
Abstract
Mesial temporal lobe epilepsy (MTLE) is a common epileptic disorder; little is known whether it is associated with peripheral epigenetic changes. Here we compared blood whole genomic DNA methylation pattern in MTLE patients (n = 30) relative to controls (n = 30) with the Human Methylation 450 K BeadChip assay, and explored genes and pathways that were differentially methylated using bioinformatics profiling. The MTLE and control groups showed significantly different (P < 1.03e-07) DNA methylation at 216 sites, with 164 sites involved hyper- and 52 sites hypo- methylation. Two hyper- and 32 hypo-methylated sites were associated with promoters, while 87 hyper- and 43 hypo-methylated sites corresponded to coding regions. The differentially methylated genes were largely related to pathways predicted to participate in anion binding, oxidoreductant activity, growth regulation, skeletal development and drug metabolism, with the most distinct ones included SLC34A2, CLCN6, CLCA4, CYP3A43, CYP3A4 and CYP2C9. Among the MTLE patients, panels of genes also appeared to be differentially methylated relative to disease duration, resistance to anti-epileptics and MRI alterations of hippocampal sclerosis. The peripheral epigenetic changes observed in MTLE could be involved in certain disease-related modulations and warrant further translational investigations.
Similar content being viewed by others

Introduction
Temporal lobe epilepsy (TLE) is a common neurological disease that may affect up to 1% population, representing a significant healthcare and financial burden to society and families1,2. Epidemiological studies estimate an approximate 0.5% morbidity of TLE in China (9 million patients nationwide), with 5–6 million patients requiring continuous medical attention and 0.4 million new patients being diagnosed each year. Youth and adults are mostly affected, with a higher prevalence in rural than urban areas3,4. Mesial temporal lobe epilepsy (MTLE) is the most common type of TLE, featured by refractory seizures worsen with time, and often become resistant to anti-epileptic therapies and require brain surgery to control symptoms5,6. The clinicopathological manifestations of MTLE appear to be largely related to lesions of the medial temporal lobe structures7,8,9. Atrophy or sclerosis of the hippocampal formation (HS) and neighboring limbic structures can be detected via brain imaging, with neuronal death, granule cell dispersion, mossy fiber sprouting, neuronal network reorganization, gliosis and inflammation found histologically in affected brain regions10,11,12,13,14,15,16,17,18,19,20,21.
While great progress has been made in the past decades in understanding of the neuropathology and pathophysiology of MTLE, the etiological and pathogenic underpinnings of this disorder remain poorly understood5,6,19,20,21. Biochemical and bioinformatics approaches are used in recent years to study MTLE, revealing broad changes in protein, mRNA and DNA expression and/or modulation in the brain22,23,24. For instance, proteomic studies have shown remarkable and dynamic changes in protein expression involving multiple cellular systems in epileptic brain25,26,27,28,29,30. Transcriptomic studies indicate that a few hundred genes are either up- or down-regulated in the ictogenic relative to unaffected brain regions31,32,33,34,35,36,37. Epigenetic changes are being recognized as a part of the molecular reconfiguration in TLE, with a wide array of genes involved in neuronal/synaptic transmission, cell survival/death and transcriptional regulation differentially methylated in the brains of patients38,39,40,41,42.
Recent epigenetics studies have shown blood DNA methylation changes in a number of neurological and psychiatric diseases, including Alzheimer’s disease, Parkinson’s disease, Down’s syndrome, infantile spasm, depressive disorders and schizophrenia43,44,45,46,47,48,49,50,51,52,53,54,55,56. Investigations into peripheral DNA methylation changes are considered important to help develop novel biomarkers for early diagnosis of some brain diseases. Here we carried out a case-control study on blood whole-genome DNA methylation pattern in 30 MTLE patients relative to sex/age-matched controls, and have identified a panel of differentially methylated genes involved in multiple interactive molecular pathways between the two groups.
Results
Sample and assay quality controls
The integrity of purified blood DNA was evaluated with agarose gel electrophoresis. In ethidium bromide stained gels, the bulk of DNA was migrated at locations relative to molecular sizes apparently greater than 15000 base-pairs (bp), arranged largely as two distinct bands. The pattern of DNA bands was comparable across all samples, without smearing of labeling signal (Supplemental Fig. 1). Several sample-independent assay controls were carried out, which verified the efficacy and specificity of the Beadchip. (1) Staining controls indicated that the signal of dinitrophenyl (DNP) (green fluorescent channel) and biotin (green fluorescent channel) attached beads was sufficient; (2) Hybridization controls indicated that the signal (green fluorescence) was intensified with the increase of biotin-tagged fluorescent probes in a concentration-dependent manner; (3) Target removal controls revealed a loss of fluorescent signal in conditions whereby the oligos were extended using the probe sequence as template; (4) Extension controls showed that the signal was enhanced with the extension of either the guanine-cytosine (GC) residues (green fluorescence) or adenine-thymine (AT) residues (red fluorescence) (Supplementary Fig. 2).
Sample-dependent assay controls were used to verify the efficacy of bisulfite sodium conversion on the Illumina 450K Beadchip system. In Infmium-I assay controls, the hybridization signal appeared properly strong as tested with three quality control probes (C1–3) for the green fluorescence (Supplementary Fig. 3A) and three control probes (C4-6) for the red fluorescence (Supplementary Fig. 3B). In Infmium-II assay control, four probes (red fluorescent) were used to verify the efficacy of bisulfite sodium conversion involving the cytosine residues, which yielded strong red fluorescence (Supplementary Fig. 3C,D).
Overall changes in blood genomic DNA methylation in MTLE
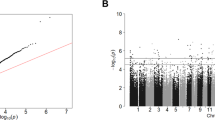
Using the Genome studio V2011 software, the β-values of 485,577 DNA methylation sites were reported for the samples from individual control (n = 30) and patient (n = 30) subjects (Fig. 1). Statistical analysis indicated that a total of 216 sites showed difference in the degree of methylation between the two groups (defined using a cutoff P value < 1.03e-07). Among these differentially methylated sites, 164 sites involved hyper-methylation, whereas 52 sites were hypo-methylated, yielding a ratio of 3.15 (Supplementary Table 1). These differentially methylated sites appeared to distribute on the 22 chromosomes without an apparent preference (Fig. 2).

