiome of coral surface mucus has an important role in mediating holobiont health20, and hence, the periodical release of mucus supposedly supports maintaining a beneficial bacterial microbiome via the removal of undesirable bacteria from the coral colony surface. Notably, mucus surfaces in many domains of multicellular life provide a protective barrier function and are assumed to constitute a common organismal feature51. This is supported by our data, as we find that mucus-associated bacteria in bleached P. lobata are similar to those in healthy P. lobata colonies, and long-term monitoring of their post-bleaching survival rates could further corroborate this assumption. In contrast, Bourne, et al.32 showed a shift of bacteria associated with the tissue of bleached corals. This indicates that mucus-associated bacterial communities may be less dynamic than those that are tissue-associated. Accordingly, the stably associated bacteria in the SML may provide a protective function, even and especially when coral health is compromised as during coral bleaching. In this regard, Lee, et al.52 showed that under heat stress the chemical composition of mucus in the coral Acropora muricata changed, which might either influence the associated bacterial community or be a consequence of it. Based on the presence of stable bacterial associates in our study, we infer that the mucus chemical composition did not change, although coral bleaching likely affected the availability of carbohydrates for mucus production23.
It is interesting to note that all mucus samples were dominated by few OTUs that were previously reported from saline environments, arguing that the high salinity of the PAG and RS might indeed comprise a structural determinant of bacteria associated with corals in these regions. Further, the consistent presence of these taxa in all coral mucus samples irrespective of site or bleaching state implies that they play an important role in the coral holobiont. Pseudomonas veronii has previously been found in fungiid corals experimentally exposed to high salinities (49 PSU)41. Dietzia sp. is found in the marine environment and in soil, human skin, and the intestinal tract of a carp, and plays a role in biodegradation, bioremediation, industrial fermentation, and carotenoid pigmentation53. The presence of Brachybacterium sp. has previously been reported in oil-contaminated coastal sand54 and salt-fermented seafood55. In contrast, site-specific bacterial taxa displayed lower abundance on average. Nevertheless, these bacteria suggest that Porites spp. have the ability to harbor flexible, and presumably locally adjusted microbiomes, which might at least in part contribute to the resilience of this coral genus56,57.
Predictive bacterial functional profiling between the PAG and the RS revealed differences in the abundance of bacteria associated with sulfur and nitrogen cycling. Differences in sulfur cycling included a scarcity of ‘Sulfur oxidizer’ and ‘Sulfate reducer’ and an increased abundance of ‘Sulfide oxidizer’ in samples from the PAG. Corals and especially their endosymbiotic algae are major producers of dimethylsulfoniopropionate (DMSP)58. Its breakdown products, such as dimethyl sulfoxide (DMSO), result mainly from bacterial metabolism and play a significant role in the scavenging of harmful reactive oxygen species (ROS)59. Importantly, Symbiodinium produce elevated levels of ROS during thermal stress, which may result in coral bleaching60,61. Consequently, the high temperatures in the PAG likely triggers increased ROS production demanding increased availability of ROS scavengers such as DMSP and DMSO, which could explain the functional differences in sulfur cycling observed in SML-associated bacteria between the PAG and the RS.
Differences in nitrogen cycling included increased abundance of ‘Dinitrogen-fixing’, ‘Ammonia oxidizer’, and ‘Nitrite reducer’ in the RS. Efficient nitrogen fixation and nitrogen recycling is essential for corals to thrive in nutrient-limited environments62. The RS constitutes a highly oligotrophic environment where nitrogen is presumably not readily available for corals. Compared to the RS, nitrogen is not a limiting nutrient in the PAG63, allowing for comparably high uptake for nitrogen sources (increased abundance of ‘Nitrogen fixation’)64 and lacking the need for efficient nitrogen recycling (decreased abundance of ‘Ammonia oxidizer’). From our analyses, functional profiling supports that environmental conditions strongly influence bacterial nitrogen fixation in corals65.
