
Abstract
The role of marginal populations for the long-term maintenance of species’ genetic diversity and evolutionary potential is particularly timely in view of the range shifts caused by climate change. The Centre-Periphery hypothesis predicts that marginal populations should bear reduced genetic diversity and have low evolutionary potential. We analysed temporal stability at neutral microsatellite and adaptive MHC genetic variation over five decades in four marginal Atlantic salmon populations located at the southern limit of the species’ distribution with a complicated demographic history, which includes stocking with foreign and native salmon for at least 2 decades. We found a temporal increase in neutral genetic variation, as well as temporal instability in population structuring, highlighting the importance of temporal analyses in studies that examine the genetic diversity of peripheral populations at the margins of the species’ range, particularly in face of climate change.
Similar content being viewed by others

Introduction
The importance of marginal populations for the long-term maintenance of species’ genetic diversity and evolutionary potential has long been discussed1,2 but it is now particularly timely in view of range shifts caused by climate change3,4. According to the Centre-Periphery hypothesis, marginal populations inhabit unstable and poorly connected habitats, and may be expected to harbour less genetic variation and have lower evolutionary potential than those at the centre of the distribution5,6. However, although genetic diversity within populations seems to decline on average from the centre of the distribution to the periphery6, there is no conclusive evidence that geographically, historically or climatically marginal populations display lower average fitness7. Understanding the dynamics of populations at the species’ range limits, including patterns of extinction and recolonization and their ability to adapt to environmental variation, is key to predict their responses to climate change8. Critically, studies looking at genetic diversity in central versus marginal populations have largely focused on contemporary patterns of genetic diversity, using primarily neutral markers6, which may not fully reflect the adaptive potential of populations9.
The genes of the major histocompatibility complex (MHC), which are some of the most studied in relation to adaptive genetic variation10,11, are useful markers to use in combination with neutral markers to reconstruct not only the genetic diversity but also the adaptive potential of marginal populations. MHC genes are central to immunity as they encode for proteins that present pathogen-derived antigens to T-cells, initiating the adaptive immune response12. MHC genes are amongst the most polymorphic genes in vertebrates and also some of the best studied13 and the variation in the MHC residues that bind antigens from pathogens is thought to be maintained by balancing selection driven by pathogens14,15 and also influenced by mate choice16,17. Evidence of selection on the MHC genes has been identified in many species as heterozygote advantage18,19, association of individual MHC alleles and/or genotypes with susceptibility to specific pathogens20,21, rare-allele advantage22 and changes in allele frequencies under experimental infections23. Comparisons between MHC loci, or markers linked to them, and neutral markers can be used to infer differences in the relative levels of neutral and adaptive variation within and among populations, which can be variable not only across closely related species24,25 but also within species depending on the spatial scale of analysis26.
Salmonids are good study models of marginal populations because most of their current species’ range was recolonised from a few refugia after the last glaciation27,28,29, which permits geographical comparisons among recently diverged populations. Most salmonid species have declining populations in at least some parts of their range30,31,32,33 and within the salmonids, the Atlantic salmon (Salmo salar) is probably the species which has suffered the most dramatic decline34. The main causes for these declines are over-exploitation, habitat fragmentation, interactions with farm escapes35, and likely climate change36. In addition, as a result of their anadromous life cycle and homing behaviour, Atlantic salmon populations tend to be locally adapted37. Atlantic salmon is also particularly suited for MHC studies as it represents the minimal-essential-MHC in fish, with only two unlinked MHC class I and class II genes expressed38,39 and evidence of balancing selection acting on potential peptide binding residues (PBRs) in class I39 and in class II loci26,38. Previous studies had shown evidence of salmon MHC-disassortative mating40, as well as an association between specific MHC alleles and pathogen resistance41,42,43 and there are microsatellite markers linked to both class I and class II loci that can be used as good proxies for functional variation at these genes44,45,46.
In Europe, the current distribution of the Atlantic salmon seems to reflect a pattern of postglacial recolonisation from multiple refugia28,47. Iberian salmon populations inhabit the southern limit of the species’ range and the Iberian glacial refugium seems to have been one of the important refugia from which the northern Atlantic salmon range was recolonised28. Some of these marginal populations have suffered severe declines over the last 100 years, particularly during the last decades48,49, but seem to maintain their genetic distinctiveness28 and a higher genetic diversity than expected from their population size50.
Here, we examined temporal and spatial patterns of neutral and adaptive genetic diversity measured by microsatellites and MHC-linked markers in four marginal salmon populations in the Iberian refugium with a complicated demographic history, to test the general hypothesis that marginal populations have temporarily unstable genetic diversity and low adaptive potential. To this end, we compared spatial and temporal patterns of variability at neutral markers and markers linked to genes under selection (MHC class I and class II).