Bead tags are listed vertically at the right side of the map. The extents of green and red represent levels of hyper-methylation, hypo-methylation, respectively, whereas black indicates no change in methylation, relative to control.

The differentially methylated sites, including the 216 sites with P < 1.03e-07 by Bonferroni correction test, appear to distribute across all 22 chromosomes without apparent preference.
Further analysis in reference to DNA functional domains, among the hyper-methylated sites there were 67 sites located at non-coding regions (67%), 65 sites at coding domains (39%), 30 sites associated with promoters (18%) and 4 involved the 3′ UTR terminal (3%). Among the hypo-methylated sites, there were 3 sites occurred in non-coding intergenic regions (6%), 14 sites at coding domains (26%), 36 sites associated with promoters (68%), and none related to the 3′ UTR end (Fig. 3).

Percentage rates for the hyper-methylated and hypo-methylated sites associated with the promoter, coding (body), non-coding (intergenic) and the 3′ UTR regions are illustrated in (A) and (B), respectively.
Among the 66 differentially methylated sites over promoter regions (Fig. 4A), the ratio of hyper- to hypo-methylated sites was 0.83 (30/36), which was lower than the overall ratio (164/52 = 3.15) of the entire set of hyper- to hypo-methylated DNA sites in the whole genome. A total of 36 differentially methylated sites were associated with the CpG island structures (Fig. 4B), mostly involved hypo-methylation (34/36). Thus, the 34 hypo-methylated sites occurred largely at the CpG islands per se (24), with the remainder at the N-shores (5), S-shores (3) and Open area (2), whereas none in the N- and S-shelves (Fig. 4C).

Panel (A) illustrates the heat map derived from clustering analysis of the microarray data from individual control (n = 30) and patient (n = 30) subjects as indicated above the heat map. Panel (B) shows the organization of the CpG island structures. Panel (C) plots the percentage distribution of the differentially methylated DNA sites at corresponding CpG island domains.
Cross-validation of methylation with pyrosequencing
In order to evaluate the accuracy of DNA methylation detected with the Methylation450 BeadChip assay, a subset of CpG loci with varying degree of methylation was selected for additional methylation analysis using the pyrosequencing method. Thus, patient (n = 15) and control (n = 15) blood samples were subjected to methylation analysis at eight loci (cg17461336, cg09248887, cg06852575, cg04358264, cg10690677, cg21593669, cg25291941, cg11582617) via PCR amplification followed by pyrosequencing. Pyrosequencing revealed methylation of cg17461336 at 45.7 ± 3.1% in the MTLE group and 43.8 ± 4.1% in the control group (Fig. 5A), which was correlated with the data from Methylation450 BeadChip test when analyzed against individually samples (P = 0.0005, R2 = 0.3548) (Fig. 5A, insert). The cg09248887 site had methylation rate at 88.2 ± 1.9% in the MTLE group and 86.6 ± 4.3% in the control group by pyrosequencing (Fig. 5B), significantly correlated with the BeadChip data relative to individual subjects (P = 0.0374, R2 = 0.1456) (Fig. 5B, insert). For cg06852575, the methylation rates were 30.2 ± 2.9% in the MTLE group and 32.0 ± 2.2% in the control group (Fig. 5C), consistent with the BeadChip data referring to individual cases (P = 0.0194, R2 = 0.1801) (Fig. 5C, insert). For cg04358264, the methylation rates were 85.9 ± 1.4% in the MTLE group and 85.3 ± 2.5% in the control group (Fig. 5D), also in line with the BeadChip data among individuals (P = 0.0207, R2 = 1769) (Fig. 5D, insert). The cg10690677 (Fig. 5E) and cg21593669 (not shown) sites showed very high (near 100%) methylation among patient and control samples with both methods, although there was no correlation between the two sets of value as analyzed against individual cases (likely due to a ceiling effect) (Fig. 5E, insert). Opposite to the above, the loci cg25291941 (Fig. 5F) and cg11582617 (not shown) had very low methylation (near 0%) by either method, and there was also no correlation between the two sets of data among individuals (likely a floor effect) (Fig. 5F, insert).

Shown are degrees pf methylation of six representative CpG loci reported by Methylation450 BeadChip (Y axis, ratio) and pyrosequencing (X axis, percentage) assays. Red and green dots in each panel plot values of methylation at a given site among individual patients (n = 15) with temporal lobe epilepsy (TLE) and controls (n = 15). Insert in the panel shows a correlation analysis between the two sets of values among individual cases regardless of subject groups, with P and R2 values labeled for each gene site. For cg17461336 (A), cg09248887 (B), cg06852575 (C) and cg04358264 (D), the degrees of methylation detected by the two methods were positively correlated (P < 0.05) in reference to individual samples. The two methods consistently reveal an exceptionally high methylation of cg10690677 (E) and minimal methylation of cg25291941 (F) among individual subjects, while there is no correlation between the two sets of values for either site in a case-based manner (likely due to ceiling and floor effects, respectively).
Relevance of differentially methylated DNA sites to functional genes
The differentially methylated sites revealed by the BeadChip assay were analyzed in relevance to known functional genes against the DAVID bioinformatics database (http://david.abcc.ncifcrf.gov/home.jsp/). Among the 216 differentially methylated sites, a sum of 130 sites was associated with known functional genes, with 87 sites being hyper-methylated, whereas 43 sites hypo-methylated. A subset of genes in either the hyper- or hypo-methylated genes was shown to have single nucleotide polymorphism (SNP) from query sites (Supplementary Table 1). The top ten genes with the greatest extent of hyper-methylation were ZNF638, CYP3A43, UNC80, AIRE, RBPJL, SCGB1D1, FAM49B, FLJ44606, PYY and NLK. The hyper-methylated loci were at the body region among eight of the above ten genes, and three of them had SNPs (Table 1). The top ten genes with the greatest extent of hypo-methylation were SERAC1, ADCY5, HOXD11, ZNF257, CLTCL1, ZNF702P, GORASP2, POP1 and TSPAN3. Among this latter set, the hypo-methylated sites occurred largely (n = 7) around the promoter region, while none was associated with SNP (Table 1).
