
Abstract
There is still an ongoing demand for a simple broad-spectrum molecular diagnostic assay for pathogenic bacteria. For this purpose, we developed a single-plex High Resolution Melt (HRM) assay that generates complex melt curves for bacterial identification. Using internal transcribed spacer (ITS) region as the phylogenetic marker for HRM, we observed complex melt curve signatures as compared to 16S rDNA amplicons with enhanced interspecies discrimination. We also developed a novel Naïve Bayes curve classification algorithm with statistical interpretation and achieved 95% accuracy in differentiating 89 bacterial species in our library using leave-one-out cross-validation. Pilot clinical validation of our method correctly identified the etiologic organisms at the species-level in 59 culture-positive mono-bacterial blood culture samples with 90% accuracy. Our findings suggest that broad bacterial sequences may be simply, reliably and automatically profiled by ITS HRM assay for clinical adoption.
Similar content being viewed by others


Introduction
Acute febrile illnesses often present with vague symptoms and are caused by a myriad of potential bacterial pathogens1,2,3. Unfortunately, accurate diagnosis of the causative agent(s) can be compromised by diagnostic assays biased to target a narrow pre-determined set of microorganisms. Broad differential diagnostics that rapidly identify unknown bacterial pathogen(s) within the acute-care timescale would enable early fine-tuning of antibiotic stewardship and reduce patient morbidity and mortality4.
Molecular approaches based on broad-range nucleic acid amplification of bacterial targets are well suited for differential diagnosis of acute febrile illness. However, downstream amplicon analysis technologies (e.g. microarray, mass spectrometry, and sequencing) for microbial identification vary in information content, complexity, speed and cost, factors which limit their practicality for clinical implementation. High Resolution Melt (HRM) interrogates an amplicon’s sequence variants through heat denaturation in the presence of an intercalating dye. This technique takes place as a rapid single-step, closed-tube process performed directly on generic PCR platforms5. Although not as information rich as sequencing, the simplicity, speed, cost, and accessibility of HRM suggest that microbiological analysis protocols that incorporate HRM for first-pass screening or diagnosis hold great promise for clinical adoption. Due to these advantages, our goal is to develop HRM-based strategies to accomplish reliable sequence fingerprinting for bacterial species identification.
In HRM, the pattern of an amplicon’s heat denaturation generates a sigmoidal melt curve. The peak of the curve’s first derivative dictates the melting temperature (Tm), which is primarily determined by the sequence %GC content and length. Previous HRM studies used short amplicons (<300 bp) and found that small (0.2 °C) shifts in Tm between samples generated reliable variant discrimination6. However, short amplicons tend to generate Tms with narrow temperature ranges (~4 °C based on our prior work7) of the full melt spectrum (i.e. 60–95 °C). If 0.2 °C is required to identify differences, a dynamic Tm range of 4 °C could only distinguish up to 20 variants. This represents a major challenge if HRM is to be expanded for large-scale fingerprinting of potentially hundreds of sequences. To overcome this limitation, we and others have designed HRM assays using multiple separate amplification reactions and primer sets to interrogate short hypervariable sequence stretches within the 16S rRNA gene (16S)7,8,9,10,11,12,13,14. This multiplex approach may moderately improve the discriminatory power, but the multiple parallel reactions complicate assay format and are impractical for samples containing few targets.
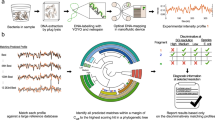
In addition to Tm-based information, HRM also observes the exact melt curve shape as a function of the actual nucleic acid sequence and strand complementarity. Multiphasic melt curves can arise depending on the number of regions with different %GC content (melt domains), which is beneficial in sequence fingerprinting15. We explored the use of a long amplicon (~1000 bp) covering six hypervariable regions in the 16S genetic locus to gather more melt domains and sequence diversity with the use of a single primer set16. Although the long amplicons yielded more biphasic melt curves, with occasional double “peaks” on the derivative curves, the narrow Tm range still constrains the profiling breadth. The 16S also has limited sequence variability to enable species-level discrimination of organisms (only 65 to 83% of cases)17. Another contributor to curve shape is the heteroduplex that forms when two strands from different amplicons with sufficient base complementarity anneal during cooling. Artificial heteroduplex formation by mixing reference DNA with unknown DNA in a 1:1 ratio has been exploited as a strategy for heterozygote and variant screening to enhance melt curve diversity18. However, this multi-step approach adds to assay complexity. Herein, we explore the use of bacterial internal transcribed spacer region (ITS) as an alternative target locus. We hypothesize that by strategically choosing this phylogenetic locus for both its higher interspecies and intragenomic sequence heterogeneity, we can generate highly complex melt curves for large-scale profiling with enhanced species-level specificity in a simple assay format (Fig. 1).

