
Abstract
Acute respiratory distress syndrome (ARDS) is a potentially devastating form of acute lung injury, which involves neutrophilic inflammation and pulmonary cell death. Reactive oxygen species (ROS) play important roles in ARDS development. New compounds for inhibiting the onset and progression of ARDS are required. Carnosine (β-alanyl-L-histidine) is a small di-peptide with numerous activities, including antioxidant effects, metal chelation, proton buffering capacity and the inhibition of protein carbonylation and glycoxidation. We have examined the preventive effects of carnosine on tissue injury, oedema and inflammation in a murine model for ARDS. Oral administration of carnosine suppressed lipopolysaccharide (LPS)-induced vascular permeability, tissue injury and inflammation in the lung. In vivo imaging analysis revealed that LPS administration increased the level of ROS and that this increase was inhibited by carnosine administration. Carnosine also suppressed LPS-induced neutrophilic inflammation (evaluated by activation of myeloperoxidase in the lung and increased extracellular DNA in bronchoalveolar lavage fluid). Furthermore, carnosine administration suppressed the LPS-induced endoplasmic reticulum stress response in vivo. These results suggest that the oral administration of carnosine suppresses LPS-induced lung injury via carnosine’s ROS-reducing activity. Therefore, carnosine may be beneficial for suppressing the onset and progression of ARDS.
Similar content being viewed by others

Introduction
Acute respiratory distress syndrome (ARDS) was first defined in 1967 by the American–European Consensus Conference and is a major clinical problem in intensive care unit patients1,2. Even though approximately 150,000 individuals per year receive a diagnosis of ARDS in the United States, a standard clinical protocol for treatment has not been established as yet, and ARDS mortality remains at 40–50%1,2.
ARDS is often associated with pneumonia (direct injury) or sepsis (indirect injury). These conditions result in the pulmonary inflammation and injury that arise from alveolar-capillary membrane damage and leakage of protein-rich oedema fluid into alveoli3,4,5. Epithelial and endothelial damage also induces severe inflammatory responses and an increase in vascular permeability, not only in the lungs but also in other organs, resulting in multiple organ failure3,4,5. Because clinical trials using steroids have not been able to show a significant improvement in patients’ mortality6,7, steroids are no longer routinely used to treat ARDS patients. Many other pharmacological therapies, such as β-2 agonists, surfactant protein C and statins have been investigated; however, these therapies have not been approved8,9,10,11. Therefore, identifying new strategies to prevent ARDS development is very important.
Reactive oxygen species (ROS) have been shown to play a major role in ARDS development12,13. In ARDS patients, ROS are produced extensively by infiltrating leukocytes, especially neutrophils. ROS damage not only the lung but also other organs, by promoting neutrophilic inflammation, increasing vascular permeability, and activating the coagulation system12,13. Moreover, ROS induce epithelial and endothelial damage, which are involved in ARDS development14. Increases in ROS levels in plasma, expired breath condensates and bronchoalveolar lavage fluid (BALF) have been reported in ARDS patients and ARDS-related animal models13,15,16,17,18.
ROS are reportedly involved in the formation and development of neutrophilic extracellular traps (NETs)19. NETs are composed of decondensed chromatin fibers and cytoplasmic protein, such as myeloperoxidase and neutrophil elastase. NETs are believed to be an important defensive system against bacterial infections. However, excessive activation of NETs is thought to be the cause of various inflammatory diseases, including lung diseases20,21. Furthermore, NETs formation has also been reported to play a major role in ARDS and a lipopolysaccharide (LPS)-induced lung injury model20,22,23.
The endoplasmic reticulum (ER) stress response results in the accumulation of unfolded and misfolded proteins and is reportedly induced by various stressors, including ROS24,25. The unfolded protein response (UPR) is an adaptive mechanism to refold unfolded and misfolded proteins in the ER. Glucose-regulated protein 78 (GRP78), a representative ER chaperone, confers protection against stressors by mediating this refolding. However, if ER stress is not resolved by adaptive UPR signaling, ER stress-dependent apoptotic signaling pathways are activated, such as phosphorylation of c-Jun N-terminal kinase and induction of CCAAT/enhancer-binding protein-homologous protein (CHOP)24,25. Therefore, the ER stress response is implicated in several diseases, such as cancer, inflammatory bowel disease and lung diseases24,26. Recent studies showed that LPS induced increases in various ER stress markers in the lung, and inhibition of ER stress ameliorated LPS-induced lung inflammation27,28. Thus, a compound that inhibits neutrophilic inflammation or the ER stress response would suppress the onset and progression of ARDS.
Carnosine (β-alanyl-L-histidine) is a small di-peptide with numerous activities, including antioxidant effects, metal ion chelation, proton buffering capacity, and inhibitory effects on protein carbonylation and glycoxidation29,30. Carnosine is abundantly present in skeletal muscles, cerebral cortex, kidney, spleen and plasma30. We have previously reported that carnosine suppresses Zn2+ -induced hypothalamic cell death and the ER stress response31,32. These results suggested that carnosine protects against Zn2+ -induced neurotoxicity by suppressing the ER stress response.
Recently, the efficacy of carnosine against lung injury has been shown in animal models. For example, carnosine ameliorates H9N2 swine influenza virus- or bleomycin-induced lung injury in vivo33,34. Ohata et al. shown that polaprezinc, a chelate compound consisting of zinc and carnosine, protects mice against intraperitoneal LPS administration-dependent septic shock35. However, the efficacy of carnosine against intratracheal LPS administration-induced lung injury (a major ARDS-related animal model) has not been shown. Furthermore, the protective mechanism of carnosine against LPS-induced lung injury is unknown.