Results
Microsatellite variability and population structuring
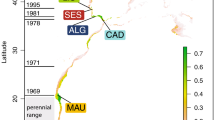
Individuals captured by anglers in the rivers Asón, Deva, Nansa and Pas (Northern Spain; Fig. 1) between 1948 and 2002 were genotyped at 13 putatively neutral microsatellite DNA markers. Deviations from HWE were only observed for loci CTAX (13 samples) and Sssp2210 (8 samples) following strict Bonferroni correction for multiple tests (Table 1). The overall results of the analyses did not change by excluding these two microsatellites (data not shown) and we opted for including them. A total of 196 alleles were observed across neutral microsatellites for the whole sample. BOTTLENECK results indicated that allelic frequency distributions did not depart from the expected L-shaped distribution. The number of alleles ranged from five at locus SsaD486 to 26 at locus SsaD144b. Significant correlations in allele frequencies between adjacent temporal samples for all comparisons could suggest stability in allele frequencies, but none of the correlations was significant after applying strict Bonferroni correction for multiple tests (Supplementary table S1). Heterozygosity (Ho) increased significantly over time in the rivers Pas (Mann-Kendall trend test s = 8 P = 0.041), Nansa (s = 6 P = 0.042) and Pas (s = 10, P = 0.008) whilst no significant temporal change in Ho was observed in the Deva (s = 6 P = 0.117). Equally, allelic richness increased temporally in the rivers Asón (s = 8 P = 0.042), Nansa (s = 6 P = 0.042) and Pas (s = 8 P = 0.042), but not in the river Deva (s = 6 P = 0.117). Following population analysis by STRUCTURE, the estimated optimal number of genetic groups was K = 6 (Fig. 2). The results indicated that the rivers Pas and Nansa, and to a lesser extent the river Ason, suffered a drastic change in population structure post-80 s, such that the genetic composition of these rivers in 2002 is rather different than that observed in 1950 and 1960. Similar results were obtained when the rivers were analysed individually (Supplementary figure S1). Genetic distance (DA) between temporal samples of the same rivers (0.057 to 0.177) were of a similar order to genetic distances between river samples (0.060 to 0.215). The NJ-phenogram, although with low statistical support, suggested that samples from the rivers Ason and Deva tended to group by river and not by decade, whereas samples from the rivers Nansa and Pas were intermingled, with a tendency to associate by decade instead of river (Fig. 3). AMOVA results also indicated significant temporal heterogeneity within the Asón and Pas samples (Table 2). Among river genetic variation was significant for each temporal sample, excluding the 1990 s samples, though there seemed to be a decrease in magnitude of FST over time. No deviation from neutrality was identified by the Ewens-Watterson test. The selection analysis implemented in LOSITAN identified outliers only in the samples from 1960 and 1980, in particular Ssa197 (P = 0.997), SsaD485 (P = 0.998), Ssa1438 (P = 0.984) in 1960, all under positive selection and Ssa197 (P = 0.997), SsaD485 (P = 0.997) in 1980, also under positive selection.

The distance between rivers varies from 3 to 50 Km. Map created using Maptool (http://www.seaturtle.org/maptool).

Each individual is represented by a vertical bar of a colour that represents its estimated membership to one of 6 genetic clusters. Labels below the plot indicate river and decade.

Neighbour-Joining (NJ) phylogenetic tree depicting the relationship between temporal samples from four different peripheral salmon populations based on (a) microsatellites, (b) Sasa-UBA-3’UTR MHC lined marker and (c) Sasa-DAA-3′UTR MHC lined marker. Numbers in the microsatellites tree indicate clusters supported by >50% of 5000 bootstrap iterations.
