
Abstract
Enumeration and especially molecular characterization of circulating tumour cells (CTCs) holds great promise for cancer management. We tested a modified type of an in vivo enrichment device (Catch&Release) for its ability to bind and detach cancer cells for the purpose of single-cell molecular downstream analysis in vitro. The evaluation showed that single–cell analysis using array comparative genome hybridization (array-CGH) and next generation sequencing (NGS) is feasible. We found array-CGH to be less noisy when whole genome amplification (WGA) was performed with Ampli1 as compared to GenomePlex (DLRS values 0.65 vs. 1.39). Moreover, Ampli1-processed cells allowed detection of smaller aberrations (median 14.0 vs. 49.9 Mb). Single-cell NGS data obtained from Ampli1-processed samples showed the expected non-synonymous mutations (deletion/SNP) according to bulk DNA. We conclude that clinical application of this refined in vivo enrichment device allows CTC enumeration and characterization, thus, representing a promising tool for personalized medicine.
Similar content being viewed by others


Introduction
Tumour metastasis is the advanced stage of cancer and accounts for approximately 90% of cancer-related deaths1. In the current model of tumour progression circulating tumour cells (CTCs) play an important role as the presence of CTCs has been correlated with metastatic relapse and disease progression2,3. In addition, the number of CTCs is a strong prognostic factor correlated with outcomes and superior to other variables such as PSA in progressive castration-resistant prostate cancer4. CTCs are thought to carry important molecular information from the primary tumour or metastatic sites5 so that CTCs harbour the potential value of a biomarker for monitoring genetic cancer progression6.
Although the diagnostic impact of CTC analysis may be considerable, their extremely low concentration makes it difficult to exploit their full potential7. Various technologies have been used for detection, enumeration, and isolation of CTCs from peripheral blood of patients8. So far, however, batch sampling of 10 ml of peripheral blood remains a limitation for many methods leading to suboptimal sensitivity for detection of CTCs9. Furthermore, batch sampling requires more or less continuous CTC turnover, which in fact might be neither continuous nor uniform, thus introducing additional bias. Also, CTCs may be quite fragile and escape CTC analyses during multi-step ex vivo isolation procedures10, this causing a process-related bias. In contrast to batch sample-based enrichment techniques, in vivo enrichment of CTCs may evade some of the bias.
The CellCollector CANCER01 (DC01, GILUPI) is a CE-approved medical device that uses antibodies against the epithelial cell adhesion molecule (EpCAM) for isolating CTCs directly from peripheral blood in vivo11. When compared in the same patient cohort the DC01 resulted in a higher number of patients positive for CTCs than CellSearch12 underlining DC01’s potential for CTC analysis. However, DC01 was not designed for molecular genetic characterization beyond CTC enumeration. To enable downstream analysis at the single-cell level we tested a novel device allowing recovery of attached cells [CellCollector CANCER03 (Catch & Release, C&R, GILUPI)] which is not yet CE-certified for clinical application, thus currently allowing for in vitro use only.
In the present study, we tested if the C&R which is also based on cell enrichment by EpCAM capture, allows isolation and recovery of single tumour cells in vitro. For this purpose, we investigated the charging and detachment performance of the CellCollector C&R in vitro using tumour cells suspended at different cell densities either in PBS or peripheral blood. In order to test the compatibility of the recovering protocol with downstream analysis for cell characterisation we amplified single cells recovered from the C&R using two whole genome amplification (WGA) strategies and analysed the amplified single-cell products using comparative genomic hybridization (array-CGH) and next generation sequencing (NGS).