Taken together, in this study we found stable bacterial communities in the SML of bleached and healthy coral colonies of P. lobata from the PAG and the RS. This underscores the barrier function of coral mucus and we argue that bleached coral colonies benefit from the stable composition and distribution of SML-associated bacteria that presumably provide protective functions. In line with this, we found several abundant and ubiquitous bacterial taxa that we identified as core bacterial microbiome members of coral mucus. Further, regional differences in the mucus bacterial microbiome between PAG and RS were represented by less abundant bacteria that could be associated with a shift in predicted bacterial functional profiles. The specific regional bacterial taxa may thus contribute to the success of P. lobata colonies across a range of environmental conditions.
Methods
Study sites and coral mucus collection
Coral mucus was collected from bleached and healthy Porites lobata colonies from two reefs in the PAG, Saadiyat (24°35′56.4″N 54°25′17.4″E; samples: PAG1–PAG10) and Ras Ghanada (24°50′53.2″N 54°41′25.1″E; samples: PAG21–PAG30), and from three reefs in the central RS, Shib Nazar (22°20′27.4″N 38°51′07.6″E; samples: RS1–RS10), Al-Fahal (22°15′06.0″N 38°57′23.2″E; RS11–RS20), and Inner Fsar (22°13′58.4″N 39°01′45.6″E; samples: RS21–RS30), in September and October 2012, respectively. For each reef, mucus from 5 bleached and 5 healthy coral colonies were collected at 6 to 8 m depth, comprising a total of 20 samples from the PAG and 30 samples from the RS. Following66, corals were considered bleached if at least 20% of the colony surface had lost coloration. Mucus samples were collected using sterile syringes by sucking up mucus from the coral surface and by irritating the surface with the syringe tip while concomitantly collecting the released mucus. Syringes were placed in sterile Whirl-Paks. Upon return to the boat, syringes were placed upside-down in order for the heavier mucus to settle. Water on top of mucus was discarded and remaining mucus was ejected into cryotubes and frozen in liquid nitrogen. Samples were stored at −80 °C. In addition to the mucus samples, 1 L of seawater from each reef was collected at 1 to 2 m depth with a cubitainer. Cubitainers were transported on ice and 500 ml of collected water samples were subsequently filtered on 0.22 μm Milipore Durapore filters (Millipore, Billerica, MA, USA). Filters were snap-frozen in liquid nitrogen and stored at −80 °C.
DNA extraction
100 μL of mucus were used for DNA extraction using the Qiagen AllPrep DNA/RNA Mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. For extraction of DNA from seawater filters, half of each filter was cut into small stripes with sterile razorblades and transferred into 2 ml test tubes. After adding 400 μL Qiagen RLT buffer, the samples were incubated on a rotating wheel for 20 minutes. Subsequent extraction steps were performed according to the manufacturer’s protocol. DNA concentrations were quantified on a NanoDrop 2000C spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
Identification of algal symbionts from mucus of bleached and healthy Porites colonies
To determine algal symbionts, the ITS2 rDNA region was amplified with primers ITSintfor2 and ITS2CLAMP following67 with the following modifications: during touch-down PCR amplification the annealing temperature was decreased by 0.5 °C every cycle for 20 cycles, followed by 27 cycles at a final annealing temperature of 52 °C. Symbiodinium types were determined by DGGE profiling and subsequent sequencing of prominent bands. Prominent bands were excised from the DGGE gel and re-amplified as described in ref. 68. PCR products were then purified with Illustra ExoStar enzyme mix (SelectScience, Bath, UK), and samples were sequenced bidirectionally at the KAUST BioScience Core Laboratory (Thuwal, Saudi Arabia). Sequences were quality trimmed in CodonCode Aligner (CodonCode Corporation, Centerville, MA). Forward and reverse sequences were assembled into contigs and aligned using ClustalW. Each contig was BLASTed against a local reference database of Symbiodinium ITS2 sequences69 and against ITS2 sequences of type C15 variants recently described from Porites spp. in the Red Sea46.