MHC-linked variability and population structuring
Considering all historical samples, the rate of false alleles was low for both Sasa-DAA-3′UTR (0.28) and Sasa-UBA-3′UTR (0.30). Allelic drop out for Sasa-DAA-3′UTR was higher than reported for the neutral microsatellites (9.63 compared to the neutral mean of 3.89), whereas the ADO rate for Sasa-UBA-3′UTR was more similar (2.96). A total of 22 different alleles were found for the Sasa-UBA-3′UTR locus, and 15 for the Sasa-DAA-3′UTR locus. The large range of Sasa-DAA-3′UTR allele sizes (207 bp-367 bp) may have contributed to the greater rate of ADO, particularly in historical samples46. Nine samples significantly deviated from Hardy-Weinberg equilibrium after Bonferroni corrections (Table 1), most of them corresponding to the oldest samples, and did not appear to have any particular bias for either the Sasa-UBA-3′UTR locus or the Sasa-DAA-3′UTR locus. Analysis of linkage disequilibrium of class I and class II linked markers for all samples revealed no linkage disequilibrium of these loci (global P-value = 0.531). Observed heterozygosities ranged from 0.17–1.00 across all samples (Table 1). Time series analyses indicated an increase in genetic diversity in the river Pas in the Sasa-DAA-3′UTR locus (Ho and Ar: s = 8 P = 0.0042) and a decrease in Sasa-UBA-3′UTR in the rivers Nansa (Ho and Ar: S = −4 P = 0) and Pas (Ho: s = −1 P = 0.009; Ar: s = −2 P = 0.006). According to LOSITAN, Sasa-UBA-3′UTR locus was under positive selection in 1960 (P = 0.995) and 1980 (P = 0.995). In contrast to neutral microsatellites, significant correlations in allele frequencies were observed between adjacent temporal samples for all comparisons, except for those involving the Deva 1960s sample (Supplementary Table S1). This indicates stability of allele frequencies over time for both Sasa-UBA-3′UTR and Sasa-DAA-3′UTR loci. Allele distributions mostly overlapped among the four populations, with some very low frequency alleles being only represented in one or two of the rivers (Supplementary Figure S2). AMOVA analysis indicated significant genetic structuring at the class I locus at all temporal periods tested, whereas for the class II locus, the 1980s and 1990s were significantly differentiated as well as samples from 2002 and the 1960s (Table 3). The phylogenetic tree for the class I-linked marker indicated a relationship among rivers very similar to that for the neutral microsatellites, clustering the samples from rivers Asón and Deva according to river of origin, whereas the samples from rivers Nansa and Pas were largely intermingled within a cluster. In contrast, the class II marker, showed no structuring of samples based on river of origin in any of the rivers.
Discussion
Peripheral (marginal) populations tend to be genetically and morphologically distinct as a consequence of their isolation and typically smaller size, and are considered particularly valuable because they can help preserve the evolutionary potential of the species2. Atlantic salmon populations in northern Spain represent peripheral populations at the southern limit of the species’ range; these have been in decline since the 1960’s and are now classified as endangered51. However, despite inhabiting the margins of the species’ range and having small effective population sizes, these populations display levels of genetic diversity comparable to those reported for larger populations at the center of the distribution50. Northern Iberian rivers are thought to have been a refugium for Atlantic salmon during the last glacial maximum, and it is possible that this is the reason why these populations appear to harbour higher than expected ancestral mitochondrial DNA variation compared to more northerly European populations28. Additionally, stocking from different sources carried out in the 80s and in the 90s could have also contributed to the temporal differentiation of these populations31. Between 1972 (when stocking records start) and the 90s, these rivers (initially the Rivers Ason and Pas and then extending to the River Nansa and to lesser extent the River Deva) were stocked with high densities of eyed ova (200,000-300,000 annually) and fry (90,000-120,000 annually) mainly from Scotland and Iceland. Stoking from the 90s was carried out from native sources, and primarily from the river of origin during the last years48,51. Our results from neutral microsatellites indicate that there has been a temporal increase in genetic diversity (heterozygosity and allelic richness) in three of the four rivers over a 50 year period, but also some temporal maintenance of genetic identity in the river Deva. In contrast, increases in neutral genetic diversity in the rivers Ason, Nansa and Pas, coupled with the strong changes in their genetic background from the 80s and a temporal decrease in genetic structuring suggest that their genetic composition could have been affected by foreign stocking, as previously indicated using mtDNA31. We found no conclusive evidence of selection in the MHC-linked markers, apart from Sasa-UBA-3′UTR, that together with three neutral microsatellites deviated from neutrality in samples from the 60s and 80s, suggesting the parallelism between the class I marker and the rest of the microsatellites. Results from neutral markers largely mirrored those of the class I MHC-linked marker (Sasa-UBA-3′UTR) but not those of Sasa-DAA-3′UTR (class II). This is perhaps not surprising given the differences in response to selection previously observed between both markers46. In this case, Sasa-DAA-3′UTR did not indicate any clustering of samples, by river or decade. Such a pattern of variation could reflect adaptation to local conditions in these marginal populations, an scenario that might be expected given the homing behavior of Atlantic salmon and their tendency to form locally adapted populations37, but also genetic drift due to low effective population size. Recently introduced salmonid populations in Chile suggested that MHC class II functional diversity of invasive populations has decreased over time, in contrast to diversity at neutral markers which has remained very high52,53 as a consequence of admixture54. Therefore, it is possible that, even if some neutral diversity has remained high in some Iberian salmon rivers, perhaps as consequence of foreign stocking, diversity at non-neutral markers may have been eroded over time due to geographical differences in selection55 and to adaptation to local conditions (e.g. parasites)56,57. Our current results, in combination with previous studies on the same populations, indicate, despite their low effective population size50, these salmon harbour high neutral genetic diversity, atypical in marginal populations, highlighting the importance of the demographic history for the maintenance of the genetic diversity. This is particularly relevant in view of the predictions of the consequences of climate change for salmonids, i.e. movement of the thermal niche of salmon towards north as well as decreased production and population extinction in the southern range of species58. Our study highlights the importance of a adopting not only a spatial but also a temporal approach, considering both neutral as well as adaptive markers, in studies that examine changes in genetic diversity of peripheral populations at the margins of the species’ range.