Results
Catch and release performance of cells enriched by the CellCollector C&R
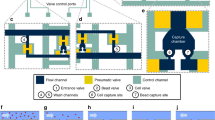
One of the disadvantages of the currently used in vivo enrichment devices is that captured cells firmly attach to the wire preventing CTCs to be recovered for further analysis. In contrast, the CellCollector C&R is coated with a polymer layer susceptible to enzymatic treatment (Fig. 1a and Supplementary Table S1). Therefore, captured cells can be detached from the wire and subjected to molecular analysis down to single cell level. When exposed to high target cell concentrations in PBS/2% BSA (i.e. 105 cells/ml) in vitro, most cells were found being captured in the groove of the C&R (Fig. 1b). We investigated the C&R’s capacity of isolating and detaching pre-stained cells (Fig. 1c) by spiking 1·102, 1·103, and 1·104 HT-29 cells per ml in 5 ml of peripheral blood (n = 3 each). Thirty minutes of exposure to the wire resulted in 7–113 isolated cells across all applied spiking protocols (for details see Supplementary Table S2). When forwarded to enzymatic treatment re-evaluation of the slides yielded average detachment efficiencies of 52%, 62%, and 74% (Fig. 2a). Control spiking experiments of 1·102, 1·103, and 1·104 HT-29 cells per ml in PBS/2% BSA (n = 2 each) resulted in average detachment rates of 65%, 75%, and 72%, respectively. Exposure of the C&R to 5·105 HT-29 (n = 5) and LNCaP cells (n = 6) in PBS/2% BSA resulted in 117–393 and 74–754 isolated cells, respectively. The detachment of cells performed similar in both cell lines with mean efficiencies of 81.3% (range 54.9%–93.2%) and 81.1% (range 63.51%–92.87), respectively (Fig. 2). Selected recovered cells (Fig. 1c) were forwarded to single-cell isolation by means of micromanipulation.

(a) Schematic composition of the C&R detector. Cells are captured by anti-EpCAM antibodies (step 1) and released by enzymatic disintegration of the polymer layer (step 2) (drawing by Georg Peinhaupt). (b) Scanning electron microscopic image of cells captured by the C&R, scale bars 200 μm (red) and 20 μm (white), respectively. (c) Captured HT-29 cells stained with Hoechst 33342 (blue) and CFSE (green), red lines indicate the borders of the functionalized wire, scale bar: 50 μm.

(a) We spiked HT-29 (circles) and LNCaP (squares) cells at densities of 100 to 100,000 cells per ml buffer (grey) or blood (red) and allowed the CellCollector to capture cells during 30 min incubation. Before and after cell detachment the cells on the C&R devices were counted and detachment efficiencies calculated. Detachment efficiencies shown as a function of spiked cell density (a) and number of captured cells before detachment (b), respectively. Horizontal bars (a) indicate mean detachment efficiencies; dashed line is to guide eye only (b).
Detachment from C&R does not affect single-cell array-CGH quality
Next, we amplified single-cell DNA of the micromanipulated cells (tumour cell lines) mentioned above by two different WGA technologies, i.e. Ampli1 and GenomePlex, and assessed their performance on multiple levels. First, we performed QC-PCR13 to identify cells suitable for downstream analysis. These data revealed that the number of cells yielding satisfying amplification products was higher when we used Ampli1 instead of GenomePlex (5/8 vs 3/8 single cells, respectively, Supplementary Fig. S1). WGA products with sufficient quality were then subjected to array-CGH. The resulting derivative log2 ratio spread (DLRS) values were used to evaluate the performance of the two WGA protocols and sampling procedures at the array level yielding a much more detailed picture. Generally, non-amplified genomic DNA scored lowest DLRS values (~0.3). Compared to that, Ampli1 and GenomePlex amplified single cells showed increased noise levels with Ampli1-processed cells yielding significantly lower DLRS values than cells processed by GenomePlex [mean DLRS 0.65 (n = 11) vs 1.39 (n = 5); unpaired t-test, p < 0.0001, Fig. 3a]. In addition, Ampli1-processed cells showed higher concordance with the genomic DNA reference sample (Supplementary Fig. S2). Moreover, we saw no significant difference between single cells directly isolated from cell suspension and those detached from C&R [DLRS mean values 0.71 (n = 5) vs. 0.60 (n = 6), p > 0.05; Fig. 3b], nor was there a cell line-specific difference (Supplementary Fig. S3). To further specify noise-related effects on the array-CGH profiles we had a closer look on HT-29 cells after GenomePlex (n = 4) and Ampli1 (n = 6) amplification. We detected a higher number of aberrations in Ampli1-processed cells (mean 37 vs 12) and significantly shorter aberrations as compared to GenomePlex-processed cells (mean 14.01 vs 49.9 Mb, p < 0.0001; Fig. 2c, Supplementary Tables S3 and S4). In addition, we observed single cell heterogeneity identifying microdeletions present in single cells but not in the bulk sample (Supplementary Figs S4 and S5, Supplementary Tables S3 and S4).