We therefore examined the effect of carnosine in LPS-induced lung injury in the present study. Oral administration of carnosine suppressed oedema, tissue injury and inflammation in the lungs of mice that were administered LPS intratracheally. We also found that administration of carnosine suppressed neutrophilic inflammation (NETs formation) and the ER stress response by decreasing ROS production in vivo. These results suggest that carnosine might be beneficial for suppressing the onset and progression of ARDS.
Results
Effect of carnosine on LPS-induced lung injury
We examined the effect of carnosine on LPS-induced lung injury, an animal model of acute lung injury. Histopathological analysis of lung sections revealed that intratracheal LPS administration caused alveolar haemorrhage, leukocyte infiltration and severe lung interstitial oedema, and that this injury was suppressed by oral carnosine administration (Fig. 1a). Intratracheal LPS administration increased vascular permeability in lung tissue, and simultaneous carnosine administration suppressed this increase (Fig. 1b). Protein concentration in BALF, an indicator of lung injury and oedema, was also increased by LPS administration, and carnosine administration suppressed this increase (Fig. 1c). Moreover, LPS administration induced expression of pro-inflammatory cytokines (tumour necrosis factor-α, TNF-α and interleukin-6, IL-6) and chemokines (chemokine (CXC motif) ligand-1, CXCL1, and CXCL2) in lung tissue, whereas carnosine administration significantly suppressed this increase, except for TNF-α (Fig. 1d). In contrast, carnosine did not affect the LPS-induced expression of Toll-like receptor 4 (TLR4) and myeloid differentiation primary response gene 88 (MyD88). Overall, the results shown in Fig. 1 suggest that oral carnosine administration protects mice against LPS-induced lung injury.

Male ICR mice were intratracheally administered with LPS (1 mg/kg) or the LPS vehicle (Control). Mice were orally administered with the indicated doses of carnosine (Car, mg/kg) or saline (Vehicle), immediately before and 24 h after LPS administration (a,b), or immediately prior to LPS administration (c,d). Sections of pulmonary tissue (a) or BALF (c) were prepared 48 h after LPS administration. Sections were subjected to histopathological examination (H&E staining) (scale bar, 500 μm) (a). Evans blue dye (30 mg/kg) was administered intravenously 6 h after LPS administration, and 2 h later, was extracted from the lung samples and quantified (b). The amount of protein present in the BALF was determined by the Bradford method (c). Total RNA was extracted from lung 24 h after LPS administration, and subjected to real-time RT-PCR using a specific primer set for each gene. Values were normalized to Hprt1 and expressed relative to the Control (d). Values are mean ± S.E.M.; #P < 0.05; ** or ##P < 0.01 (*, vs Control; #, vs Vehicle).
In addition to the LPS-induced lung injury model, we also examined the effect of carnosine in a zymosan (a TLR2 ligand)-induced lung injury model. As shown in Supplementary Fig. S1, zymosan administration induced lung injury, increasing the protein concentration and the number of neutrophils in BALF. These effects were suppressed by the oral carnosine administration.
Effect of carnosine on LPS-induced ROS increase in vivo
ROS are involved in the onset and progression of acute lung injury12,13. We therefore monitored ROS levels using an in vivo imaging system. As shown in Fig. 2a and b, intratracheal LPS administration increased ROS levels in the lung. Conversely, simultaneous carnosine administration clearly suppressed this LPS-dependent ROS increase. We further examined the ROS-reducing activity of carnosine in vitro. As shown in Fig. 2c, treatment of RAW264 cells with phorbol myristic acid (PMA) induced ROS production. Addition of carnosine significantly suppressed the production of ROS. These results suggest that carnosine suppressed LPS-induced lung injury by suppressing increases in ROS levels.

Male ICR mice were intratracheally administered with LPS (1 mg/kg) or the LPS vehicle (Control). Mice were orally administered with the indicated doses of carnosine (Car, mg/kg) or saline (Vehicle) immediately prior to LPS administration. Luminescent probe (L-012, 75 mg/kg) was administered 6 h after the LPS administration. Isolated lungs were imaged using a Lumazone in vivo imaging system (a). The summed pixel intensity of the ROS signal was determined using SlideBook 6 software (b). RAW264 cells were pre-incubated with the indicated concentrations of carnosine (Car, mM) for 30 min. They were then incubated with PMA (100 nM) for 30 min in the presence of dihydroethidium (DHE), an indicator of superoxide. DHE fluorescence was measured using a fluorescence microplate reader (c). Values represent mean ± S.E.M. #P < 0.05; ** or ##P < 0.01 (b: vs Control; #, vs Vehicle, c: * vs Control, # vs PMA).
Effect of carnosine on LPS-induced inflammatory responses
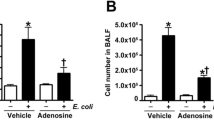
ROS are an exacerbating factor in neutrophilic inflammation19,36. We therefore monitored LPS-induced neutrophilic inflammatory responses by measuring the number of leucocytes in BALF 48 h after LPS administration. As shown in Fig. 3a,b, the total number of leucocytes, and especially the number of neutrophils, was increased by the LPS treatment, and these effects were suppressed by simultaneous carnosine administration. In contrast, LPS administration slightly increased the number of macrophages, and these increases were not suppressed by carnosine administration. We also examined the effect of carnosine on myeloperoxidase (MPO) activity (a marker of neutrophilic inflammation) in lung tissues. As shown in Fig. 3c, MPO activity in lung tissues was increased by LPS administration and this increase was suppressed by oral carnosine administration. Moreover, as shown in Supplementary Fig. S2, LPS treatment increased neutrophil elastase-positive cells in lung tissue, and this increase was clearly suppressed by carnosine administration.