16S rRNA gene sequencing
DNA isolated from coral mucus and seawater was used for PCR amplification of a portion of the 16S rRNA gene. Five to 35 ng DNA from mucus samples and 1 to 3 ng DNA from seawater samples were used to amplify variable regions 5 and 6 of the 16S rRNA gene with the primers 784 F [5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGAGGATTAGATACCCTGGTA-3′] and 1061 R [5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGCRRCACGAGCTGACGAC-3′]70, which have been shown to amplify well with coral DNA71. Illumina MiSeq adaptor overhangs (underlined above; Illumina, San Diego, CA, USA) were used for subsequent library indexing. All PCRs were run in triplicates per sample using Qiagen Multiplex PCR Kit with 0.2 μM of each primer and a total reaction volume of 25 μL. Cycling conditions were as follows: 95 °C for 15 min, followed by 30 cycles of 95 °C for 30 s, 55 °C for 90 s, 72 °C for 30 s, and a final extension cycle of 72 °C for 10 min. Successful amplification was checked via 1% agarose gel electrophoresis and sample triplicates were pooled in equimolar ratios. Pooled samples were cleaned with Agencourt AMPpure XP magnetic beads system (Beckman Coulter, Brea, CA, USA) and subsequently underwent an indexing PCR to add Nextera XT indexing and sequencing adapters (Illumina) following the manufacturer’s protocol. PCR products were sequenced on the Illumina MiSeq platform at the KAUST BioScience Core Laboratory. Sequence data determined in this study are available under NCBI’s BioProject ID PRJNA352338, accessible at https://www.ncbi.nlm.nih.gov/bioproject/PRJNA352338/.
Bacterial community analysis
Raw sequencing data were analyzed using mothur v.1.36.372. Sequence reads were split according to barcodes, assembled to contigs, and quality trimmed. Identical sequences were merged using the ‘unique.seqs’ command to save computation time, and the command ‘count.seqs’ was used to keep a count of the number of sequences over samples represented by the remaining representative sequence. Sequences that occurred only once across the entire dataset were removed. The remaining sequences were aligned against SILVA database release 11973 and pre-clustered (3-bp difference)74. Chimeric sequences were removed using UCHIME75. Mitochondria, chloroplast, archaea, eukaryote, and unknown sequences were removed. Sequences were classified with the Greengenes database76 using a 60% bootstrap cut-off. For subsequent OTU (Operational Taxonomic Unit)-based analyses, samples were subsampled to 2,827 sequences as determined by the sample with the lowest number of sequences; a 97% similarity cut-off was then applied to obtain OTUs. Chao1 index, Simpson evenness, and Inverse Simpson Index were calculated as implemented in mothur. To assess differences between bacterial communities associated with mucus and seawater, analysis of MOlecular VAriance (AMOVA) was performed in mothur. AMOVA was further used to test for differences in bacterial communities between bleached and healthy colonies per region and for differences between regions. To determine OTUs that were associated with bleached and healthy colonies of P. lobata from either the PAG or the RS (and combinations thereof), the statistical package IndicSpecies77 was used using OTU abundances with a significance threshold of P ≤ 0.01. Due to the design of the IndicSpecies analysis, the identified bacterial OTUs are exclusive to the specific health state ∗ region combination tested.
For determination of the core bacterial microbiome, all OTUs were considered that were presented in >75% of all coral samples based on OTU abundance counts. Notably, more abundant taxa are potentially more likely to be consistently identified across samples, hence, the choice of the >75% cutoff. We categorized mucus core OTUs as ‘exclusive’ when they were not present in any of the water samples, as ‘mucus-dominant’ when the ratio of mean sequence counts in mucus over water samples was >1, and as ‘environmental’ when the ratio of mean sequence counts in mucus over water samples was <1.