Plots of derivative log ratio spread (DLRS) values indicating noisiness in log ratio data of single cells are shown. (a) Single cells amplified with GenomePlex yielded higher DLRS values as cells amplified with Ampli1indicating poor signal-to-noise properties associated with less accurate CNV detection (unpaired t-test, p < 0.0001). (b) Using Ampli1, no difference (p = 0.16) was seen between single cells obtained from cell suspensions and cells that were recovered from C&R. All single cells amplified with Ampli1 have DLRS values < 1 (dashed grey line). (c) Single cells amplified by means of Ampli1 (white circles) yielded a higher number of gains and losses at low sizes as compared to WGA4-amplified cells (grey triangles). This was highly significant when we analyzed the aberration detection performance based on the WGA-method (white bar: Ampli1; grey bar: GenomePlex) used (p < 0.0001, Mann-Whitney U test).
Detection of non-synonymous mutations of C&R recovered single cells
Next, we forwarded non-amplified genomic DNA (gDNA) and C&R-treated and Ampli1-processed single cells (LNCaP and HT-29 cell line cells, ten and five cells, respectively) to targeted NGS. Sequencing yielded at minimum 1 million reads across all single cells with most abundant reads in the expected range between 130 and 139 bp (87–93% of reads on target; >90% above AQ20) and only a few amplicons to drop out (Supplementary Fig. S6).
For LNCaP cells, sequencing data unveiled a codon 6 frameshift mutation in PTEN at 100% mutant allele frequency and the TP53 P72R polymorphism for all ten single cells. Furthermore, we found a mutation in SMAD4 in eight of ten cells at mutant allele frequency rates ranging from 19% to 37%. Two single cells showed mutations in additional three genes (CTNNB1, FBXW7, SMO) at low mutation frequency (range 13% to 15%).
All five sequenced single HT-29 cell samples yielded homozygous TP53 (R273H) and SMAD4 (Q311) mutation at 100% mutant allele frequency. We detected KIT (M541L) and APC (E1554E) in all as well as BRAF (V600E) mutations in four of five single cells with their mutation frequencies being similar to HT-29 bulk DNA. One cell presented with a second SMAD4 (H530R) mutation albeit at a low allele mutation frequency of 18%. Additionally, we found noncoding SNPs in single cells of both cell lines. Tables 1 and 2 summarise the sequencing data of LNCaP and HT-29 cells, respectively.
Discussion
Recent clinical data14 and our own data (work in EU-FP7-consortium CTC-SCAN, to be reported elsewhere) suggest that in vivo enrichment of CTCs using CellCollector DC01 results in detection of higher CTC numbers and increased sensitivity for detection in patients as compared to CellSearch, which is the current gold standard for CTC enumeration. Based on the promising data regarding CTC isolation we investigated if we can successfully link the in vivo isolation approach with single-cell downstream analysis. In this study we report our data regarding a new version of an anti-EpCAM-coated detector, called C&R (for catch and release), designed (but not yet clinically certified) for CTC enrichment directly from peripheral blood. It resembles the CellCollector DC01 regarding its CTC capture principle but, in addition, comes with some advantages (summarized in Supplementary Table S1) with its cell detachment option being the most important. The rationale of this study was to examine if this setting would technically allow CTC characterisation beyond enumeration ideally; such a characterisation could be indicative for treatment decision15.