Male ICR mice were intratracheally administered with LPS (1 mg/kg) or LPS vehicle (Control). Mice were orally administered with the indicated doses of carnosine (Car, mg/kg) or saline (Vehicle), immediately before and 24 h after LPS administration. BALF and lung homogenates were prepared 48 h after LPS administration. BALF cells were stained with Diff-Quik reagents after centrifugation with a Cytospin® 4 (scale bar, 100 μm) (a). The total cell numbers and the neutrophil numbers were determined (b). MPO activity in lung homogenates was measured using an MPO assay kit according to the manufacturer’s protocol (c). Values are mean ± S.E.M.; * or #P < 0.05; ** or ##P < 0.01. (*, vs Control; #, vs Vehicle).
Neutrophils release their DNA into the extracellular space to produce NETs20,22,23. As indicators of NETs formation, we monitored dsDNA levels in BALF, and citrullinated histone H3 (Cit-H3) levels in BALF and lung sections. As shown in Fig. 4a, dsDNA levels were increased by the LPS treatment and suppressed by simultaneous carnosine administration. The expression of Cit-H3 in BALF was also induced by the LPS treatment, and was suppressed by carnosine administration (Fig. 4b,c). Moreover, the expression of Cit-H3 in lung tissue was induced by LPS treatment, and again was clearly suppressed by carnosine administration (Fig. 4d). The results in Figs 3 and 4 suggest that carnosine administration protects against LPS-induced lung injury by suppressing neutrophilic inflammation.

Male ICR mice were intratracheally administered with LPS (1 mg/kg) or LPS vehicle (Control). Mice were orally administered with the indicated doses of carnosine (Car, mg/kg) or saline (Vehicle), immediately before and 24 h after LPS administration. BALF (a–c) and sections of pulmonary tissue (d) were prepared 48 h after LPS administration. The amount of double-stranded DNA (dsDNA) present in the BALF was determined using the Quant-iT™ PicoGreen® dsDNA Reagent and Kits according to the manufacturer’s protocol (a). BALF samples (2 μL) were analyzed by immunoblotting with an antibody against citrullinated histone H3 (Cit-H3) (b). The Cit-H3 band intensity was determined using Image J software. In the Control group, Cit-H3 expression was not detected (n.d.) (c). Immunohistochemical analysis of pulmonary tissue with an antibody against Cit-H3 was performed (scale bar, 100 μm) (d). Values are mean ± S.E.M.; #P < 0.05; ** or ##P < 0.01 (*, vs Control; #, vs Vehicle).
Effect of carnosine on LPS-induced pulmonary cell death and the ER stress response
ROS are thought to induce pulmonary cell death14. We therefore examined the effect of carnosine on LPS-induced pulmonary cell death using a terminal deoxynucleotidyl transferase dUTP nick-end labelling (TUNEL) assay. As shown in Fig. 5a,b, intratracheal LPS administration increased TUNEL-positive cells in the lung. Conversery, simultaneous carnosine administration suppressed LPS-induced pulmonary cell death. Next, we examined the cytoprotective effects of carnosine in vitro. As shown in Fig. 5c,d, treatment of A549 cells with menadione (to induce oxidative stress) decreased viable cell numbers.

Male ICR mice were intratracheally administered with LPS (1 mg/kg) or LPS vehicle (Control). Mice were orally administered with carnosine (Car; 100 mg/kg) or saline (Vehicle), immediately before and 24 h after LPS administration. Sections of pulmonary tissue were prepared 48 h after LPS administration. Sections were subjected to the TUNEL assay and DAPI staining (scale bar, 100 μm) (a). The numbers of TUNEL-positive cells were counted (b). A549 cells were incubated with menadione (Mena, 4 μM) for 24 h in the presence of the indicated concentrations (mM) of carnosine (Car). Viable cell numbers were determined using a WST-based cell counting kit (c) or CellTiter-Glo® 2.0 (d). Values are mean ± S.E.M.; ** or ##P < 0.01 (b: *, vs Control; #, vs Vehicle, c,d: # vs menadione).
Carnosine is thought to suppress the ER stress response31. Therefore, we monitored the expression of ER stress-related genes in lung tissue using real-time RT-PCR. LPS treatment induced the expression of Grp78, Gadd34, Edem and Pdi mRNA; carnosine administration significantly suppressed the expression of Grp78, Chop, Gadd34, Edem and Pdi mRNA (Fig. 6). These results suggest that carnosine administration protects against LPS-induced pulmonary cell death by suppressing the ER stress response.

Male ICR mice were intratracheally administered with LPS (1 mg/kg) or LPS vehicle (Control). Mice were orally administered with carnosine (Car; 100 mg/kg) or saline (Vehicle) immediately before LPS administration. Total RNA was extracted from lung and subjected to real-time RT-PCR using a specific primer set for each gene, 24 h after LPS administration. Values were normalized to Hprt1 and expressed relative to the Control. Values are mean ± S.E.M.; #P < 0.05; ** or ##P < 0.01 (*, vs Control; #, vs Vehicle).
Discussion
Carnosine is abundantly present in muscle and brain, and has various beneficial effects such as antioxidant activity, metal chelating effects, proton buffering capacity, anti-tumour cell growth activity and the inhibition of protein carbonylation and glycoxidation. Hence, carnosine is thought to be a good candidate for anti-aging or neuroprotective therapy37,38,39. Recently, we reported that carnosine suppresses Zn2+ -induced neuronal cell death, suggesting that carnosine could be a candidate for preventing or treating vascular-type dementia31,32. In the present study, we were interested in examining the effects of carnosine on ARDS development using an LPS-induced lung injury model. We showed that oral carnosine administration suppresses LPS-induced lung injury and inflammation, and we propose that carnosine may be beneficial for preventing ARDS development.