Methods
Origin of the samples and DNA extraction
Adipose fins from dead adult Atlantic salmon, captured by anglers in the rivers Asón, Deva, Nansa and Pas (Northern Spain; Fig. 1) were collected in 2002 and stored in 95% ethanol at 4 °C prior to genetic analysis. Dried scales from the same rivers collected from adult fish caught by anglers since 1948 were also included in the analyses. Due to their limited availability, historical scales were pooled across four decades following Ciborowski et al.31: 1948–1957, 1960–1963, 1980–1989, 1990–1996. No scale samples were available for any river from the 1970s, or for the river Nansa pre-1960. Therefore, 19 groups of samples, stratified by decade and river were generated for analysis (Table 1).
Total DNA was extracted using the Promega™ Wizard SV 96 Genomic DNA Purification System. Manufacturer’s protocols were adhered to for modern adipose fin samples, but for historical scales we increased the incubation time during the elution steps to five minutes and decreased the elution volume to 80–100 μl. Between one and three historical scale extractions were carried out in a dedicated ancient DNA laboratory, physically separated from PCR procedures. A blank control was extracted concurrently and subsequently amplified in PCR reactions. All eluted DNA was stored at −20 °C.
PCR amplification and microsatellite genotyping
All individuals were genotyped at 13 putatively neutral microsatellite DNA loci (Ssa85, Ssa171, Ssa197, Ssa20259; SSsp1605, SSsp221060; SsaA124, SsaD144, SsaD48661; SsoSL43862; CTAX, EST47, HSP63; Table 1) and two MHC linked markers, Sasa-UBA-3′UTR and Sasa-DAA-3′UTR64, in three multiplex reactions. Each individual was repeatedly genotyped at all loci, and 10% of the samples were genotyped in triplicate. Reactions were carried out according to the QIAGEN Multiplex PCR Kit reaction protocol in 8 ul volume. Each reaction included 4 ul of QIAGEN Multiplex PCR Kit reaction mixture, 2 mM of each primer and 2 ul of the extracted DNA solution. For the scale samples, 0.2 uM BSA was added to each reaction. The thermocycler profile consisted of 95 °C for 15 min, either 30 or 35 (tissue or scale DNA extraction, respectively) cycles of 94 °C 30 sec, 58 °C 90 sec, 72 °C for 30 sec and a final hold of 60 °C for 30 min. PCR products were run on a 3100 ABI Prism capillary sequencer using the Genescan-500 LIZ size standard. Alleles were scored using Genemapper V3.5 software (Applied Biosystems) and genotypes were manually checked.
Data analysis
Individuals with fewer than eight successfully genotyped loci were discarded from analysis (final sample sizes in Table 1). All loci were tested for conformity with Hardy-Weinberg equilibrium using the randomization test implemented in GENEPOP65 and were also tested for neutrality using the Ewens-Watterson test66 in ARLEQUIN v367. Rates of allelic dropout (ADO) and false alleles (FA) were estimated according to Broquet and Petit (2004)68. Allelic richness (Ar) was calculated using FSTAT69. Statistical significance of temporal trends was tested using the Mann-Kendall trend test70 implemented in PAST71. Pairwise differences in allelic frequencies between decades were estimated for each river using GENEPOP. FST values of genetic differentiation were estimated using GENETIX v.4.0472 and significances were determined with 1000 permutations To correct for simultaneous tests, strict Bonferroni corrections were applied73.
STRUCTURE v2.3.374 was used to test how many genetic populations were represented by all individuals caught in each of the four rivers. We followed the methodology outlined in74. First, we constructed phylograms for all individuals from each river based on individual distance matrices calculated with the program POPULATIONS75 using an allele sharing distance (ASD) method76 to visualise whether there was any clustering of individuals into discrete population units. Following this, all individuals from each river were modelled in STRUCTURE. The program was run applying the admixture model, as this model was likely to be closer to the true nature of the history of these populations compared with a non-admixture model. The parameters of the simulations were burn-in length of 50,000 iterations; 100,000 MCMC repetitions; testing for K (the number of populations) between 2 and 8 over 10 repeated simulations. We estimated the correct value of K using the Evanno method77 as implemented in STRUCTURE HARVESTER (http://users.soe.ucsc.edu/~dearl/software/struct_harvest/). We then used CLUMPP78 and DISTRUCT79 to summarise and represent the results. Spatial and temporal structuring was also analysed using AMOVA as implemented in ARLEQUIN and POPULATIONS was used to generate a consensus unrooted neighbour-joining tree (10,000 bootstrapped replicated) of the samples based Nei’s DA distance80, which was visualized using TREEVIEW81.