Functional differences based on bacterial 16S community composition (OTU taxonomy and abundance), were assessed with METAGENassist78. Input files were created in mothur using the ‘make.shared’ and ‘classify.otu’ commands based on all coral samples. 1,978 distinct OTUs were assigned, mapped, condensed into 500 functional taxa, and filtered based on interquantile range79. After filtering, 375 functional taxa remained and were normalized over samples by sum and over taxa by Pareto scaling. These data were analyzed for ‘metabolism by phenotype’, and Euclidean distance measure (single clustering algorithm) was used to visualize the results in a heatmap.
Relative abundance of nifH and 16S rRNA genes using qPCR
To confirm the increased functional abundance of ‘Dinitrogen-fixing’ in coral colonies from the RS (as inferred from METAGENassist), abundance of the nifH gene relative to abundance of the 16S rRNA gene was measured using quantitative PCR (qPCR). Reactions were run in triplicate per mucus sample on a 7900HT Fast Real-Time PCR System (Applied Biosystems, USA) using a reaction volume of 20 μL containing 2 μL of DNA (approximately 1 ng), 10 μL of Platinum SYBR green qPCR Supermix-UDG (Invitrogen, USA), and 0.4 μL of ROX reference dye. For amplification of the nifH gene, the primers F2 and R680 were used; for amplification of the 16S rRNA gene, primers 784F and 1061R were used70. Amplification reactions were performed with a primer concentration of 0.2 μM and with an initial polymerase activation step at 50 °C for 2 min and a denaturation step at 94 °C for 1 min followed by 50 cycles of 94 °C for 30 sec, 51 °C for 60 s, 72 °C for 60 s, and a final step of 72 °C for 3 min with a subsequent melting curve analysis. Triplicate cycle threshold (Ct) values for each sample were averaged. Relative abundance differences based on differences in gene copy numbers were calculated using the equation ΔCt = (CtnifH − Ct16S) for all mucus samples from the PAG and RS. Fold-change (FC) difference of nifH between PAG and RS was calculated as FC = 2−ΔΔCt with ΔΔCt = (ΔCtRS − ΔCtPAG) and tested for differences between regions using a T-test. Efficiency of qPCR was 82.12% based on the formula E = 10(−1/slope) − 181 and the R2 of the standard curve was >0.99.
Additional Information
Accession codes: Sequence data determined in this study are available under NCBI’s BioProject ID PRJNA352338, accessible at https://www.ncbi.nlm.nih.gov/bioproject/PRJNA352338/. Bacterial core microbiome OTU reference sequences are available under GenBank Accession numbers KY565430-KY565470.
How to cite this article: Hadaidi, G. et al. Stable mucus-associated bacterial communities in bleached and healthy corals of Porites lobata from the Arabian Seas. Sci. Rep. 7, 45362; doi: 10.1038/srep45362 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Knowlton, N. & Rohwer, F. Multispecies Microbial Mutualisms on Coral Reefs: The Host as a Habitat. Am Nat 162, S51–S62 (2003).
Rosenberg, E., Koren, O., Reshef, L., Efrony, R. & Zilber-Rosenberg, I. The role of microorganisms in coral health, disease and evolution. Nat Rev Micro 5, 355–362 (2007).
Rohwer, F., Seguritan, V., Azam, F. & Knowlton, N. Diversity and distribution of coral-associated bacteria. Marine Ecology Progress Series 243 (2002).
Muscatine, L., Falkowski, P. G., Porter, J. W. & Dubinsky, Z. Fate of photosynthetic fixed carbon in light-adapted and shade-adapted colonies of the symbiotic coral Stylophora pistillata . Proc Roy Soc Ser B Biol Sci 222, 181–202 (1984).
Mouchka, M. E., Hewson, I. & Harvell, C. D. Coral-Associated Bacterial Assemblages: Current Knowledge and the Potential for Climate-Driven Impacts. Integr Comp Biol 50, 662–674 (2010).