We focused on neutrophilic inflammation and the ER stress response as mechanisms of carnosine’s preventive effect on ARDS development. As shown in Figs 3 and 4, carnosine administration inhibited LPS-induced neutrophilic inflammation. Furthermore, as shown in Figs 5 and 6, carnosine administration inhibited the LPS-dependent ER stress response. ROS are reportedly thought to induce neutrophilic inflammation and the ER stress response19,24,26,36. For example, extracellular superoxide induces NETs, and treatment with diphenyleneiodonium, an inhibitor of NADPH oxidase, inhibits this NETs induction40. Regarding ER stress, cigarette smoke extract induces the apoptosis of bronchial epithelial cells through a superoxide anion-triggered signaling pathway mediated by protein kinase RNA-like endoplasmic reticulum kinase–eukaryotic initiation factor 2 alpha kinase (PERK-eIF2a, one of the ER stress sensors)41. We showed that carnosine administration suppressed the LPS-dependent ROS increase in lung tissue (Fig. 2). Considering these results, we suggest that carnosine suppressed both neutrophilic inflammation and the ER stress response by suppressing LPS-dependent ROS increases.
ARDS is often associated with not only pneumonia but also sepsis, and caecal puncture ligation (CLP) in mice induces phenomena similar to those observed in ARDS patients42. Moreover, mechanical ventilation (MV), a life-saving intervention for ARDS patients, also causes ventilator-induced lung injury (VILI), which increases mortality43. ROS have been shown to play a major role in sepsis and VILI in both clinical and animal models. In contrast, carnosine administration clearly suppressed the LPS-dependent ROS increase (Fig. 2). Although we have not examined the effects of carnosine in mice subjected to CLP or MV, we assume that carnosine administration may have preventive effects against CLP- or MV-induced lung injury by suppressing the associated ROS increases.
Considering the clinical application of carnosine, it is important to examine the effect of carnosine after the onset of lung injury, and the effect of carnosine on prognosis. As shown in Supplementary Fig. S3, even when carnosine was administered after LPS, carnosine suppressed the LPS-induced increase in the number of neutrophils and the protein concentration in BALF. Intratracheal administration of LPS and hydrochloric acid, a newly reported ARDS-related model44, as shown in Supplementary Fig. S4, this was lethal to mice, but carnosine administration increased their survival rate (P = 0.036). These results support our view that carnosine has the beneficial effects for preventing ARDS development.
We found that oral carnosine administration clearly suppressed LPS-induced ROS increases. To our knowledge, this is the first time that the ROS-reducing activity of carnosine has been confirmed by in vivo imaging analysis. However, the mechanism by which carnosine administration suppressed ROS production needs to be clarified. Carnosine treatment was previously reported to induce copper/zinc superoxide dismutase (Cu/Zn-SOD) and glutathione peroxidase enzymatic activities in a rat experimental subarachnoid haemorrhage model45. Furthermore, other groups showed that carnosine has a hydroxyl radical scavenging effect46,47. We suggest that carnosine suppresses LPS-dependent ROS increases by these mechanisms.
The effects of other antioxidant molecules on preventing ARDS have already been reported. For example, Cu/Zn-SOD or lecithinized Cu/Zn-SOD administration is also protective in ARDS-related animal models48,49. The new vitamin E derivative, ETS-GS, protected against CLP-induced systemic inflammation in rats50. Early administration of lipoic acid provided protective effects against LPS-induced oxidative stress in the lung51. Moreover, simvastatin reduced LPS-induced lung injury by decreasing neutrophil recruitment and radical formation52. Some investigations suggested beneficial effects of statins therapy in patients with sepsis and ARDS53,54, although stains have not been approved for this use. Therefore, it is necessary to compare the protective effects of carnosine against LPS-induced lung injury with those of other antioxidant molecules. Furthermore, combination effects of other antioxidants with carnosine have been reported in other organs. For example, carnosine plus vitamin E treatment more strongly suppressed LPS-induced liver injury compared with carnosine or vitamin E alone55. Moreover, α-lipoic acid and carnosine supplementation increased antioxidant activity in the serum, liver and skin of rats56. Examining combination effects of other antioxidants with carnosine in the LPS-induced lung injury model would be highly worthwhile.
In conclusion, we revealed that oral administration of carnosine suppresses LPS-dependent lung injury and inflammation. These results suggest that carnosine may be beneficial for suppressing the onset and progression of ARDS. Moreover, carnosine is used in anti-aging supplements in the United States. Therefore, carnosine could be useful not only as a prophylactic drug but also as a supplement preventing the onset and progression of ARDS.
Materials and Methods
Chemicals and animals
LPS from Escherichia coli (055:B5) was obtained from Sigma (St. Louis, MO). Diff-Quik was from Sysmex (Kobe, Japan). An antibody against actin was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against histone H3 (citrulline R2 + R8 + R17) and neutrophil elastase were purchased from Abcam (Cambridge, UK). L-carnosine, luminal-based chemiluminescent probe (L-012), Evans Blue Dye, zymosan, isoflurane and formalin neutral buffer solution were from WAKO Pure Chemicals (Tokyo, Japan). 4,6-diamidino-2-phenylindole (DAPI) was purchased from Dojindo (Kumamoto, Japan). RNeasy® kit was obtained from Qiagen (Hilden, Germany), PrimeScript® 1st strand cDNA Synthesis Kit was from Takara Bio (Ohtsu, Japan), and SsoFast™ EvaGreen Supermix was from Bio-Rad (Hercules, CA). Mounting medium for immunohistochemical analysis (VECTASHIELD™) was purchased from Vector Laboratories (Burlingame, CA). Mayer’s haematoxylin, 1% eosin alcohol solution and mounting medium for histological examination (malinol) were from MUTO Pure Chemicals (Tokyo, Japan). Novo-Heparin (5000 units) for injection was from Mochida Pharmaceutical (Tokyo, Japan). A549 cells or RAW264 cells were purchased from the American Type Culture Collection (Manassas, VA) or RIKEN BioResource Center (Tsukuba, Japan). ICR mice (6–7 weeks old, male) were purchased from Charles River (Yokohama, Japan). The experiments and procedures described here were carried out in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health, and were approved by the Animal Care Committee of Musashino University.