Ontological profiling of differentially methylated blood genes in MTLE
With bioinformatics analysis per the DAVID and GO databases, the 216 differential DNA methylation sites could be linked to multiple genes arranged in 4 groups in context of cellular component (CC) pathways. These included (1) GO:0030529~ribonucleoprotein complex (involving 8 differentially methylated genes, P = 0.077 between MTLE and control groups by t-test, same data presenting format below); (2) GO:0005792~microsome (5 genes, P = 0.092); (3) GO:0031981~nuclear lumen (16 genes, P = 0.094) and (4) GO:0042598~vesicular fraction (5 genes, P = 0.094). Statistical analyses indicated that the extent of methylation appeared to approach significant differences between the MTLE and control groups for each of the above CC systems, with the fold of enrichment ranging from 1.5 to 3 (Supplementary Table 2).
The differentially methylation sites could be linked to 9 groups in context of biological processing systems (BP pathways). These included (1) GO:0040008~regulation of growth (involving 8 differentially methylated genes, P = 0.010); (2) GO:0001501~skeletal system development (7 genes, P = 0.025); (3) GO:0048705~skeletal system morphogenesis (4 genes, P = 0.044); (4) the GO:0016055~Wnt receptor signaling pathway (4 genes, P = 0.067); (5) GO:0009952~anterior/posterior pattern formation (4 genes, P = 0.076); (6) GO:0009123~nucleoside monophosphate metabolic process (3 genes, P = 0.078); (7) GO:0010720~positive regulation of cell development (3 genes, P = 0.085); (8) GO:0006164~purine nucleotide biosynthetic process (4 genes, P = 0.086) and (9) GO:0006396~RNA processing (8 genes, P = 0.090). The extent of methylation changes showed statistically significant differences for groups (1), (2) and (3) between the MTLE and control groups, with the fold of enrichment ranged from 2 to 6 (Supplementary Table 3). Multiple genes participated in the above significantly altered BP pathways, including TMX1, MAPT, PPP2CA, MAEL, NPR1, WRN, E4F1 and AFG3L2 in group (1); TAPT1, ALOX15, HOXB2, ANKRD11, DLL3, COL11A2 and HOXD11 in group (2); and HOXB2, ANKRD11, COL11A2 and HOXD11 in group (3).
In context of molecular function (e.g., biochemical cascade), the differentially methylated genes were organized into 11 groups, including (1) GO:0043168~anion binding (5 genes, P = 0.006); (2) GO:0043167~ion binding (44 genes, P = 0.019); (3) GO:0031404~chloride ion binding (4 genes, P = 0.022); (4) GO:0016712~oxidoreductase activity (3 genes, P = 0.022); (5) GO:0004886~retinoid-X receptor activity (2 genes, P = 0.067); (6) GO:0046872~metal ion binding (40 genes, P = 0.079); (7) GO:0030375~thyroid hormone receptor coactivator activity (2 genes, P = 0.082); (8) GO:0010861~thyroid hormone receptor activator activity (2 genes, P = 0.082); (9) GO:0003708~retinoic acid receptor activity (2 genes, P = 0.089); (10) GO:0043169~cation binding (40 genes, P = 0.089) and (11) GO:0005506~iron ion binding (6 genes, P = 0.089). The extent of differential methylation showed significant differences between the patients and controls for groups (1) to (4), including genes such as GABRB1, SLC34A2, CLCN6, CLCA4, CYP3A43, CYP3A4, CYP2C9 that are critically involved in ion channel functions and oxidoreductant activity. The fold of enrichment varied greatly from over 1 to nearly 30 (Supplementary Table 4).
Genomic network profiling of differential methylated blood genes in MTLE
With bioinformatics assessment against the KEGG database (http://www.genome.jp/kegg/), a total of 6 genomic networks/pathways were generated in context of potential biological interplays between/among the genes with differential methylation in MTLE (Table 2). These pathways were consolidated as (1) hsa00591: Linoleic acid metabolism; (2) hsa00983: Drug metabolism. (3) hsa00830: Retinol metabolism; (4) hsa00980: Metabolism of xenobiotics by cytochrome P450; (5) hsa00982: Drug metabolism; and (6) hsa00140:Steroid hormone biosynthesis. Statistically, the extent of differential methylation was significantly different between the patient and control groups for pathways (1) through (5), but not for pathway (6), with the differentially methylated pathways (hsa00980, hsa00983, hsa00982) involved in drug metabolism. The fold of enrichment among these sets of pathways ranged from over 7 to more than 16 (Table 2). Further bioinformatics analysis indicated that the hsa00980 pathway plays a critical role in drug metabolism (Supplementary Fig. 3), especially for the processing of the commonly used antiepileptics in the clinic, including carbamazepine, oxcarbazepine and valproic acid (Fig. 6).

This pathway participates in the metabolism of multiple drugs (see Supplementary Fig. 4). Shown are the parts related to the metabolism of several commonly used antiepileptics, i.e., carbamazepine, oxcarbazepine and valproic acid. Images are obtained from KEGG (http://www.kegg.jp/kegg/kegg1.html) with permission (Ref. #: 16835).
Preliminary analysis of differential DNA methylation relative to disease course, anti-epileptics resistance and MRI abnormality
The patient group available for the present study was not large; we carried out preliminary analyses on DNA methylation relative to some diseases sub-characteristics (Table 3; Fig. 7). Two-third cases of the group had a disease course for 10 years and longer; we found 9 genes in these patients with significantly differential methylation relative to the cases with disease duration less than 10 years, including the solute carrier family 45 member 4 and low density lipoprotein adaptor protein 1. Among the top 10 hypermethylated and top 10 hypomethylated DNA loci, each involved 9 genes (Supplementary Table 5). The extent of methylation was different at five functional genes between patients that were clinically assessed as being anti-epileptics resistant and sensitive, including the microtubule associated protein 1 light chain 3b, active BCR-related, kallikrein peptidase 14, allantoises and MAGI family member. Except for the first gene, this group represented the known gene-coding sites of the top 10 hypermethylated loci. Among the 10 hypomethylated loci, nine of which involved DNA coding areas (Supplementary Table 6). MRI abnormality was reported among a half of the patients, while 9 cases of them had hippocampal sclerosis (Table 3; Fig. 7C). Give that hippocampal sclerosis is pathologically characteristic of MTLE, we analyzed methylation profile between patients with hippocampal sclerosis relative to those lacked MRI detected brain abnormality (n = 14) (Table 3). In the top 10 differentially methylated sites, there were 7 gene-coding DNA loci, of which 3 were among the most hyper-methylated genes, while 4 belonged to the most hypo-methylated genes, respectively (Supplementary Table 7). While we attempted to profile the above differential methylation against the DAVID, GO and KEGG databases, no specifically remarkable associations were found between disease course, anti-epileptics resistance or MRI abnormality with particular GO pathways or genomic networks (data not shown).