Sasa-UBA-3′UTR and Sasa-DAA-3′UTR data were analysed separately (as in64). GENEPOP on the web82 was used to estimate observed and expected heterozygosities (Ho and He) for each locus in each sample. Allelic richness was calculated at each locus for each sample using FSTAT version 2.9.383. Statistical significance of the temporal trends of genetic diversity (heterozygosity and allelic richness) was tested using the Mann-Kendall trend test70 implemented in PAST71.
All markers, neutral and MHC-linked microsatellites, were tested for neutrality using Lositan84,85, under 50,000 simulations, estimated neutral mean FST, infinite alleles mutation model, 99% confidence interval and false discovery rate of 0.1%. All populations were tested for recent bottlenecks using BOTTLENECK v.1.2.0286.
Additional Information
How to cite this article: Ciborowski, K. et al. Temporal and spatial instability in neutral and adaptive (MHC) genetic variation in marginal salmon populations. Sci. Rep. 7, 42416; doi: 10.1038/srep42416 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Kirkpatrick, M. & Barton, N. H. Evolution of a species’ range. Am Nat 150, 1–23 (1997).
Lesica, P. & Allendorf, F. W. When are peripheral populations valuable for conservation? Conserv Biol 9, 753–760 (1995).
Hampe, A. & Petit, R. J. Conserving biodiversity under climate change: the rear edge matters. Ecol Lett 8, 461–467 (2005).
Thomas, C. D. et al. Extinction risk from climate change. Nature 427, 145–148 (2004).
Vucetich, J. A. & Waite, T. A. Spatial patterns of demography and genetic processes across the species’ range: Null hypotheses for landscape conservation genetics. Conserv Genet 4, 639–645 (2003).
Eckert, C., Samis, K. & Lougheed, S. Genetic variation across species’ geographical ranges: the central–marginal hypothesis and beyond. Mol Ecol 17, 1170–1188 (2008).
Pironon, S., Villellas, J., Morris, W. F., Doak, D. F. & García, M. B. Do geographic, climatic or historical ranges differentiate the performance of central versus peripheral populations? Glob Ecol Biogeogr 24, 611–620, doi: 10.1111/geb.12263 (2015).
Buckley, L. B. & Kingsolver, J. G. Functional and phylogenetic approaches to forecasting species’ responses to climate change. Ann Rev Ecol Evol Syst 43, 205–226 (2012).
McKay, J. K. & Latta, R. G. Adaptive population divergence: markers, QTL and traits. Trends Ecol Evol 17, 285–291, doi: 10.1016/S0169-5347(02)02478-3 (2002).
Hedrick, P., Parker, K. & Lee, R. Using microsatellite and MHC variation to identify species, ESUs, and MUs in the endangered Sonoran topminnow. Mol Ecol 10, 1399–1412 (2001).
Hedrick, P. & Kim, T. Genetics of complex polymorphisms: parasites and maintenance of MHC variation. (Harvard University Press, 2000).
Janeway, C. A., Travers, P., Walport, D. & Shlomchik, M. J. Immunobiology: The Immune System in Health and Disease. (2004).
Hughes, A. L. & Yeager, M. Natural selection at major histocompatibility complex loci of vertebrates. Annual review of genetics 32, 415–435, doi: 10.1146/annurev.genet.32.1.415 (1998).
Doherty, P. C. & Zinkernagel, R. M. Enhanced immunological surveillance in mice heterozygous at the H-2 gene complex. Nature 256, 50–52 (1975).
Slade, R. W. & McCallum, H. I. Overdominant vs. frequency-dependent selection at MHC loci. Genetics 132, 861–864 (1992).
Apanius, V., Penn, D., Slev, P. R., Ruff, L. R. & Potts, W. K. The nature of selection on the major histocompatibility complex. Crit Rev Immunol 17, 179 - 224 (1997).
Consuegra, S. & Garcia de Leaniz, C. MHC-mediated mate choice increases parasite resistance in salmon. Proceedings of the Royal Society of London, Series B 275, 1397–1403, doi: 10.1098/rspb.2008.0066 (2008).
Kurtz, J. et al. Major histocompatibility complex diversity influences parasite resistance and innate immunity in sticklebacks. Proc R Soc Lond Ser B-Biol Sci 271, 197-204 (2004).
Wegner, K. M., Reusch, T. B. H. & Kalbe, M. Multiple parasites are driving major histocompatibility complex polymorphism in the wild. J Evol Biol 16, 224–232 (2003).