Treatment of mice with LPS and carnosine
Mice anesthetized with isoflurane were given a single intratracheal injection of LPS (1 mg/kg) in 0.9% NaCl (2 ml/kg), zymosan (1 mg/kg) in 0.9% NaCl (2 ml/kg) or 0.2 M hydrochloric acid (2 ml/kg) using a P200 micropipette via the mouth. During administration, the nostrils of the mice were blocked with a finger, so that the solutions were inhaled from the mouth into the respiratory tract as the mice breathed.
Mice were orally administered carnosine (10, 50, 100 mg/kg) in 0.9% NaCl by syringe using a sonde needle. The first administration of carnosine was given immediately before LPS administration (except for Supplementary Fig. S3). In control experiments, we examined the effect of administering carnosine alone, and found that it did not affect the lung histology, the protein concentrations or number of leukocytes in BALF, the plasma levels of malondialdehyde (an indicator of ROS), or the number of neutrophils in blood (Supplementary Fig. S5). The levels of malondialdehyde in the plasma were determined using TBARS Assay Kit (Cayman Chemical, Ann Arbor, MI). Measurement of neutrophil number was performed by the LSI Medience Corporation Central Laboratory (Tokyo, Japan), using a Sysmex XT-2000iV™ automated haematology analyser (Sysmex, Kobe, Japan).
Evaluation of lung permeability
To quantitatively examine lung permeability, Evans blue dye (30 mg/kg) was intravenously administered 2 h before the mice were sacrificed. Tissue samples were cut into pieces and incubated with formamide solution at 60 °C for 24 h. Samples were centrifuged to obtain supernatants, the absorbances of which were measured at 620 nm to determine the amount of Evans blue dye present.
Preparation of BALF
BALF was collected by cannulating the trachea and lavaging the lung twice with 1 ml of sterile 0.9% NaCl containing 50 units/ml heparin. Approximately 1.8 ml of BALF was routinely recovered from each mouse and the total cell number was counted using a hemocytometer. After centrifugation with a Cytospin®4 (Thermo Fisher Scientific, Waltham, MA), cells were stained with Diff-Quik reagents and the ratio of neutrophils to total cell number was determined. The amount of protein and double-stranded DNA (dsDNA) present in the BALF was evaluated by the Bradford method and by using a Quant-iT™ PicoGreen® dsDNA Assay Kit (Thermo Fisher Scientific).
Measurement of ROS by in vivo imaging analysis
In vivo imaging of ROS in mice was performed as described previously49,57, with some modifications. We used an imaging system (Lumazone in vivo imaging system; Shoshin Em, Okazaki, Japan), which contains a chamber equipped with an electron-multiplying CCD camera. Mice were intravenously administered with the luminescent probe, L-012, in saline (75 mg/kg). At 5 min after the L-012 injection, mice were euthanized and the lungs were rapidly dissected and imaged (5 min exposure). All data were analyzed using SlideBook 6 software (Intelligent Imaging Innovations, Inc., Denver, CO).
Real-time reverse transcription (RT) polymerase chain reaction (PCR) analysis
Total RNA was extracted from lung tissue using an RNeasy kit according to the manufacturer’s protocol. Samples were reverse-transcribed using the PrimeScript® kit described above. The synthesized cDNA was used in real-time PCR experiments with SsoFast EvaGreen Supermix and analyzed with a Bio-Rad (Hercules, CA) CFX96™ real-time system and CFX Manager™ software. Specificity was confirmed by electrophoretic analysis of reaction products and by the inclusion of template- or reverse transcriptase-free controls. To normalize the amount of total RNA present in each reaction, hypoxanthine phosphoribosyltransferase 1 (HPRT1) cDNA was used as an internal standard. Primers were designed using Primer3 or Primer-BLAST websites. Primers sequences will be provided upon request.
Immunoblotting analysis and measurement of MPO activity
BALF or homogenized lung samples were prepared. BALF samples (2 μL) were then applied to NuPAGE® Novex 4–12% Bis-Tris Gel (Thermo Fisher Scientific) and subjected to electrophoresis. After western blotting, proteins were detected with their respective antibodies and chemiluminescent staining using SuperSignal™ West Dura Extended Duration Substrate (Thermo Fisher Scientific). Band intensities were quantitated by using ImageJ software (version 1.39 u). The MPO activity of homogenized lung samples was measured using an MPO assay kit (BioVision, Milpitas, CA) according to the manufacturer’s protocol.
Histological and immunohistochemical analyses and TUNEL assay
Tissue samples were fixed in 10% neutral buffered formalin for 24 h, and then embedded in paraffin before being cut into 4 μm thick sections. Sections were stained first with Mayer’s hematoxylin and then with 1% alcoholic eosin (H&E staining). Slides were mounted with malinol and visualized with a microscope and digital camera (Olympus DP71; Tokyo, Japan).