First, we tested the efficiency of the C&R to isolate and detach target cells expressing EpCAM. Exposing the device to high cell densities (i.e. 105 cells/ml) resulted in cells covering the device across the whole length (Fig. 1b). Detachment efficiencies were >80% for HT-29 and LNCaP cell line cells indicating, as expected, no difference between cells of different EpCAM expression levels. When exposed to less target cells (500–50 000) spiked into 5 ml of peripheral blood, the isolated number of cells ranged from 7 to 113 cells (see Supplementary Table S2) showing no correlation with spiked numbers of HT-29 cells. Towards lower target cell densities the detachment efficiency decreased to range between >50% and 75% (Fig. 2a).
We were able to perform single-cell DNA analysis after recovering LNCaP and HT-29 cells from the C&R device by enzymatically disintegrating its polymer layer (Fig. 1). Importantly, the procedure of recovering single cells from the C&R for downstream analysis did not affect the DNA quality (Fig. 3b). However, we found DNA quality to be significantly affected by the choice of the WGA method used for single cell amplification. This was evident across analysed cell lines as DLRS values, which reflect noisiness in log ratio data (where increased values correlate with poor signal-to-noise properties) hampering accurate detection of aberrations: Single cells amplified with linker adapter-based Ampli1 showed >2.5-fold reduction of background noise (Fig. 3a, Supplementary Fig. S2). As a consequence of this we achieved a significantly higher resolution in array-CGH profiles when using Ampli1 compared to GenomePlex (Fig. 3c, Supplementary Tables S3 and S4). This probably points at a shortcoming of the heat-fragmentation based GenomePlex procedure not seen in earlier experiments done on low-template analysis (containing several nuclei) of syncytial nuclear aggregates16.
Using Ampli1-based WGA in NGS we were able to add another layer of high resolution molecular genetic analysis to the array-CGH screening. We detected a number of sequence variants in the single cells, which were matching the data obtained from non-amplified bulk cell DNA (Tables 1 and 2). Notably, the frame shift mutation in PTEN turned out to be homozygous in all our single LNCaP cells (Table 1), whereas it was previously reported to be heterozygous17. Some cells showed mutations in additional genes (CTNNB1, FBXW7, SMO) at low mutation frequency (range 13 to 15%), which may be partly due to a true mutation event occurring at one allele in the hypertriploid/hypotetraploid cells, polymerase error during WGA or sequencing errors based on erroneous target enrichment18.
Growing knowledge in the field of cancer genetics will be constantly used to improve disease monitoring and treatment19 with CTC analysis evolving from mere enumeration towards molecular characterization. Promising targets driving cancer progression, drug resistance, and relapse such as androgen receptor variants, loss of PTEN, and ETS gene rearrangement have already been identified and liquid biopsy might provide a feasible non-invasive and low-risk access to optimized treatment20,21,22.
Our data show that target cells can be efficiently detached from the CellCollector C&R. Moreover, processed single cells yielded high quality DNA suitable for a combined screening-based (array-CGH) and target-specific (NGS) analysis approach. The data from our in vitro study provide evidence of C&R’s potential for clinical application, hence, its clinical application contributing to drive cancer treatment towards personalized medicine.
Methods
Sample collection and processing
This study was approved by the ethics committee of the Medical University of Graz (25–240 ex 12/13). Peripheral blood for spiking experiments was sampled from healthy controls. All methods were performed in accordance with the relevant guidelines and regulations.