Dot graphs plot the age of onset (A) and duration (B) of epilepsy among the 30 patients listed from 1–30. Panel (C) shows a cranial T2 weighted Magnetic Resonance Imaging (MRI) film from patient #22, with the arrow pointing to enhanced T2 signal suggestive of sclerosis of the right-side hippocampus. Panel (D) shows slow and spike waves recorded on regions representing the right temporal lobe of this patient during awake and sleep, especially evident at anterior temporal area.
Discussion
DNA methylation serves an important epigenetic mechanism for regulation of gene expression. It may occur at the coding, promoter or other regions of genomic sequence in a tissue and cell specific manner, and might be significantly altered in disease conditions57. In particular, methylation of the CpG island regions, which is often associated with promoters in mammalian genome, may play a key role in controlling the expression of corresponding downstream genes58. Although the relationship between peripheral epigenetic profile and central nervous system conditions could be very complex, exploring peripheral epigenetic alterations in brain disorders is considered a direction with translational significance, given that in most clinical settings only peripheral samples, especially blood, are available. Differential central and peripheral DNA methylation is reported lately in a variety of neurological and psychiatric disorders, with some studies showing a parallel pattern change in brain and blood derived samples43,44,45,46,47,48,49,51,52,53,54,55,56. For TLE, emerging data point to a potential role for aberrant epigenetic modulation in its etiology, pathogenesis or ictal pathophysiology37,38,39,40,41,42,59,60, while less is known about the peripheral epigenetic change in this disorder.
The present study extends a first set of human data demonstrating differential DNA methylation in blood genome in MTLE. We have identified 216 differential DNA methylation sites, with 164 sites being significantly hyper-methylated and 52 sites hypo-methylated, in the MTLE patient relative to healthy control groups, according to Methylation450 BeadChip analysis. We cross-validated DNA methylation with the pyrosequencing method at a subset (8) of CpG loci. Overall, methylation data reported by the two methods are consistent among this set of loci that have varying degrees of methylation. In fact, the methylation data reported by BeadChip and the pyrosequencing methods are positively correlated in a case-based manner (regardless of experimental groups) for four sites.
The Beadchip assay system can theoretically report methylation changes at 485,577 sites across the whole genome. Thus, the differential DNA methylation appears to occur at a fairly low rate (i.e., 4/10000) in relative to the whole blood genome (using a cutoff P value < 1.03e-07 for multi-factorial statistical testing). De- or hypo-methylation in the promoter/CpG island regions is generally considered to enhance the expression of downstream genes50,58. In this study, the overall ratio of hyper- to hypo-methylated sites around the promoter regions was 0.83, with that for the CpG islands and surrounding structures even lower (2/34 or 0.0625), suggestive of a predominantly down-regulated methylation at these structures. One may expect that such changes could enhance the expression of some corresponding genes. Of note, the incidence of methylation at the promoter regions as assessed in brain samples has been shown to be increased (about 81.5%) in patients with MTLE42, but reduced (over 90%, assessed in hippocampal tissue) in a mouse model of TLE60.
The bioinformatics profile of our data indicates that the differentially methylated regions can be correlated to 130 known functional genes. Among the top ten hyper- and top ten hypo-methylated genes (Table 2), a subset (i.e., GABRB1, LC34A2, CLCN6, CLCA4, CYP3A43, CYP3A4, CYP2C9) has been related to epilepsy in epidemiological, pathophysiological or clinical context (to be discussed further). Thus, our current data suggest that DNA methylation changes in MTLE can occur in many genes that are not (yet) known to be associated with epilepsy or seizure disorders. This finding is in line with the notion that multiple genetic/epigenetic factors could be involved in TLE or seizure disorders42.
Ion channel dysfunction represents a fundamental pathophysiological element underlying abnormal neuronal discharge in seizure disorders, with evidence supporting an involvement of genetic and epigenetic factors in epileptic channelopathies37,39,61,62. Mutations of the genes (SCN1A, SCN1B, SCN2A) coding some sodium channel subunits cause enhanced excitability of glutamatergic neurons and/or reduced excitability of GABAergic neurons in familial seizure disorders63,64,65,66,67,68. Mutation of the gene coding the chloride channel protein (CLCN2) might also potentiate neuronal excitability in idiopathic epilepsy69,70. In the present study, genes coding the chloride channel 6 (CLCN6), chloride channel accessory 4 (CLCA4), sodium carrier (SLC34A2) and GABA receptor subunit (GABRB1) proteins show differential methylation in the MTLE relative to control groups, based on blood sample testing.
Resistance to antiepileptic drugs in MTLE can lead to progressive neuropathology and poor prognosis among many patients. Although the underlying mechanism remains unclear, changes in the enzyme systems responsible for drug metabolism could be a modulatory factor. The cytochrome P450 superfamily of proteins (CYPs) plays a major role in drug metabolism, accounting for about 75% of the total metabolism, especially deactivation and clearance of many drugs71,72,73. In the present study, several genes coding the members of CYPs, including CYP3A4, CYP3A43 and CYP2C9, are among the top ten differentially methylated genes in the MTLE relative to control groups. Our GO and KEGG bioinformatics analyses further indicate that they are part of the drug metabolic pathways responsible for some commonly used antiepileptic drugs, such as carbamazepine, oxcarbazepine and valproic acid. It should be noted that the epigenome-wide association analysis in a recent study with brain samples from TLE patients shows no significant methylation changes of the CYPs42. Therefore, the finding of differential methylation in CYP3A43, CYP3A4 and CYP2C9 in blood DNA samples in the present study appears to more likely reflect a peripheral epigenetic modulation.