For immunohistochemical analysis, sections were incubated with Tris–EDTA buffer (pH 9.0) or proteinase K (20 μg/ml) for antigen retrieval and then incubated with DAKO® peroxidase blocking reagent for removal of endogenous peroxidase activity. Sections were blocked with 3% goat serum or 5% goat serum for 10 min, incubated overnight with rabbit anti-histone H3 antibody (1:100 dilution) or for 2 h with rabit anti-neutrophil elastase antibody (1:100 dilution) in DAKO® Antibody Diluent, and then incubated with a DAKO® EnVision peroxidase-labelled polymer–goat anti-rabbit immunoglobulin conjugate for 1 h. Then, 3,3′-diaminobenzidine was applied to the sections for colour development. The sections were finally counterstained with Mayer’s haematoxylin. Slides were mounted with malinol and visualized with a microscope and digital camera (Olympus DP71; Tokyo, Japan).
For the TUNEL assay, sections were incubated first with proteinase K (20 μg/ml) for 15 min at 37 °C, then with terminal deoxynucleotide transferase and biotin-14-ATP for 1 h at 37 °C, and finally with an Alexa Fluor 488–streptavidin conjugate and DAPI (5 μg/ml) for 2 h. Slides were mounted with Vectashield and inspected with the aid of a microscope and digital camera (Olympus DP71).
Cell culture
A549 cells (a human type II pulmonary epithelial cell line) and RAW264 cells (a mouse macrophage-like cell line) were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. All cells were cultured in a humidified atmosphere of 95% air with 5% CO2 at 37 °C. Viable cell number was quantified using a WST-based cell counting kit (Dojindo, Kumamoto, Japan) or CellTiter-Glo® 2.0 (Promega Corporation, Madison, WI). The levels of ROS in vitro were quantified using dihydroethidium (DHE), an indicator of superoxide (Thermo Fisher Scientific).
Statistical analysis
All values are expressed as the mean ± S.E.M. Two-way ANOVA followed by Dunnett’s test or the Student’s t-test for unpaired results was used to evaluate differences between three or more groups or between two groups, respectively. Kaplan–Meier plots were used to describe survival data and log-rank tests was performed to assess statistical differences. SPSS24 software was used for all statistical analyses. Differences were considered to be significant for values of P < 0.05.
Additional Information
How to cite this article: Tanaka, K.-I. et al. Preventive Effects of Carnosine on Lipopolysaccharide-induced Lung Injury. Sci. Rep. 7, 42813; doi: 10.1038/srep42813 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Rubenfeld, G. D. et al. Incidence and outcomes of acute lung injury. N Engl J Med 353, 1685–1693, doi: 353/16/1685 [pii]10.1056/NEJMoa050333 (2005).
Force, A. D. T. et al. Acute respiratory distress syndrome: the Berlin Definition. JAMA 307, 2526–2533, doi: 10.1001/jama.2012.5669 (2012).
Dushianthan, A., Grocott, M. P., Postle, A. D. & Cusack, R. Acute respiratory distress syndrome and acute lung injury. Postgrad Med J 87, 612–622, doi: 10.1136/pgmj.2011.118398 (2011).
Han, S. & Mallampalli, R. K. The acute respiratory distress syndrome: from mechanism to translation. J Immunol 194, 855–860, doi: 10.4049/jimmunol.1402513 (2015).
Baron, R. M. & Levy, B. D. Recent advances in understanding and treating ARDS. F1000Res 5, doi: 10.12688/f1000research.7646.1 (2016).
Steinberg, K. P. et al. Efficacy and safety of corticosteroids for persistent acute respiratory distress syndrome. N Engl J Med 354, 1671–1684, doi: 10.1056/NEJMoa051693 (2006).
Lamontagne, F. et al. Corticosteroid therapy for acute lung injury, acute respiratory distress syndrome, and severe pneumonia: a meta-analysis of randomized controlled trials. J Crit Care 25, 420–435, doi: 10.1016/j.jcrc.2009.08.009 (2010).
Gao Smith, F. et al. Effect of intravenous beta-2 agonist treatment on clinical outcomes in acute respiratory distress syndrome (BALTI-2): a multicentre, randomised controlled trial. Lancet 379, 229–235, doi: 10.1016/S0140-6736(11)61623-1 (2012).
McAuley, D. F. et al. Simvastatin in the acute respiratory distress syndrome. N Engl J Med 371, 1695–1703, doi: 10.1056/NEJMoa1403285 (2014).
Spragg, R. G. et al. Effect of recombinant surfactant protein C-based surfactant on the acute respiratory distress syndrome. N Engl J Med 351, 884–892, doi: 10.1056/NEJMoa033181 (2004).
National Heart, L. et al. Rosuvastatin for sepsis-associated acute respiratory distress syndrome. N Engl J Med 370, 2191–2200, doi: 10.1056/NEJMoa1401520 (2014).
Sarma, J. V. & Ward, P. A. Oxidants and redox signaling in acute lung injury. Compr Physiol 1, 1365–1381, doi: 10.1002/cphy.c100068 (2011).
Tasaka, S., Amaya, F., Hashimoto, S. & Ishizaka, A. Roles of oxidants and redox signaling in the pathogenesis of acute respiratory distress syndrome. Antioxidants & Redox Signaling 10, 739–753, doi: 10.1089/Ars.2007.1940 (2008).
Lucas, R., Verin, A. D., Black, S. M. & Catravas, J. D. Regulators of endothelial and epithelial barrier integrity and function in acute lung injury. Biochem Pharmacol 77, 1763–1772, doi: 10.1016/j.bcp.2009.01.014 (2009).
Lamb, N. J., Gutteridge, J. M., Baker, C., Evans, T. W. & Quinlan, G. J. Oxidative damage to proteins of bronchoalveolar lavage fluid in patients with acute respiratory distress syndrome: evidence for neutrophil-mediated hydroxylation, nitration, and chlorination. Crit Care Med 27, 1738–1744 (1999).