Cell lines and evaluation of CellCollector C&R
Human cancer cell lines HT-29 (colon, kindly provided by GILUPI), LNCaP (prostate, kindly provided by Martine Mazel, Laboratory of Rare Circulating Human Cells, Montpellier, France), and PC-3 (prostate, obtained from ATCC) were cultured as recommended by the distributors. Cells were pre-stained with carboxyfluorescein succinimidyl ester (CFSE, Life Technologies; 5 μM in DMSO) and Hoechst 33342 and rinsed twice in 1x PBS. Stained cells were resuspended at different concentrations ranging from 100 to 100,000 cells/ml in 5 ml of PBS/2% BSA or peripheral blood. The latter was rinsed and restored to initial blood volume with PBS to avoid clotting of blood group antigen expressing HT-29 cells. The CellCollectors were charged with target cells by incubation with 5 ml spiked cell suspension at room temperature (RT) in a 9 ml reaction tube (Venosafe) on a horizontal roller mixer (SRT6D, Stuart Scientific) at 5 rpm for 30 min. For cell detachment, the C&R was incubated in pre-warmed 1x release buffer (GILUPI) at 37 °C for 10 min followed by agitation at 200 rpm on a plate shaker (DELFIA, Perkin-Elmer) at RT for 15 min. After centrifugation at 300× g (Heraeus Megafuge 40R, Thermo Fisher) for 10 min we gently removed the C&R device and centrifuged again. Collected cells were rinsed twice in 1x PBS/2% BSA, resuspended in 50 μl and transferred to chamber slides (Nunc). We counted the cells on the C&R before and after the detachment treatment under florescent microscope (Observer Z1, Zeiss) to evaluate cell detachment efficiency. We micromanipulated recovered cells using an inverted microscope (Axiovert M 200, Zeiss) as previously reported23. In short, we transferred 1 μl 1x PBS containing single cells or cell pools into caps of 200 μl PCR tubes for subsequent WGA using Ampli1™ WGA Kit (Silicon Biosystems) or GenomePlex Kit (Sigma). For the latter the PCR caps were preloaded with 9 μl of nuclease-free water. Samples were immediately centrifuged shortly, forwarded to WGA by Ampli1 or stored at −80 °C for later amplification using GenomePlex. Genomic DNA was extracted from cell lines using the DNeasy Blood and Tissue Kit (Qiagen) as described by the manufacturer. DNA concentration and purity were determined using a Nanodrop spectrophotometer (Thermo Fisher).
Single-cell whole genome amplification
Amplification using GenomePlex was performed as previously described with minor modifications24,25. Briefly, cells were digested in 1x single cell lysis and fragmentation buffer containing proteinase K (Sigma), followed by GenomePlex library preparation. Amplification was performed by adding 7.5 μl of 10 × amplification master mix, 48.5 μl nuclease-free water, and 5 μl WGA DNA polymerase.
Ampli1 amplification was performed according to the manufacturer’s protocol and published before26. Briefly, samples collected in 1 μl of PBS were lysed with 2 μl lysis reaction mix (42 °C, overnight). DNA was digested after adding 2 μl of digestion reaction mix at 37 °C for 3 h. Following enzyme inactivation (65 °C for 5 min), 5 μl of ligation mix containing pre-annealed adapter nucleotides were ligated to the fragmented DNA at 15 °C overnight. Finally, 40 μl of Primary PCR Reaction Mix was added to each sample and amplified as published26.
WGA products were purified using GenElute PCR Clean-Up Kit (Sigma) and subjected to a quality control multiplex PCR (QC-PCR) as described previously13,27. Samples not passing the QC criteria (<3 of 4 PCR products) were excluded from array-CGH and NGS analysis. Ampli1-processed samples were re-amplified for array-CGH and NGS as published recently28.
Array comparative genomic hybridization (array-CGH)
Each purified sample was labelled with the Bioprime Array CGH Genomic Labeling System (Life Technologies) as described previously24. Briefly, dCTP-Cy5 labelled sample DNA and dCTP-Cy3 labelled reference DNA (both GE Healthcare) were purified with Amicon Ultracel-30 filters (Millipore) and mixed in equal amounts of 250 ng for hybridization. We ran array-CGH on oligonucleotide-based SurePrint G3 Human CGH 8 × 60 K Microarrays (Agilent Technologies), processed the images with Feature Extraction software and performed data analysis using Agilent Genomic Workbench Lite Edition 7.0.4.0 (Agilent). Aberrations were detected using the ADM-2 algorithms with a threshold of eight and Centralization Algorithm set to a threshold of 6.0 with a bin size of 10. Calls needed to span at least three consecutive probes; the minimum absolute average was set to log2 ratio of 0.25. We used derivative log ratio spread (DLRS) values to assess the quality of array-CGH profiles29.