Bonneaud, C., Pérez-Tris, J., Federici, P., Chastel, O. & Sorci, G. Major Histocompatibility alleles associated with local resistance to malaria in a passerine. Evolution 60, 383–389, doi: 10.1111/j.0014-3820.2006.tb01114.x (2006).
Gómez, D., Conejeros, P., Marshall, S. & Consuegra, S. MHC evolution in three salmonid species: a comparison between class II alpha and beta genes. Immunogenetics 62, 531–542, doi: 10.1007/s00251-010-0456-x (2010).
Schwensow, N., Fietz, J., Dausmann, K. H. & Sommer, S. Neutral versus adaptive genetic variation in parasite resistance: importance of major histocompatibility complex supertypes in a free-ranging primate. Heredity 99, 265–277 (2007).
Eizaguirre, C., Lenz, T. L., Kalbe, M. & Milinski, M. Rapid and adaptive evolution of MHC genes under parasite selection in experimental vertebrate populations. Nat Commun 3, 621 (2012).
Hambuch, T. M. & Lacey, E. A. Enhanced selection for MHC diversity in social tuco-tucos. Evolution 56, 841–845, doi: 10.1554/0014-3820(2002)056[0841:ESFMDI]2.0.CO;2 (2002).
Jarvi, S. I., Tarr, C. L., Mcintosh, C. E., Atkinson, C. T. & Fleischer, R. C. Natural selection of the major histocompatibility complex (Mhc) in Hawaiian honeycreepers (Drepanidinae). Mol Ecol 13, 2157–2168 (2004).
Landry, C. & Bernatchez, L. Comparative analysis of population structure across environments and geographical scales at major histocompatibility complex and microsatellite loci in Atlantic salmon (Salmo salar). Mol Ecol 10, 2525–2539 (2001).
Verspoor, E., McCarthy, E. M. & Knox, D. The phylogeography of European Atlantic salmon (Salmo salar L.) based on RFLP analysis of the ND1/16sRNA region of the mtDNA. Biol J Linn Soc 68, 129 - 146 (1999).
Consuegra, S. et al. Mitochondrial DNA variation in Pleistocene and modern Atlantic salmon from the Iberian glacial refugium. Mol Ecol 11, 2037–2048 (2002).
King, T. L. et al. In The Atlantic Salmon 117–166 (Blackwell Publishing Ltd, 2007).
Stouder, D. J., Bisson, P. A. & Naiman, R. Pacific Salmon & Their Ecosystems: Status and Future Options. (Springer Science & Business Media, 2012).
Ciborowski, K. et al. Stocking may increase mitochondrial DNA diversity but fails to halt the decline of endangered Atlantic salmon populations. Conserv Genet 8, 1355–1367 (2007).
Lackey, R. T., Lach, D. H. & Duncan, S. L. Policy options to reverse the decline of wild Pacific salmon. FISHERIES-BETHESDA 31, 344 (2006).
Winfield, I. et al. Population trends of Arctic charr (Salvelinus alpinus) in the UK: assessing the evidence for a widespread decline in response to climate change. Hydrobiologia 650, 55–65 (2010).
Parrish, D. L., Behnke, R. J., Gephard, S. R., McCormick, S. D. & Reeves, G. H. Why aren’t there more Atlantic salmon (Salmo salar)? Can J Fish Aquat Sci 55, 281 - 287 (1998).
Skaala, Ø., Wennevik, V. & Glover, K. A. Evidence of temporal genetic change in wild Atlantic salmon, Salmo salar L., populations affected by farm escapees. ICES Journal of Marine Science: Journal du Conseil 63, 1224–1233 (2006).
Friedland, K. D., Reddin, D. G. & Castonguay, M. Ocean thermal conditions in the post-smolt nursery of North American Atlantic salmon. ICES Journal of Marine Science: Journal du Conseil 60, 343–355 (2003).
Garcia de Leaniz, C. et al. A critical review of adaptive genetic variation in Atlantic salmon: implications for conservation. Biological Reviews 82, 173–211, doi: 10.1111/j.1469-185X.2006.00004.x (2007).
Consuegra, S., Megens, H. J., Leon, K., Stet, R. J. M. & Jordan, W. C. Patterns of variability at the major histocompatibility class II alpha locus in Atlantic salmon contrast with those at the class I locus. Immunogenetics 57, 16–24 (2005).
Consuegra, S. et al. Rapid Evolution of the MH Class I Locus Results in Different Allelic Compositions in Recently Diverged Populations of Atlantic Salmon. Mol Biol Evol 22, 1095–1106, doi: 10.1093/molbev/msi096 (2005).
Landry, C., Garant, D., Duchesne, P. & Bernatchez, L. ‘Good genes as heterozygosity’: the major histocompatibility complex and mate choice in Atlantic salmon (Salmo salar). Proc. R. Soc. Lond. B 268, 1279–1285 (2001).