Bourne, D. G., Morrow, K. M. & Webster, N. S. Insights into the Coral Microbiome: Underpinning the Health and Resilience of Reef Ecosystems. Annu Rev Microbiol 70, 317–340 (2016).
Koh, E. G. L. Do Scleractinian Corals Engage in Chemical Warfare Against Microbes? J Chem Ecol 23, 379–398 (1997).
Ritchie. Regulation of microbial populations by coral surface mucus and mucus-associated bacteria. Mar Ecol Prog Ser 322, 1–14 (2006).
Rypien, K. L., Ward, J. R. & Azam, F. Antagonistic interactions among coral‐associated bacteria. Env Microbiol 12, 28–39 (2010).
Wegley, L., Edwards, R., Rodriguez‐Brito, B., Liu, H. & Rohwer, F. Metagenomic analysis of the microbial community associated with the coral Porites astreoides . Env Microbiol 9, 2707–2719 (2007).
Roder, C., Bayer, T., Aranda, M., Kruse, M. & Voolstra, C. R. Microbiome structure of the fungid coral Ctenactis echinata aligns with environmental differences. Mol Ecol 24, 3501–3511 (2015).
Ziegler, M. et al. Coral microbial community dynamics in response to anthropogenic impacts near a major city in the central Red Sea. Mar Pollut Bull 105, 629–640 (2016).
Roder, C. et al. Bacterial profiling of White Plague Disease in a comparative coral species framework. ISME J 8, 31–39 (2014).
Roder, C., Arif, C., Daniels, C., Weil, E. & Voolstra, C. R. Bacterial profiling of White Plague Disease across corals and oceans indicates a conserved and distinct disease microbiome. Molecular Ecology 23, 965–974 (2014).
Bourne, D. G. & Munn, C. B. Diversity of bacteria associated with the coral Pocillopora damicornis from the Great Barrier Reef. Env Microbiol 7, 1162–1174 (2005).
Koren, O. & Rosenberg, E. Bacteria Associated with Mucus and Tissues of the Coral Oculina patagonica in Summer and Winter. Appl Environ Microb 72, 5254–5259 (2006).
Li, J. et al. Bacterial dynamics within the mucus, tissue and skeleton of the coral Porites lutea during different seasons. Sci Rep 4, 7320 (2014).
Sweet, M. J., Croquer, A. & Bythell, J. C. Bacterial assemblages differ between compartments within the coral holobiont. Coral Reefs 30, 39–52 (2011).
Brown, B. & Bythell, J. Perspectives on mucus secretion in reef corals. Mar Ecol Prog Ser 296, 291–309 (2005).
Glasl, B., Herndl, G. J. & Frade, P. R. The microbiome of coral surface mucus has a key role in mediating holobiont health and survival upon disturbance. ISME J 10, 2280–2292 (2016).
Stabili, L., Schirosi, R., Parisi, M. G., Piraino, S. & Cammarata, M. The Mucus of Actinia equina (Anthozoa, Cnidaria): An Unexplored Resource for Potential Applicative Purposes. Mar Drugs 13, 5276–5296 (2015).
Shnit-Orland, M. & Kushmaro, A. Coral mucus-associated bacteria: a possible first line of defense. FEMS Microb Ecol 67, 371–380 (2009).
Coffroth, M. A. Mucous sheet formation on poritid corals: An evaluation of coral mucus as a nutrient source on reefs. Mar Biol 105, 39–49 (1990).
Marshall, A. T. & Wright, O. P. Confocal laser scanning light microscopy of the extra-thecal epithelia of undecalcified scleractinian corals. Cell and Tissue Research 272, 533–543 (1993).
Wild, C. et al. Coral mucus functions as an energy carrier and particle trap in the reef ecosystem. Nature 428, 66–70 (2004).
Maynard, J. et al. Projections of climate conditions that increase coral disease susceptibility and pathogen abundance and virulence. Nature Clim. Change 5, 688–694 (2015).