The current study also reveals differential DNA methylation at genomic loci related to some other biological, molecular or signaling systems. Specifically, the genes or genomic interplaying pathways involved in cell/tissue morphogenesis and development, steroid hormone biosynthesis and metabolism as well as skeletal system development exhibit significantly differential methylation in the current MTLE relative to control groups. Further studies will be needed to understand these epigenetic changes in biological and clinical perspectives. Nonetheless, it is noteworthy that many clinical studies have suggested that chronic epilepsy or perhaps the long-term use of antiepileptic drugs may cause growth/developmental problems in children, while abnormal steroid hormone metabolism and certain forms of osteopathy are noted in patients under drug treatments74,75,76,77,78. In the current cohort of male and female patients and controls, the mean ages were 25 to 35 years, while one-third of the patients with epilepsy onset before 12 year-old (Table 3).
TLE in general and MTLE in specific are complex in clinical setting, and may present with a great spectrum of neurological symptoms, location/extent of cerebral lesions in imaging study, pattern of EEG alterations. Upon receiving medical attention, they are treated with anti-epileptics and other drugs accordingly. This was the case for the epileptic patients enrolled in our current study. While we have attempted to profile the DNA methylation data from the patient group relative to demographic, clinical and brain imaging features using bioinformatics tools, the results do not yet allow a clear conclusion whether specific blood DNA methylation changes are related to several subtype clinical features (disease duration, anti-epileptics resistance and hippocampal sclerosis). Thus, studies with larger patient cohort would be needed for stratified analysis to determine if unique differential blood DNA methylation can be linked to particular subclinical features of MTLE, therefore allowing further development as peripheral biomarkers of this disorder.
Conclusion
In summary, the present study presents a preliminary set of human data indicating differential DNA methylation at multiple loci of whole blood genome in mesial temporal lobe epilepsy relative to healthy control. Based on bioinformatics analysis, some of the differentially methylated sites are associated with promoters, genes and molecular pathways important for biological and metabolic regulation of anion binding/ion channel activity, oxidoreductant activity, drug metabolism, growth regulation and skeletal system development. These epigenetic changes might reflect some of the pathogenic and/or compensative biological modulations in mesial temporal lobe epilepsy or epilepsy/seizure disorders in general.
Methods
Subjects
Patients and controls were enrolled into the current study following written informed consent obtained from all subjects. All research protocols were approved by the Ethics Committee of Central South University, Xiangya School of Medicine and the affiliated Xiangya Hospital, with experiments carried out in accordance with the guideline for study involving human subjects. A total of 30 patients (18 males and 12 females) and 30 sex- and age-matched healthy controls were recruited through our epilepsy clinic from October, 2010 to October, 2014. Patients and controls were all Han Chinese, without previous history of seizure disorders or familial history of other neurological diseases (excluding secondary seizure disorders). All patients met the criteria for the diagnosis of MTLE set by the International League Against Epilepsy (ILAE), including clinical characteristics of simple or complex partial seizures and ictal scalp electroencephalogram (EEG) findings of spike, sharp, slow-spike and slow-sharp waves related to medial temporal lobe regions. Abnormal brain magnetic resonance image (MRI) findings, including hippocampal sclerosis, encephalomal acia foci, lacunar infarction or encephalomal atrophy, were present in approximately a half of the patients. Patients were treated with common anti-epileptics including carbamazepine, oxcarbazepine and valproic acid to control symptoms according to an individually-managed principle. The demographic, clinical, brain imaging and EEG profiles of the subjects are summarized as Table 3, with some metric data (age at disease onset and duration of epilepsy) and representative MRI and EEG presented as Fig. 7.
Whole blood DNA extraction
Frozen blood samples from patients and control subjects were thawed at 37 °C, followed immediately by whole blood DNA extraction using a commercial kit (Beijing Adly biological company, Beijing, China) according to manufacturer’s instructions. DNA concentration and purity were determined in a NanoDrop spectrophotometer by measuring the optic density ratio of maximal absorbent wavelengths at 260 nm to 280 nm (OD260/280) using salmon sperm DNA as an internal standard. Samples with DNA concentration greater than 50 ng/μl and OD260/280 ratio from 1.60 to 2.10 were used for further experimentations. Integrity of the extracted DNA from all samples was checked by agarose gel electrophoresis with ethidium bromide stain.
BeadChip DNA methylation assay
The pattern and extent of DNA methylation were assayed using the Infinium HumanMethylation450 BeadChip Kit (Illumina, Inc., San Diego, CA, USA). The kit contained 12332 probes, reporting sample-dependent and independent DNA methylation at specific genomic sites or for assay quality control. DNA samples were processed via a series of steps, including DNA denaturation, whole genome amplification, fragmentation, precipitation, resuspension and hybridization, according to manufacturer’s protocols. Signal (grey scale) of the final hybridization products was captured with the iScan system (Illumina, Inc.). The extent of methylation at a given site was determined by the β value, calculated as β = max (signal B, 0)/[max (signal A, 0) + max (signal B, 0) + 100], ranging from 0–1, representing completely not to totally methylated.
Pyrosequencing characterization of selected DNA methylation loci
After obtained information about the differentially methylated sites from the Methylation450 BeadChip analysis, we selected eight CpG loci with varying extent of methylation (cg17461336, cg09248887, cg06852575, cg04358264, cg10690677, cg21593669, cg25291941 and cg11582617) for evaluation by means of Pyrosequencing, as a measure of assay cross-validation. To this end, blood DNA samples from 15 MTEL cases and 15 sex and age-matched control cases were bisulfited, followed by PCR amplification of the regions of interest using the PyroMark PCR kit (Qiagen, CA, USA) according manufacturer’s instruction. Nucleotide probes included a biotinylated version allowing detection by streptavidin sepharose, as listed below: (1) cg17461336: GGTTAATTAGGGTATGATATAGAGTAAGA (F), ACCCAAATATACACCACCTAAAT (R-bio) and GGGTATGATATAGAGTAAGATT (S); (2) cg09248887: TGAGGTTATTGTTAGGGAATAGGAGAT (F), CCTTCATCAATCATCCTCCCTTCAAA (R-bio) and CACAATACTTAATTTTTTAACTTC (S); (3) cg06852575: GATTTGTAGGTATTGGGAGATTTT (F), CAAATTCCAACCACCCCCTTC (R-bio) and AGAAATGGTGAGAGTG (S); (4) cg04358264: AATGGGTAAAGTTATAGTGGATTAATT (F), AACCACTTTACAAAAATATTATCAAATACT (R-bio) and AATTAAATTTTAGTTTTGATAAAGG (S); (5) cg10690677: AGTTGATTGTTTTAAAAGGGTAAAGG (F), AACAAAAATCCTCTATTCCAACAAACTATC (R-bio) and GGTGGAGTAGGTTGAGTAGA (S); (6) cg21593669: AGATTGTAGGAGTAGGAGGTATAGTATTA (F), AACCAAAAAACTAAACAACATCTCTAC (R-bio) and GGAGGTATAGTATTAGGATG (S); (7) cg25291941: TGAAGGGGTTGTAAGAGGG (F), ACTCCCAACTCTCTATAAATCTT (R-bio) and TGTAAGAGGGAAGGT (S); and (8) cg11582617: GGAAGTTATTGTGGGTGGAGTTAG (F), ACCCCCTTATCTAAAAAACCCTACTCTT (R-bio) and GTGGGTGGAGTTAGT (S). Pyrosequencing assay was carried out on a PyroMark Q24 instrument using the PyroMark Gold Q24 Reagents (Qiagen, CA, USA). Purification and subsequent processing of the biotinylated single-stranded DNA were performed following the manufacturer’s recommendations.