Langefors, Lohm, J. Grahn, M., Andersen, O. & von Schantz, T. Association between major histocompatibility complex class IIB alleles and resistance to Aeromonas salmonicida in Atlantic salmon. Proc. R. Soc. Lond. B 268, 479–485 (2001).
Lohm, J. et al. Experimental evidence for major histocompatibility complex-allele-specific resistance to a bacterial infection. Proc. R. Soc. Lond. B 269, 2029–2033 (2002).
Grimholt, U. et al. MHC polymorphism and disease resistance in Atlantic salmon (Salmo salar); facing pathogens with single expressed major histocompatibility class I and class II loci. Immunogenetics 55, 210–219 (2003).
Grimholt, U., Drabløs, F., Jørgensen, S., Høyheim, B. & Stet, R. The major histocompatibility class I locus in Atlantic salmon (Salmo salar L.): polymorphism, linkage analysis and protein modelling. Immunogenetics 54, 570–581 (2002).
Stet, R. et al. Unique haplotypes of co-segregating major histocompatibility class II A and class II B alleles in Atlantic salmon (Salmo salar) give rise to diverse class II genotypes. Immunogenetics 54, 320–331 (2002).
Consuegra, S., de Eyto, E., McGinnity, P., Stet, R. J. M. & Jordan, W. C. Contrasting responses to selection in class I and class II[alpha] major histocompatibility-linked markers in salmon. Heredity 107, 143–154, doi: http://www.nature.com/hdy/journal/v107/n2/suppinfo/hdy2010177s1.html (2011).
Bernatchez, L. & Wilson, C. C. Comparative phylogeography of nearctic and palearctic fishes. Mol Ecol 7, 431–452 (1998).
Garcia de Leániz, C. & Martinez, J. J. In Atlantic Salmon 179–209 (Springer, 1988).
Parrish, D. L., Behnke, R. J., Gephard, S. R., McCormick, S. D. & Reeves, G. H. Why aren’t there more Atlantic salmon (Salmo salar)? Can J Fish Aquat Sci 55, 281–287 (1998).
Consuegra, S., Verspoor, E., Knox, D. & de Leaniz, C. G. Asymmetric gene flow and the evolutionary maintenance of genetic diversity in small, peripheral Atlantic salmon populations. Conserv Genet 6, 823–842, doi: 10.1007/s10592-005-9042-4 (2005).
Garcıa de Leániz, C., Serdio, A. & Consuegra, S. Present status of Atlantic salmon in Cantabria. El salmón, joya de nuestros rıos. 55–82 (2001).
Monzón-Argüello, C., Garcia de Leaniz, C., Gajardo, G. & Consuegra, S. Less can be more: loss of MHC functional diversity can reflect adaptation to novel conditions during fish invasions. Ecol Evol 3, 3359–3368 (2013).
Monzon-Arguello, C., Garcia de Leaniz, C., Gajardo, G. & Consuegra, S. Eco-immunology of fish invasions: the role of MHC variation. Immunogenetics in press (2014).
Consuegra, S., Phillips, N. C. & Gajardo, G. & Garcia de Leaniz, C. Winning the invasion roulette: escapes from fish farms increase admixture and facilitate establishment of nonnative rainbow trout. Evolutionary Applications (2011).
Miller, K. M., Kaukinen, K. H., Beacham, T. D. & Withler, M. E. Geographic heterogeneity in natural selection on an MHC locus in sockeye salmon. Genetica 111, 237–257 (2001).
Spurgin, L. G. & Richardson, D. S. How pathogens drive genetic diversity: MHC, mechanisms and misunderstandings. Proc R Soc Biol Sci Ser B 277, 979–988 (2010).
Consuegra, S. & Garcia de Leaniz, C. MHC-mediated mate choice increases parasite resistance in salmon. Proc R Soc Biol B 275, 1397–1403, doi: 10.1098/rspb.2008.0066 (2008).
Jonsson, B. & Jonsson, N. A review of the likely effects of climate change on anadromous Atlantic salmon Salmo salar and brown trout Salmo trutta, with particular reference to water temperature and flow. J Fish Biol 75, 2381–2447 (2009).
O’Reilly, P. T., Hamilton, L. C., McConnell, S. K. & Wright, J. M. Rapid analysis of genetic variation in Atlantic salmon (Salmo salar) by PCR multiplexing of dinucleotide and tetranucleotide microsatellites. Can J Fish Aquat Sci 53, 2292–2298 (1996).
Paterson, S., Piertney, S., Knox, D., Gilbey, J. & Verspoor, E. Characterization and PCR multiplexing of novel highly variable tetranucleotide Atlantic salmon (Salmo salar L.) microsatellites. Mol Ecol Notes 4, 160–162 (2004).