Hoegh-Guldberg, O. et al. Coral reefs under rapid climate change and ocean acidification. Science 318, 1737–1742 (2007).
Hughes, T. P. et al. Phase shifts, herbivory, and the resilience of coral reefs to climate change. Curr Biol 17, 360–365 (2007).
Carpenter, K. E. et al. One-third of reef-building corals face elevated extinction risk from climate change and local impacts. Science 321, 560–563 (2008).
Kushmaro, A., Loya, Y., Fine, M. & Rosenberg, E. Bacterial infection and coral bleaching. Nature 380, 396–396 (1996).
Ainsworth, T. D., Fine, M., Roff, G. & Hoegh-Guldberg, O. Bacteria are not the primary cause of bleaching in the Mediterranean coral Oculina patagonica. ISME J 2, 67–73 (2007).
Bourne, D., Iida, Y., Uthicke, S. & Smith-Keune, C. Changes in coral-associated microbial communities during a bleaching event. ISME J 2, 350–363 (2008).
van Hooidonk, R., Maynard, J. A. & Planes, S. Temporary refugia for coral reefs in a warming world. Nature Clim. Change 3, 508–511 (2013).
Cacciapaglia, C. & van Woesik, R. Climate-change refugia: shading reef corals by turbidity. Global Change Biology 22, 1145–1154 (2016).
Fine, M., Gildor, H. & Genin, A. A coral reef refuge in the Red Sea. Glob Chang Biol 19, 3640–3647 (2013).
Burt, J., Al-Harthi, S. & Al-Cibahy, A. Long-term impacts of coral bleaching events on the world’s warmest reefs. Mar Environ Res 72, 225–229 (2011).
Roik, A., Roder, C., Röthig, T. & Voolstra, C. R. Spatial and seasonal reef calcification in corals and calcareous crusts in the central Red Sea. Coral Reefs 35, 681–693 (2016).
Roik, A. et al. Year-Long Monitoring of Physico-Chemical and Biological Variables Provide a Comparative Baseline of Coral Reef Functioning in the Central Red Sea. PLoS One 11, e0163939 (2016).
Zvyagintseva, I. S., Poglazova, M. N., Gotoeva, M. T. & Belyaev, S. S. Effect on the Medium Salinity on Oil Degradation by Nocardioform Bacteria. Microbiol 70, 652–656 (2001).
Lo, J. R., Lang, J. M., Darling, A. E., Eisen, J. A. & Coil, D. A. Draft Genome Sequence of an Actinobacterium, Brachybacterium muris Strain UCD-AY4. Genome Announcements 1, e00086–00013 (2013).
Röthig, T., Ochsenkuhn, M. A., Roik, A., van der Merwe, R. & Voolstra, C. R. Long-term salinity tolerance is accompanied by major restructuring of the coral bacterial microbiome. Mol Ecol 25, 1308–1323 (2016).
D. Ainsworth, T. et al. The coral core microbiome identifies rare bacterial taxa as ubiquitous endosymbionts. ISME J 9, 2261–2274 (2015).
Ziegler, M. et al. Biogeography and molecular diversity of coral symbionts in the genus Symbiodinium around the Arabian Peninsula. Journal of Biogeography, doi: 10.1111/jbi.12913 (2017).
Hume, B. C. C. et al. Ancestral genetic diversity associated with the rapid spread of stress-tolerant coral symbionts in response to Holocene climate change. Proc Nat Acad Sci USA 113, 4416–4421 (2016).
Hume, B. C. et al. Symbiodinium thermophilum sp. nov., a thermotolerant symbiotic alga prevalent in corals of the world’s hottest sea, the Persian/Arabian Gulf. Sci Rep 5, 8562 (2015).
Ziegler, M., Roder, C., Büchel, C. & Voolstra, C. R. Niche acclimatization in Red Sea corals is dependent on flexibility of host-symbiont association. Mar Ecol Prog Ser 533, 149–161 (2015).