Circos plots showing the aberrations of single cells from LNCaP and PC-3 cell lines were generated using open access circos software (http://www.circos.ca).
Library preparation and Ion Torrent targeted next generation sequencing (NGS)
We amplified 10 ng DNA in Ion AmpliseqHiFi Master Mix using the multiplexed Ampli1 CHPCustom Beta primer panel covering 2265 COSMIC hot spot regions across 315 amplicons of 50 cancer-related genes (kindly provided by Silicon Biosystems; Supplementary Table S5). Quality and concentration of the amplicon pools were determined using the High Sensitivity DNA Kit on a 2100 Bioanalyzer (both Agilent). The amplicons were ligated to adapters containing Ion Express Barcode Adaptors Kit (Life Technologies) according to the manufacturer’s instructions. Prepared libraries were quantified by qPCR using the Ion Library Quantitation Kit (Life Technologies). Clonal amplification of the barcoded DNA libraries was carried out using Ion OneTouch 200 kit and libraries were sequenced on an Ion Proton sequencer using the Ion P1 chip (PGM sequencer; Ion Torrent).
All samples were sequenced to a depth of >1 million reads, with reads in the expected range between 90 and 130 bp, 87–93% of reads on target and more than 90% above AQ20. Mapping to hg19 and variant calling was performed using the open source Ion Torrent Suite Software (Life Technologies). All called variants were annotated using open source software (ANNOVAR30; SnpEff 31) and custom Perl scripts. All coding, non-synonymous mutations were further evaluated and visually inspected in Integrated Genome Viewer32 and variant calls resulting from technical read errors or sequence effects were excluded from the analysis. The threshold of detected variant frequency was set to 10% mutated alleles among all samples.
Scanning electron microscopy (SEM) images of cells captured by CellCollector C&R
A CellCollector C&R loaded with HT-29 cells was subjected to SEM sample processing as previously described33,34. Briefly, the devices were rinsed in 1x PBS, fixed in 0.1 M sodium cacodylate buffer (pH 7.4) containing 2% formaldehyde and 2.5% glutaraldehyde, and rinsed 1x in PBS. The cells were post-fixed in the 0.1 M sodium cacodylate buffer containing 2% osmium tetroxide, rinsed again, and dehydrated in an ascending ethanol series followed by acetone. The samples were dried using a BalTec CPD 030 critical point dryer, sputter coated using a Leica SCD 500 sputter coater, and visualized using a Zeiss DSM 950 scanning electron microscope.
Statistical methods
Unpaired two-tailed t test (DLRS) and Mann-Whitney U test (aberration size) were performed using GraphPad Prism 6.0.
Additional Information
How to cite this article: Chen, S. et al. Catch and Release: rare cell analysis from a functionalised medical wire. Sci. Rep. 7, 43424; doi: 10.1038/srep43424 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Weigelt, B., Peterse, J. L. & van ‘t Veer, L. J. Breast cancer metastasis: markers and models. Nat. Rev. Cancer 5, 591–602, doi: 10.1038/nrc1670 (2005).
Cristofanilli, M. et al. Circulating tumor cells: a novel prognostic factor for newly diagnosed metastatic breast cancer. J. Clin. Oncol. 23, 1420–1430, doi: 10.1200/JCO.2005.08.140 (2005).
Hayes, D. F. et al. Circulating tumor cells at each follow-up time point during therapy of metastatic breast cancer patients predict progression-free and overall survival. Clin. Cancer Res. 12, 4218–4224, doi: 10.1158/1078-0432.CCR-05-2821 (2006).
Scher, H. I. et al. Circulating tumour cells as prognostic markers in progressive, castration-resistant prostate cancer: a reanalysis of IMMC38 trial data. Lancet Oncol. 10, 233–239, doi: 10.1016/S1470-2045(08)70340-1 (2009).