King, T. L., Eackles, M. S. & Letcher, B. H. Microsatellite DNA markers for the study of Atlantic salmon (Salmo salar) kinship, population structure, and mixed‐fishery analyses. Mol Ecol Notes 5, 130–132 (2005).
Slettan, A., Olsaker, I. & Lie, Ø. Polymorphic Atlantic salmon, Salmo salar L., microsatellites at the SSOSL438, SSOSL439 and SSOSL444 loci. Anim Genet 27, 57–58 (1996).
Vasemägi, A., Nilsson, J. & Primmer, C. R. Expressed sequence tag-linked microsatellites as a source of gene-associated polymorphisms for detecting signatures of divergent selection in Atlantic salmon (Salmo salar L.). Mol Biol Evol 22, 1067–1076 (2005).
Grimholt, U. et al. MHC polymorphism and disease resistance in Atlantic salmon (Salmo salar); facing pathogens with single expressed major histocompatibility class I and class II loci. Immunogenetics 55, 210-219 (2003).
Raymond, M. & Rousset, F. GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86, 248–249 (1995).
Ewens, W. J. The sampling theory of selectively neutral alleles. Theor Popul Biol 3, 87–112 (1972).
Excoffier, L., Laval, G. & Schneider, S. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform Online 1, 47 (2005).
Broquet, T. & Petit, E. Quantifying genotyping errors in noninvasive population genetics. Mol Ecol 13, 3601–3608 (2004).
Goudet, J. FSTAT (version 1.2): a computer program to calculate F-statistics. J Hered 86, 485–486 (1995).
Gilbert, R. O. Statistical methods for environmental pollution monitoring. (Van Nostrand Reinhold, 1987).
Hammer, O., Harper, D. A. T. & Ryan, P. D. PAST: Paleontological Statistics Software Package for Education and Data Analysis. Palaeontol Electron 4, 9 (2001).
Belkhir, K., Borsa, P., Chikhi, L., Raufaste, N. & Bonhomme, F. GENETIX 4.05, Population genetics software for Windows TM. Université de Montpellier II. Montpellier (2004).
Rice, W. R. Analyzing tables of statistical tests. Evolution. 223–225 (1989).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Langella, O. POPULATIONS 1.2. 28. Population genetic software (individuals or populations distances, phylogenetic trees). CNRS, France (2002).
Goldstein, D. & Pollock, D. Mutation processes and methods of phylogenetic inference. J. Hered 88, 335–342 (1997).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the softwarestructure: a simulation study. Mol Ecol 14, 2611–2620 (2005).
Jakobsson, M. & Rosenberg, N. A. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23, 1801–1806 (2007).
Rosenberg, N. A. distruct: a program for the graphical display of population structure. Mol Ecol Notes 4, 137–138 (2004).
Nei, M., Tajima, F. & Tateno, Y. Accuracy of Estimated Phylogenetic Trees from Molecular-Data. 2. Gene-Frequency Data. J. Mol. Evol. 19, 153–170 (1983).
Page, R. TREEVIEW, tree drawing software for Apple Macintosh and Microsoft Windows. Division of Environmental and Evolutionary Biology, Instituteo Biomedical and Life Sciences, University of Glasgow: Glasgow, Scotland, UK, (1996).
Raymond, M. & Rousset, F. Genepop (Version-1.2) - Population-Genetics Software for Exact Tests and Ecumenicism. J. Hered. 86, 248–249 (1995).
FSTAT A program to estimate and test gene diversities and fixation indices (version 2.9.3). Available from http://www.unil.ch/izea/softwares/fstat.html. (2001).
Beaumont, M. A. & Nichols, R. A. Evaluating loci for use in the genetic analysis of population structure. Proc R Soc Biol Sci Ser B 263, 1619–1626 (1996).
Antao, T., Lopes, A., Lopes, R., Beja-Pereira, A. & Luikart, G. LOSITAN: A workbench to detect molecular adaptation based on a Fst-outlier method. BMC Bioinformatics 9, 323 (2008).
Piry, S., Luikart, G. & Cornuet, J. M. BOTTLENECK: A computer program for detecting recent reductions in the effective population size using allele frequency data. J Hered 90, 502–503 (1999).
Author information
Authors and Affiliations
Contributions
S.C., W.C.J. and C.G.L. designed the study, K.C. and S.C. carried out the analyses, S.C., K.C. and C.G.L. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Ciborowski, K., Jordan, W., Garcia de Leaniz, C. et al. Temporal and spatial instability in neutral and adaptive (MHC) genetic variation in marginal salmon populations. Sci Rep 7, 42416 (2017). https://doi.org/10.1038/srep42416
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep42416
This article is cited by
-
Alternated selection mechanisms maintain adaptive diversity in different demographic scenarios of a large carnivore
BMC Evolutionary Biology (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