Baker, A. C., Starger, C. J., McClanahan, T. R. & Glynn, P. W. Coral reefs: Corals’ adaptive response to climate change. Nature 430, 741–741 (2004).
Röthig, T. et al. Distinct bacterial communities associated with the coral model Aiptasia in aposymbiotic and symbiotic states with Symbiodinium. Frontiers in Marine Science 3 (2016).
Bourne, D. G. et al. Coral reef invertebrate microbiomes correlate with the presence of photosymbionts. ISME J 7, 1452–1458 (2013).
Littman, R. A., Willis, B. L. & Bourne, D. G. Bacterial communities of juvenile corals infected with different Symbiodinium (dinoflagellate) clades. Mar Ecol Prog Ser 389, 45–59 (2009).
Bythell, J. C. & Wild, C. Biology and ecology of coral mucus release. Journal of Experimental Marine Biology and Ecology 408, 88–93 (2011).
Lee, S. T. M., Davy, S. K., Tang, S.-L. & Kench, P. S. Mucus Sugar Content Shapes the Bacterial Community Structure in Thermally Stressed Acropora muricata . Front Microbiol 7, 371 (2016).
Gharibzahedi, S. M. T., Razavi, S. H. & Mousavi, S. M. Characterization of bacteria of the genus Dietzia: an updated review. Ann Microbiol 64, 1–11 (2014).
Chou, J.-H. et al. Brachybacterium phenoliresistens sp. nov., isolated from oil-contaminated coastal sand. Int J Syst Micr 57, 2674–2679 (2007).
Park, S.-K. et al. Brachybacterium squillarum sp. nov., isolated from salt-fermented seafood. Int J Syst Micr 61, 1118–1122 (2011).
Jessen, C. et al. In-situ Effects of Eutrophication and Overfishing on Physiology and Bacterial Diversity of the Red Sea Coral Acropora hemprichii . Plos One 8, e62091 (2013).
Hernandez-Agreda, A., Leggat, W., Bongaerts, P. & Ainsworth, T. D. The Microbial Signature Provides Insight into the Mechanistic Basis of Coral Success across Reef Habitats. mBio 7 (2016).
Raina, J.-B. et al. DMSP biosynthesis by an animal and its role in coral thermal stress response. Nature 502, 677–680 (2013).
Sunda, W., Kieber, D., Kiene, R. & Huntsman, S. An antioxidant function for DMSP and DMS in marine algae. Nature 418, 317–320 (2002).
Lesser, M. P. Oxidative stress in marine environments: Biochemistry and Physiological Ecology. Annu Rev Physiol 68, 253–278 (2006).
Weis, V. M. Cellular mechanisms of Cnidarian bleaching: stress causes the collapse of symbiosis. J Exp Biol 211, 3059–3066 (2008).
Fiore, C. L., Jarett, J. K., Olson, N. D. & Lesser, M. P. Nitrogen fixation and nitrogen transformations in marine symbioses. Trends Microbiol 18, 455–463 (2010).
Banerjee, P. & Prasanna Kumar, S. Dust-induced episodic phytoplankton blooms in the Arabian Sea during winter monsoon. J Geophys Res Oceans 119, 7123–7138 (2014).
Grover, R., Maguer, J. F., Allemand, D. & Ferrier-Pages, C. Uptake of dissolved free amino acids by the scleractinian coral Stylophora pistillata . J Exp Biol 211, 860–865 (2008).
Rädecker, N., Pogoreutz, C., Voolstra, C. R., Wiedenmann, J. & Wild, C. Nitrogen cycling in corals: the key to understanding holobiont functioning? Trends Microbiol 23, 490–497 (2015).
Monroe, A. et al. In-situ observations of coral bleaching in the central Saudi Arabian Red Sea during the 2015/2016 global coral bleaching event. PLoS ONE (2017).
LaJeunesse, T. C. & Trench, R. K. Biogeography of two species of Symbiodinium (Freudenthal) inhabiting the intertidal sea anemone Anthopleura elegantissima (Brandt). Biol Bull 199, 126–134 (2000).