Ni, X. et al. Reproducible copy number variation patterns among single circulating tumor cells of lung cancer patients. Proc. Natl. Acad. Sci. USA 110, 21083–21088, doi: 10.1073/pnas.1320659110 (2013).
Scher, H. I. et al. Circulating tumor cell biomarker panel as an individual-level surrogate for survival in metastatic castration-resistant prostate cancer. J. Clin. Oncol. 33, 1348–1355, doi: 10.1200/JCO.2014.55.3487 (2015).
Lianidou, E. S. Circulating tumor cell isolation: a marathon race worth running. Clin. Chem. 60, 287–289, doi: 10.1373/clinchem.2013.216010 (2014).
Alix-Panabieres, C. & Pantel, K. Technologies for detection of circulating tumor cells: facts and vision. Lab Chip 14, 57–62, doi: 10.1039/c3lc50644d (2014).
Andreopoulou, E. et al. Comparison of assay methods for detection of circulating tumor cells in metastatic breast cancer: AdnaGen AdnaTest BreastCancer Select/Detect versus Veridex CellSearch system. Int. J. Can. 130, 1590–1597, doi: 10.1002/ijc.26111 (2012).
Paterlini-Brechot, P. Circulating Tumor Cells: Who is the Killer? Cancer Microenvironment 7, 161–176, doi: 10.1007/s12307-014-0164-4 (2014).
Saucedo-Zeni, N. et al. A novel method for the in vivo isolation of circulating tumor cells from peripheral blood of cancer patients using a functionalized and structured medical wire. Int. J. Oncol. 41, 1241–1250, doi: 10.3892/ijo.2012.1557 (2012).
Gorges, T. M. et al. Enumeration and molecular characterization of tumor cells in lung cancer patients using a novel in vivo device for capturing circulating tumor cells. Clin. Cancer Res. 22, 2197–2206, doi: 10.1158/1078-0432.CCR-15-1416 (2016).
El-Heliebi, A., Chen, S. & Kroneis, T. Using multiplex PCR for assessing the quality of whole genome amplified DNA. Methods Mol. Biol. 1347, 119–128, doi: 10.1007/978-1-4939-2990-0_9 (2015).
Mandair, D. et al. A comparison of CellCollector with CellSearch in patients with neuroendocrine tumours. Endocr.-Relat. Cancer 23, L29–32, doi: 10.1530/ERC-16-0201 (2016).
Maheswaran, S. et al. Detection of mutations in EGFR in circulating lung-cancer cells. N. Engl. J. Med. 359, 366–377 (2008).
Holland, O. et al. Detection of fetal sex, aneuploidy and a microdeletion from single placental syncytial nuclear aggregates. Fetal Diagn. Ther., doi: 10.1159/000445112 (2016).
Spans, L. et al. Variations in the exome of the LNCaP prostate cancer cell line. Prostate 72, 1317–1327, doi: 10.1002/pros.22480 (2012).
McCall, C. M. et al. False positives in multiplex PCR-based next-generation sequencing have unique signatures. J. Mol. Diagn. 16, 541–549, doi: 10.1016/j.jmoldx.2014.06.001 (2014).
Heitzer, E. et al. Complex tumor genomes inferred from single circulating tumor cells by array-CGH and next-generation sequencing. Cancer Res. 73, 2965–2975, doi: 10.1158/0008-5472.CAN-12-4140 (2013).
Antonarakis, E. S. et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 371, 1028–1038, doi: 10.1056/NEJMoa1315815 (2014).
Sun, S. et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J. Clin. Invest. 120, 2715–2730, doi: 10.1172/JCI41824 (2010).
McMenamin, M. E. et al. Loss of PTEN expression in paraffin-embedded primary prostate cancer correlates with high Gleason score and advanced stage. Cancer Res. 59, 4291–4296 (1999).