Quinlan, G. J., Lamb, N. J., Tilley, R., Evans, T. W. & Gutteridge, J. M. Plasma hypoxanthine levels in ARDS: implications for oxidative stress, morbidity, and mortality. Am J Respir Crit Care Med 155, 479–484, doi: 10.1164/ajrccm.155.2.9032182 (1997).
Han, W., Li, H., Segal, B. H. & Blackwell, T. S. Bioluminescence imaging of NADPH oxidase activity in different animal models. J Vis Exp, doi: 10.3791/3925 (2012).
Papaiahgari, S. et al. Genetic and pharmacologic evidence links oxidative stress to ventilator-induced lung injury in mice. Am J Respir Crit Care Med 176, 1222–1235, doi: 10.1164/rccm.200701-060OC (2007).
Stoiber, W., Obermayer, A., Steinbacher, P. & Krautgartner, W. D. The Role of Reactive Oxygen Species (ROS) in the Formation of Extracellular Traps (ETs) in Humans. Biomolecules 5, 702–723, doi: 10.3390/biom5020702 (2015).
Porto, B. N. & Stein, R. T. Neutrophil Extracellular Traps in Pulmonary Diseases: Too Much of a Good Thing? Front Immunol 7, 311, doi: 10.3389/fimmu.2016.00311 (2016).
Yang, H. et al. New Insights into Neutrophil Extracellular Traps: Mechanisms of Formation and Role in Inflammation. Front Immunol 7, 302, doi: 10.3389/fimmu.2016.00302 (2016).
Czaikoski, P. G. et al. Neutrophil Extracellular Traps Induce Organ Damage during Experimental and Clinical Sepsis. PLoS One 11, e0148142, doi: 10.1371/journal.pone.0148142 (2016).
Jiang, S. et al. Human resistin promotes neutrophil proinflammatory activation and neutrophil extracellular trap formation and increases severity of acute lung injury. J Immunol 192, 4795–4803, doi: 10.4049/jimmunol.1302764 (2014).
Grootjans, J., Kaser, A., Kaufman, R. J. & Blumberg, R. S. The unfolded protein response in immunity and inflammation. Nat Rev Immunol 16, 469–484, doi: 10.1038/nri.2016.62 (2016).
Cao, S. S. & Kaufman, R. J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid Redox Signal 21, 396–413, doi: 10.1089/ars.2014.5851 (2014).
Wei, J., Rahman, S., Ayaub, E. A., Dickhout, J. G. & Ask, K. Protein misfolding and endoplasmic reticulum stress in chronic lung disease. Chest 143, 1098–1105, doi: 10.1378/chest.12-2133 (2013).
Kim, H. J. et al. Inhibition of endoplasmic reticulum stress alleviates lipopolysaccharide-induced lung inflammation through modulation of NF-kappaB/HIF-1alpha signaling pathway. Sci Rep 3, 1142, doi: 10.1038/srep01142 (2013).
Kim, S. R. et al. Blockade of Interplay between IL-17A and Endoplasmic Reticulum Stress Attenuates LPS-Induced Lung Injury. Theranostics 5, 1343–1362, doi: 10.7150/thno.11685 (2015).
Sale, C. et al. Carnosine: from exercise performance to health. Amino Acids 44, 1477–1491, doi: 10.1007/s00726-013-1476-2 (2013).
Boldyrev, A. A., Aldini, G. & Derave, W. Physiology and pathophysiology of carnosine. Physiol Rev 93, 1803–1845, doi: 10.1152/physrev.00039.2012 (2013).
Mizuno, D. et al. Protective activity of carnosine and anserine against zinc-induced neurotoxicity: a possible treatment for vascular dementia. Metallomics 7, 1233–1239, doi: 10.1039/c5mt00049a (2015).
Kawahara, M., Konoha, K., Nagata, T. & Sadakane, Y. Protective substances against zinc-induced neuronal death after ischemia: carnosine as a target for drug of vascular type of dementia. Recent Pat CNS Drug Discov 2, 145–149 (2007).
Xu, T. et al. Carnosine markedly ameliorates H9N2 swine influenza virus-induced acute lung injury. J Gen Virol 96, 2939–2950, doi: 10.1099/jgv.0.000238 (2015).
Cuzzocrea, S. et al. Protective effect of orally administered carnosine on bleomycin-induced lung injury. Am J Physiol Lung Cell Mol Physiol 292, L1095–1104, doi: 10.1152/ajplung.00283.2006 (2007).
Ohata, S. et al. Polaprezinc Protects Mice against Endotoxin Shock. J Clin Biochem Nutr 46, 234–243, doi: 10.3164/jcbn.09-125 (2010).
Grommes, J. & Soehnlein, O. Contribution of neutrophils to acute lung injury. Mol Med 17, 293–307, doi: 10.2119/molmed.2010.00138 (2011).
Gariballa, S. E. & Sinclair, A. J. Carnosine: physiological properties and therapeutic potential. Age Ageing 29, 207–210 (2000).
Hipkiss, A. R. Carnosine and its possible roles in nutrition and health. Adv Food Nutr Res 57, 87–154, doi: 10.1016/S1043-4526(09)57003-9 (2009).
Hipkiss, A. R. & Gaunitz, F. Inhibition of tumour cell growth by carnosine: some possible mechanisms. Amino Acids 46, 327–337, doi: 10.1007/s00726-013-1627-5 (2014).
Al-Khafaji, A. B. et al. Superoxide induces Neutrophil Extracellular Trap Formation in a TLR-4 and NOX-dependent mechanism. Mol Med 22, doi: 10.2119/molmed.2016.00054 (2016).