Data and bioinformatics analyses
Differential DNA methylation sites were selected and analyzed with the BeadStudio Methylation Module v3.2 software (Illumina, Inc.) (http://www.illumina.com). The Pyrosequencing data were analyzed using the PyroMark Q24 software (Qiagen, CA, USA). R language (https://www.r-project.org/) was used to draw Manhattan scatterplot, and the MultiExperiment Viewer (MeV) was applied to perform microarray clustering-analysis. Methylation involving gene promoters was defined if it occurred at regions of TSS1500 (upstream), TSS200 (upstream), 5′UTR and the first exon. Methylation involving the CpG island structures was defined as to occur at either CpG island per se or the N-Shore, S-Shore, N-Shelve and S-Shelve regions, with Shores occupying 0–2 kb from CpG island and Shelves covering 2–4 kb from the CpG island. Differential methylation genes were selected using the Database for Annotation, Visualization and Integrated Discovery (DAVID) bioinformatics database (http://david.abcc.ncifcrf.gov/home.jsp/). Differentially methylated genes were selected based on a P < 1.03e-07 for multi-factorial statistical comparison. Differential gene pathways were determined using the gene ontology (GO) (http://geneontology.org/) and Kyoto Encyclopedia of Genes and Genomes (KEGG) (http://www.genome.jp/kegg/) databases. Means of designated comparing groups were statistically analyzed with Student-t test (e.g., age comparison) or nonparametric test with Bonferroni correction (SPSS21.0), with the minimal significant level of difference set at P < 0.05. Correlation analyses and curve fitting were performed using the Prism 4 program (Prism GraphPad, San Diego, CA). Figure panels were assembled with Photoshop 7.1.
Additional Information
How to cite this article: Long, H.-Y. et al. Blood DNA methylation pattern is altered in mesial temporal lobe epilepsy. Sci. Rep. 7, 43810; doi: 10.1038/srep43810 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Strzelczyk, A., Reese, J. P., Dodel, R. & Hamer, H. M. Cost of epilepsy: a systematic review. Pharmacoeconomics 26, 463–476 (2008).
Téllez-Zenteno, J. F. & Hernández-Ronquillo, L. A review of the epidemiology of temporal lobe epilepsy. Epilepsy Res Treat 2012, 630853 (2012).
Chang, L. & Wang, X. S. Progress in epigemiological investigation of epilepsia in China. Intl J Neurol Neurosurg 2, 161–164 (2012).
Pi, X. et al. Prevalence and clinical characteristics of active epilepsy in southern Han Chinese. Seizure 23, 636–640 (2012).
Berg, A. T. The natural history of mesial temporal lobe epilepsy. Curr Opin Neurol 21, 173–178 (2008).
Thom, M., Mathern, G. W., Cross, J. H. & Bertram, E. H. Mesial temporal lobe epilepsy: How do we improve surgical outcome? Ann Neurol 68, 424–434 (2010).
Woermann, F. G. & Vollmar, C. Clinical MRI in children and adults with focal epilepsy: a critical review. Epilepsy Behav 15, 40–49 (2009).
Richardson, M. Update on neuroimaging in epilepsy. Expert Rev Neurother 10, 961–73 (2010).
Bonilha, L. et al. The brain connectome as a personalized biomarker of seizure outcomes after temporal lobectomy. Neurology 84, 1846–1853 (2015).
Spigelman, I. et al. Dentate granule cells form novel basal dendrites in a rat model of temporal lobe epilepsy. Neuroscience 86, 109–120 (1998).
Houser, C. R. Neuronal loss and synaptic reorganization in temporal lobe epilepsy. Adv Neurol 79, 743–761 (1999).
Neves, R. S. et al. Granule cell dispersion is associated with memory impairment in right mesial temporal lobe epilepsy. Seizure 21, 685–690 (2012).
Yan, X. X. et al. BACE1 elevation is associated with aberrant limbic axonal sprouting in epileptic CD1 mice. Exp Neurol 235, 228–237 (2012).
Thom, M. Review: Hippocampal sclerosis in epilepsy: a neuropathology review. Neuropathol Appl Neurobiol 40, 520–543 (2014).
Bedner, P. et al. Astrocyte uncoupling as a cause of human temporal lobe epilepsy. Brain 138(Pt 5), 1208–1222 (2015).
Lucchi, C., Vinet, J., Meletti, S. & Biagini, G. Ischemic-hypoxic mechanisms leading to hippocampal dysfunction as a consequence of status epilepticus. Epilepsy Behav 49, 47–54 (2015).
Szokol, K. et al. Augmentation of Ca (2+) signaling in astrocytic endfeet in the latent phase of temporal lobe epilepsy. Front Cell Neurosci 9, 49 (2015).
Buckmaster, P. S. Does mossy fiber sprouting give rise to the epileptic state? Adv Exp Med Biol 813, 161–168 (2014).
Patterson, K. P., Baram, T. Z. & Shinnar, S. Origins of temporal lobe epilepsy: febrile seizures and febrile status epilepticus. Neurotherapeutics 11, 242–250 (2014).