El-Heliebi, A. et al. Resolving tumor heterogeneity: genes involved in chordoma cell development identified by low-template analysis of morphologically distinct cells. PLoS One 9, e87663, doi: 10.1371/journal.pone.0087663 (2014).
El-Heliebi, A. et al. Are morphological criteria sufficient for the identification of circulating tumor cells in renal cancer? J. Transl. Med. 11, 214, doi: 10.1186/1479-5876-11-214 (2013).
Kroneis, T. & El-Heliebi, A. Whole genome amplification of labeled viable single cells suited for array-comparative genomic hybridization. Methods Mol. Biol. 1347, 233–243, doi: 10.1007/978-1-4939-2990-0_16 (2015).
Klein, C. A. et al. Genetic heterogeneity of single disseminated tumour cells in minimal residual cancer. Lancet 360, 683–689, doi: 10.1016/S0140-6736(02)09838-0 (2002).
Van Beers, E. et al. A multiplex PCR predictor for aCGH success of FFPE samples. Br. J. Cancer 94, 333–337 (2006).
Czyz, Z. T., Hoffmann, M., Schlimok, G., Polzer, B. & Klein, C. A. Reliable single cell array CGH for clinical samples. PLoS One 9, e85907, doi: 10.1371/journal.pone.0085907 (2014).
Mohlendick, B. et al. A robust method to analyze copy number alterations of less than 100 kb in single cells using oligonucleotide array CGH. PLoS One 8, e67031, doi: 10.1371/journal.pone.0067031 (2013).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164, doi: 10.1093/nar/gkq603 (2010).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92, doi: 10.4161/fly.19695 (2012).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26, doi: 10.1038/nbt.1754 (2011).
Devetak, D., Klokocovnik, V., Lipovsek, S., Bock, E. & Leitinger, G. Larval morphology of the antlion Myrmecaelurus trigrammus (Pallas, 1771) (Neuroptera, Myrmeleontidae), with notes on larval biology. Zootaxa 3641, 491–500 (2013).
Lipovsek, S., Janzekovic, F., Leitinger, G. & Rupnik, M. S. Rab3a ablation related changes in morphology of secretory vesicles in major endocrine pancreatic cells, pituitary melanotroph cells and adrenal gland chromaffin cells in mice. Gen. Comp. Endocrinol. 185, 67–79, doi: 10.1016/j.ygcen.2013.01.007 (2013).
Acknowledgements
This study was funded by the Austrian Science Fund (FWF), project-no. I 1220-B19 (to P.S.) as part of the ERA-NET project in Translational Cancer Research (TRANSCAN) “Circulating Tumour Cells as Biomarker for Minimal Residual Disease in Prostate Cancer (CTC-SCAN)”. PhD candidate S.C. was trained within the frame of the PhD Program Molecular Medicine of the Medical University of Graz. The authors gratefully acknowledge Nina Schlögl, Daniel Kummer, Elisabeth Pritz, Babara Ramler, Gabi Wendt, Claudia Chudak, and Andrea Thüringer for their expert technical assistance as well as Dr. Linda Maria Schmölzer for collection of patient samples. The authors thank the project collaborators of CTC-SCAN for their advice and experience on CTC identification and Georg Peinhaupt for additional support.
Author information
Authors and Affiliations
Contributions
S.C., A.E-H., T.K. and P.S. designed the experiments. S.C. and A.E-H. performed the experiments. G.T., T.L., M.P., S.R., A.K. and K.P. contributed to staining evaluation for on-wire enumeration. Z.T.C. and B.T. helped with linker-adapter based single cell WGA and contributed reference samples for array-CGH. G.L. helped with design and coordination of SEM experiment. K.K. helped with NGS, including data processing, interpretation and discussion. S.C., A.E.-H., T.K. and P.S. interpreted the data and wrote the manuscript. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Chen, S., El-Heliebi, A., Tauber, G. et al. Catch and Release: rare cell analysis from a functionalised medical wire. Sci Rep 7, 43424 (2017). https://doi.org/10.1038/srep43424
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep43424
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