LaJeunesse, T. Diversity and community structure of symbiotic dinoflagellates from Caribbean coral reefs. Mar Biol 141, 387–400 (2002).
Arif, C. et al. Assessing Symbiodinium diversity in scleractinian corals via next-generation sequencing-based genotyping of the ITS2 rDNA region. Mol Ecol 23, 4418–4433 (2014).
Andersson, A. F. et al. Comparative Analysis of Human Gut Microbiota by Barcoded Pyrosequencing. PLoS One 3, e2836 (2008).
Bayer, T. et al. The microbiome of the Red Sea coral Stylophora pistillata is dominated by tissue-associated Endozoicomonas bacteria. Appl Environ Microb 79, 4759–4762 (2013).
Schloss, P. D. et al. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl Environ Microb 75, 7537–7541 (2009).
Pruesse, E. et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35, 7188–7196 (2007).
Huse, S. M., Welch, D. M., Morrison, H. G. & Sogin, M. L. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Env Microbiol 12, 1889–1898 (2010).
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C. & Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200 (2011).
McDonald, D. et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6, 610–618 (2012).
Cáceres, M. D. & Legendre, P. Associations between species and groups of sites: indices and statistical inference. Ecology 90, 3566–3574 (2009).
Arndt, D. et al. METAGENassist: a comprehensive web server for comparative metagenomics. Nucleic Acids Res 40, W88–95 (2012).
Hackstadt, A. J. & Hess, A. M. Filtering for increased power for microarray data analysis. BMC Bioinf 10, 1–12 (2009).
Gaby, J. C. & Buckley, D. H. A Comprehensive Evaluation of PCR Primers to Amplify the nifH Gene of Nitrogenase. PLoS One 7, e42149 (2012).
Atallah, Z. K., Bae, J., Jansky, S. H., Rouse, D. I. & Stevenson, W. R. Multiplex Real-Time Quantitative PCR to Detect and Quantify Verticillium dahliae Colonization in Potato Lines that Differ in Response to Verticillium Wilt. Phytopathology 97, 865–872 (2007).
Acknowledgements
We would like to thank the Coastal and Marine Resources Core Lab (CMOR) in KAUST and the Marine Biology Lab at NYUAD for their assistance and support in field operations. We thank Nils Rädecker for assistance with qPCR experiments and analysis, and Craig Michell for support with amplicon sequencing. This publication is based upon work supported by the King Abdullah University of Science and Technology (KAUST) Office of Sponsored Research (OSR) under Award Nos FCC/1/1973-21-01 and FCC/1/1973-22-01.
Author information
Authors and Affiliations
Contributions
C.R.V. and C.R. conceived the study and designed experiments; G.H., C.A., L.K.Y. performed experiments; G.H., T.R., L.K.Y., M.Z., C.R.V. analyzed the data; C.R.V., G.H., T.R., M.Z. wrote the manuscript. J.B. and C.R.V. contributed material/reagents/tools.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Hadaidi, G., Röthig, T., Yum, L. et al. Stable mucus-associated bacterial communities in bleached and healthy corals of Porites lobata from the Arabian Seas. Sci Rep 7, 45362 (2017). https://doi.org/10.1038/srep45362
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep45362
This article is cited by
-
The coral microbiome in sickness, in health and in a changing world
Nature Reviews Microbiology (2024)
-
Experimental mining plumes and ocean warming trigger stress in a deep pelagic jellyfish
Nature Communications (2023)
-
Microbial shifts associated to ENSO-derived thermal anomalies reveal coral acclimation at holobiont level
Scientific Reports (2023)
-
The microbiome of the endosymbiotic Symbiodiniaceae in corals exposed to thermal stress
Hydrobiologia (2023)
-
The microbiomes of two Singaporean corals show site-specific differentiation and variability that correlates with the seasonal monsoons
Coral Reefs (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