Dossani, R. H., Missios, S. & Nanda, A. The Legacy of Henry Molaison (1926–2008) and the Impact of His Bilateral Mesial Temporal Lobe Surgery on the Study of Human Memory. World Neurosurg 84, 1127–35 (2015).
Jin, S. H., Jeong, W. & Chung, C. K. Mesial temporal lobe epilepsy with hippocampal sclerosis is a network disorder with altered cortical hubs. Epilepsia 56, 772–779 (2015).
Aronica, E. & Gorter, J. A. Gene expression profile in temporal lobe epilepsy. Neuroscientist 13, 100–108 (2007).
Winden, K. D. et al. A systems level, functional genomics analysis of chronic epilepsy. PLoS One 6, e20763 (2011).
Hessel, E. V. et al. Identification of Srp9 as a febrile seizure susceptibility gene. Ann Clin Transl Neurol 1, 239–2350 (2014).
Eun, J. P., Choi, H. Y. & Kwak, Y. G. Proteomic analysis of human cerebral cortex in epileptic patients. Exp Mol Med 36, 185–191 (2004).
He, S. et al. Proteomic analysis and comparison of the biopsy and autopsy specimen of human brain temporal lobe. Proteomics 6, 4987–2996 (2006).
Yang, J. W., Czech, T., Felizardo, M., Baumgartner, C. & Lubec, G. Aberrant expression of cytoskeleton proteins in hippocampus from patients with mesial temporal lobe epilepsy. Amino Acids 30, 477–493 (2006).
Huang, Z. L. et al. Proteomic screening of postsynaptic density proteins related with temporal lobe epilepsy. Zhonghua Yi Xue Za Zhi 88, 3205–3209 (2008).
Li, A. et al. Proteomic profiling of the epileptic dentate gyrus. Brain Pathol 20, 1077–1089 (2010).
Mériaux, C. et al. Human temporal lobe epilepsy analyses by tissue proteomics. Hippocampus 24, 628–642 (2014).
Pirker, S. et al. Dynamic up-regulation of prodynorphin transcription in temporal lobe epilepsy. Hippocampus 19, 1051–1054 (2009).
Venugopal, A. K. et al. Transcriptomic Profiling of Medial Temporal Lobe Epilepsy. J Proteomics Bioinform 5, doi: 10.4172/jpb.1000210 (2012).
Bando, S. Y. et al. Complex network analysis of CA3 transcriptome reveals pathogenic and compensatory pathways in refractory temporal lobe epilepsy. PLoS One 8, e79913 (2013).
Eid, T. et al. Gene expression of glutamate metabolizing enzymes in the hippocampal formation in human temporal lobe epilepsy. Epilepsia 54, 228–238 (2013).
Teocchi, M. A. et al. Hippocampal gene expression dysregulation of Klotho, nuclear factor kappa B and tumor necrosis factor in temporal lobe epilepsy patients. J Neuroinflammation 10, 53 (2013).
Zucchini, S. et al. Identification of miRNAs differentially expressed in human epilepsy with or without granule cell pathology. PLoS One 9, e105521 (2014).
Van Liefferinge, J. et al. Altered vesicular glutamate transporter expression in human temporal lobe epilepsy with hippocampal sclerosis. Neurosci Lett 590, 184–188 (2015).
Kobow, K. et al. Increased reelin promoter methylation is associated with granule cell dispersion in human temporal lobe epilepsy. J Neuropathol Exp Neurol 68, 356–364 (2009).
Kobow, K., El-Osta, A. & Blümcke, I. The methylation hypothesis of pharmacoresistance in epilepsy. Epilepsia 54 (Suppl 2), 41–47 (2013).
Zhu, Q. et al. Increased expression of DNA methyltransferase 1 and 3a in human temporal lobe epilepsy. J Mol Neurosci 46, 420–426 (2012).
Belhedi, N. et al. Increased CPA6 promoter methylation in focal epilepsy and in febrile seizures. Epilepsy Res 108, 144–148 (2014).
Miller-Delaney, S. F. et al. Differential DNA methylation profiles of coding and non-coding genes define hippocampal sclerosis in human temporal lobe epilepsy. Brain 138(Pt 3), 616–631 (2015).
Bacalini, M. G. et al. Identification of a DNA methylation signature in blood cells from persons with Down Syndrome. Aging (Albany NY) 7, 82–96 (2015).
Sabunciyan, S. et al. Genome-wide DNA methylation scan in major depressive disorder. PLoS One 7, e34451 (2012).
Azakli, H. et al. Whole mitochondrial DNA variations in hippocampal surgical specimens and blood samples with high-throughput sequencing: a case of mesial temporal lobe epilepsy with hippocampal sclerosis. Gene 529, 190–194 (2013).
Masliah, E., Dumaop, W., Galasko, D. & Desplats, P. Distinctive patterns of DNA methylation associated with Parkinson disease: identification of concordant epigenetic changes in brain and peripheral blood leukocytes. Epigenetics 8, 1030–1038 (2013).
Tylee, D. S., Kawaguchi, D. M. & Glatt, S. J. On the outside, looking in: a review and evaluation of the comparability of blood and brain “-omes”. Am J Med Genet B Neuropsychiatr Genet 62B, 595–603 (2013).
Ladd-Acosta, C. et al. Common DNA methylation alterations in multiple brain regions in autism. Mol Psychiatry 19, 862–871 (2014).
Mather, K. A., Kwok, J. B., Armstrong, N. & Sachdev, P. S. The role of epigenetics in cognitive ageing. Int J Geriatr Psychiatry 29, 1162–1171 (2014).
Li, H. J. et al. Alteration of Scn3a expression is mediated via CpG methylation and MBD2 in mouse hippocampus during postnatal development and seizure condition. Biochim Biophys Acta 1849, 1–9 (2015).
Di Francesco, A. et al. Global changes in DNA methylation in Alzheimer’s disease peripheral blood mononuclear cells. Brain Behav Immun 45, 139–144 (2015).
Tannorella, P. et al. Methylation analysis of multiple genes in blood DNA of Alzheimer’s disease and healthy individuals. Neurosci Lett 600, 143–147 (2015).