Tagawa, Y. et al. Induction of CCAAT/enhancer-binding protein-homologous protein by cigarette smoke through the superoxide anion-triggered PERK-eIF2alpha pathway. Toxicology 287, 105–112, doi: 10.1016/j.tox.2011.06.005 (2011).
Matute-Bello, G., Frevert, C. W. & Martin, T. R. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol 295, L379–399, doi: 10.1152/ajplung.00010.2008 (2008).
Ricard, J. D., Dreyfuss, D. & Saumon, G. Ventilator-induced lung injury. Curr Opin Crit Care 8, 12–20 (2002).
Puig, F. et al. A new experimental model of acid- and endotoxin-induced acute lung injury in rats. Am J Physiol Lung Cell Mol Physiol 311, L229–237, doi: 10.1152/ajplung.00390.2015 (2016).
Zhang, Z. Y. et al. Carnosine attenuates early brain injury through its antioxidative and anti-apoptotic effects in a rat experimental subarachnoid hemorrhage model. Cell Mol Neurobiol 35, 147–157, doi: 10.1007/s10571-014-0106-1 (2015).
Tamba, M. & Torreggiani, A. Hydroxyl radical scavenging by carnosine and Cu(II)-carnosine complexes: a pulse-radiolysis and spectroscopic study. Int J Radiat Biol 75, 1177–1188 (1999).
Babizhayev, M. A. et al. L-carnosine (beta-alanyl-L-histidine) and carcinine (beta-alanylhistamine) act as natural antioxidants with hydroxyl-radical-scavenging and lipid-peroxidase activities. Biochem J 304 (Pt 2), 509–516 (1994).
Su, C. L. et al. Amelioration of superoxide dismutase on ventilator-induced lung injury by suppressing leukocyte in the lungs and systemic circulation. Chin J Physiol 56, 219–229, doi: 10.4077/CJP.2013.BAB106 (2013).
Tanaka, K. T. F., Sugizaki, T., Kawahara, M., Kuba, K., Imai, Y. & Mizushima, T. Evaluation of lecithinized superoxide dismutase for prevention of acute respiratory distress syndrome in animal models. Am J Respir Cell Mol Biol. in press (2016).
Koga, H. et al. The new vitamin E derivative, ETS-GS, protects against cecal ligation and puncture-induced systemic inflammation in rats. Inflammation 35, 545–553, doi: 10.1007/s10753-011-9344-2 (2012).
Goraca, A. & Jozefowicz-Okonkwo, G. Protective effects of early treatment with lipoic acid in LPS-induced lung injury in rats. J Physiol Pharmacol 58, 541–549 (2007).
Grommes, J. et al. Simvastatin reduces endotoxin-induced acute lung injury by decreasing neutrophil recruitment and radical formation. PLoS One 7, e38917, doi: 10.1371/journal.pone.0038917 (2012).
Kruger, P. S. & Terblanche, M. Statins in patients with sepsis and ARDS: is it over? No. Intensive Care Med, doi: 10.1007/s00134-016-4564-4 (2016).
Mansur, A. et al. Impact of statin therapy on mortality in patients with sepsis-associated acute respiratory distress syndrome (ARDS) depends on ARDS severity: a prospective observational cohort study. BMC Med 13, 128, doi: 10.1186/s12916-015-0368-6 (2015).
Kalaz, E. B. et al. Protective effects of carnosine alone and together with alpha-tocopherol on lipopolysaccharide (LPS) plus ethanol-induced liver injury. Environ Toxicol Pharmacol 42, 23–29, doi: 10.1016/j.etap.2015.12.018 (2016).
Kim, M. Y., Kim, E. J., Kim, Y. N., Choi, C. & Lee, B. H. Effects of alpha-lipoic acid and L-carnosine supplementation on antioxidant activities and lipid profiles in rats. Nutr Res Pract 5, 421–428, doi: 10.4162/nrp.2011.5.5.421 (2011).
Kielland, A. et al. In vivo imaging of reactive oxygen and nitrogen species in inflammation using the luminescent probe L-012. Free Radic Biol Med 47, 760–766, doi: 10.1016/j.freeradbiomed.2009.06.013 (2009).
Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research (C) from the Ministry of Education, Culture, Sports, Science and Technology, Japan. Finally, I would like to express my greatest appreciation to Dr. Tohru Mizushima.
Author information
Authors and Affiliations
Contributions
Participated in research design: Ken-ichiro Tanaka and Masahiro Kawahara. Conducted experiments: Ken-ichiro Tanaka, Toshifumi Sugizaki, Yuki Kanda and Tomomi Niino. Contributed new reagents or analytic tools: Ken-ichiro Tanaka and Fumiya Tamura. Performed data analysis: Ken-ichiro Tanaka, Toshifumi Sugizaki and Yuki Kanda. Wrote or contributed to the writing of the manuscript: Ken-ichiro Tanaka, Toshifumi Sugizaki, Yuki Kanda, Fumiya Tamura, Tomomi Niino and Masahiro Kawahara.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Tanaka, KI., Sugizaki, T., Kanda, Y. et al. Preventive Effects of Carnosine on Lipopolysaccharide-induced Lung Injury. Sci Rep 7, 42813 (2017). https://doi.org/10.1038/srep42813
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep42813
This article is cited by
-
Pulmonary inflammation, oxidative stress, and fibrosis in a mouse model of cholestasis: the potential protective properties of the dipeptide carnosine
Naunyn-Schmiedeberg's Archives of Pharmacology (2023)
-
Carnosine alleviates kidney tubular epithelial injury by targeting NRF2 mediated ferroptosis in diabetic nephropathy
Amino Acids (2023)
-
Safety Evaluation and Physiological Function of Dietary Balenine Derived From Opah Lampris guttatus on Skeletal Muscle of Mice
International Journal of Peptide Research and Therapeutics (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