Numata, S. et al. Blood diagnostic biomarkers for major depressive disorder using multiplex DNA methylation profiles: discovery and validation. Epigenetics 10, 135–41 (2015).
Yang, G. et al. Detection of global DNA hypomethylation of peripheral blood lymphocytes in patients with infantile spasms. Epilepsy Res 109, 28–33 (2015).
Walton, E. et al. Correspondence of DNA Methylation Between Blood and Brain Tissue and its Application to Schizophrenia Research. Schizophr Bull 42, 406–14 (2015).
Graff, J. et al. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 483, 222–226 (2012).
Attwood, J. T., Yung, R. L. & Richardson, B. C. DNA methylation and the regulation of gene transcription. Cell Mol Life Sci 59, 241–257 (2002).
Takai, D. & Jones, P. A. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci USA 99, 3740–3745 (2002).
Qureshi, I. A. & Mehler, M. F. Epigenetic mechanisms underlying human epileptic disorders and the process of epileptogenesis. Neurobiol Dis 39, 53–60 (2010).
Miller-Delaney, S. F. et al. Differential DNA methylation patterns define status epilepticus and epileptic tolerance. J Neurosci 32, 1577–1588 (2012).
Avanzini, G., Franceschetti, S. & Mantegazza, M. Epileptogenic channelopathies: experimental models of human pathologies. Epilepsia 48 (Suppl 2), 51–64 (2007).
Helbig, I., Scheffer, I. E., Mulley, J. C. & Berkovic, S. F. Navigating the channels and beyond: unravelling the genetics of the epilepsies. Lancet Neurol 7, 231–45 (2008).
Kauffman, M. A., Levy, E. M., Consalvo, D., Mordoh, J. & Kochen, S. GABABR1 (G1465A) gene variation and temporal lobe epilepsy controversy: new evidence. Seizure 17, 567–571 (2008).
Mantegazza, M. et al. Identification of an Nav1.1 sodium channel (SCN1A) loss-of-function mutation associated with familial simple febrile seizures. Proc Natl Acad Sci USA 102, 18177–18182 (2005).
Meadows, L. S. et al. Functional and biochemical analysis of a sodium channel beta1 subunit mutation responsible for generalized epilepsy with febrile seizures plus type 1. J Neurosci 22, 10699–709 (2002).
Scalmani, P. et al. Effects in neocortical neurons of mutations of the Na (v) 1.2 Na+ channel causing benign familial neonatal-infantile seizures. J Neurosci 26, 10100–10109 (2006).
Lv, N. et al. Association study between polymorphisms in the CACNA1A, CACNA1C, and CACNA1H genes and drug-resistant epilepsy in the Chinese Han population. Seizure 30, 64–69 (2015).
Zhou, L. et al. ABCB1, ABCC2, SCN1A, SCN2A, GABRA1 gene polymorphisms and drug resistant epilepsy in the Chinese Han population. Pharmazie 70, 416–420 (2015).
Kleefuss-Lie, A. et al. CLCN2 variants in idiopathic generalized epilepsy. Nat Genet 41, 954–945 (2009).
Niemeyer, M. I. et al. No evidence for a role of CLCN2 variants in idiopathic generalized epilepsy. Nat Genet 42, 3 (2010).
Guengerich, F. P. Cytochrome p450 and chemical toxicology. Chem Res Toxicol 21, 70–83 (2008).
Tavira, B. et al. A search for new CYP3A4 variants as determinants of tacrolimus dose requirements in renal-transplanted patients. Pharmacogenet Genomics 23, 445–448 (2013).
Kang, Y. S. et al. The CYP3A4*18 genotype in the cytochrome P450 3A4 gene, a rapid metabolizer of sex steroids, is associated with low bone mineral density. Clin Pharmacol Ther 85, 312–318 (2009).
Kruse, R. Osteopathies in antiepileptic long-term therapy (preliminary report). Monatsschr Kinderheilkd 116, 378–381 (1968).
Okabe, R. et al. Significance of serum CrossLaps as a predictor of changes in bone mineral density during estrogen replacement therapy; comparison with serum carboxyterminal telopeptide of type I collagen and urinary deoxypyridinoline. J Bone Miner Metab 22, 127–131 (2004).
Babayigit, A., Dirik, E., Bober, E. & Cakmakci, H. Adverse effects of antiepileptic drugs on bone mineral density. Pediatr Neurol 35, 177–181 (2006).
Bauer, S. et al. Early detection of bone metabolism changes under different antiepileptic drugs (ED-BoM-AED)–a prospective multicenter study. Epilepsy Res 106, 417–422 (2013).
Vestergaard, P. Effects of antiepileptic drugs on bone health and growth potential in children with epilepsy. Paediatr Drugs 17, 141–150 (2015).
Acknowledgements
This study was supported by the National Natural Science Foundation of China (NNSFC), grant #81401078 to H.Y.L. and #81371435 to B.X.; X.X.Y. is funded by NNSFC grant #31371095.
Author information
Authors and Affiliations
Contributions
B.X. and L.Z. conceived project idea and designed the experiments; H.Y.L., L.F., J.K., Z.H.L., W.B.X., L.L.L. and L.Z. recruited human subjects, collected clinical data and blood samples; H.Y.L. and J.K. performed DNA experiments and data analysis; and drafted the manuscript; X.X.Y. finalized the manuscript; All authors discussed the results, reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Long, HY., Feng, L., Kang, J. et al. Blood DNA methylation pattern is altered in mesial temporal lobe epilepsy. Sci Rep 7, 43810 (2017). https://doi.org/10.1038/srep43810
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep43810
This article is cited by
-
DNA Methylation Description of Hippocampus, Cortex, Amygdala, and Blood of Drug-Resistant Temporal Lobe Epilepsy
Molecular Neurobiology (2023)
-
Circulating cell-free DNA methylation mirrors alterations in cerebral patterns in epilepsy
Clinical Epigenetics (2022)
-
Utilization of machine learning for identifying symptom severity military-related PTSD subtypes and their biological correlates
Translational Psychiatry (2021)
-
Epigenome-wide association study of seizures in childhood and adolescence
Clinical Epigenetics (2020)
-
Dynamic Change of Shanks Gene mRNA Expression and DNA Methylation in Epileptic Rat Model and Human Patients
Molecular Neurobiology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