The ITS HRM assay is performed on DNA extracted from clinical samples such as positive blood culture samples. Our novel classification algorithm allows generated melt curves to identify the bacterial pathogens by matching against a reference database. Same-colored dots represent replicates from the same species classified within their boundaries and black dots outside all boundaries represent replicates from species that is not in our reference database, whereas the unknown species in the test sample (X) is identified by the classification algorithm.
HRM sensitivity to subtle differences in experimental conditions such as inconsistencies in instrumental operation and pipetting often causes run-to-run melt curves variability. The current HRM data analysis, performed with the accompanying instrument software or a commercially available one such as ScreenClust19, is not capable of compensating for these fluctuations and therefore diminish the HRM assay’s discriminatory power. To address this issue, we have developed an adaptive Naïve Bayes algorithm which has the capability to 1) enable automated melt curve classification with trained tolerance for variations in experimental conditions, 2) use a database of melt curves from reference bacterial organisms for subsequent curve-matching analysis of unknown samples, 3) discover unanticipated organisms when no match is found in the melt curve database, and 4) provide statistical interpretation. These features will yield clinically relevant information through unbiased automated interrogation of our HRM assay results.
Results
Targeting ITS generates high complexity melt curves
The 16S–23S ribosomal DNA (rDNA) internal transcribed spacer region (ITS) of the ribosomal operon rrn was used as an alternative to traditional 16S rDNA ribotyping for phylogenetic classification. ITS is known to have substantially greater interspecies polymorphisms yet high intraspecies conservation20. It is approximately 60–1,500 bp in length with up to 15 copies per genome and intragenomic variations in terms of length and sequence. Since ITS features may affect melt curve conditions, we hypothesized that ITS is an ideal target for HRM analysis that will generate melt curves of complex shapes.
To evaluate this, we generated a library of both long 16S and ITS amplicons for 89 available and characterized bacterial species archived from our prior studies. The library included mostly one strain per species with the exception of several species as listed and noted in Supplementary Table S1. These 89 species covered common pathogenic and commensal bacteria across multiple phylogenetic families and genera. Representative derivative curves for each species are available in Supplementary Fig. S1. All species exhibit high diversity in curve shapes within a given genus (i.e. Bacillus) (Fig. 2a) as well as across genera (Fig. 2b). Increased diversity of ITS curve shape as compared to 16S is clearly evident, with the ITS curves showing multiple peaks, and a much wider Tm range (15 °C compared to 5 °C for 16S) (Fig. 2a–d). We also observed that in our library, some species of the same genus that were indistinguishable by their 16S curves before can now be differentiated by their ITS curves (Fig. 2c). Multiple strains for several species included in our library generated identical melt curves, suggesting strain inclusivity at the species-level for identification (Fig. 2d). We also confirmed run-to-run and inter-operator reproducibility.

(a) Representative 16S and ITS melt curves of seven (7) species of the Bacillus genus. (b) 16S and ITS composite melt curves of all 89 bacterial organisms in our library. (c) ITS melt curves from a single strain of B. anthracis, B. mycoides, Pseudomonas aeruginosa, P. putida are compared to the their undistinguishable 16S counterparts. (d) ITS melt curves from two (2) strains of B. anthracis, 2 strains of Y. pestis, and five (5) strains of S. pneumoniae are used for the strain inclusivity test.
Template concentration on ITS curve integrity
To assess the impact of template concentration on ITS curve integrity, we serially diluted E. coli genomic DNA based on it genome copies down to extinction and performed either 40-cycle or 50-cycle PCR targeting the ITS region. At template concentrations below 102 genome copies per reaction, peak height of melt curves decreased in 40-cycle PCR HRM but was retained down to 1 genome copy in 50-cycle PCR HRM (Supplementary Fig. S2a and b).
Heteroduplex analysis
In order to determine if heteroduplex amplicon formation due to intragenomic sequence variants is the primary contributing factor to complex ITS melt curves observed, we first examined the entire genome of a representative E. coli strain (strain ATCC 25922, GenBank Accession number CP009072), and found 7 ITS copies characterized by 6 copies containing the same shorter sequence (ITS short) and 1 copy containing a longer sequence (ITS long). Alignment of these two sequences using Clustal Omega21 in silico revealed multiple regions of heterologous internal sequence (Fig. 3a). We then amplified the ITS region of E. coli ATCC 25922 with increasing cycle number. A slower migrating band observed on the gel electrophoresis beyond 25 cycles suggested increasing presence of heteroduplex structures (Fig. 3b). This was confirmed through subsequent treatment of ITS amplicons with mung bean nuclease, which cleaves unpaired DNA strands of heteroduplex products, resulting in loss of the larger band (Fig. 3c). We also performed ITS HRM analysis on 20 E. coli ITS clones from which we observed two distinct melt curve profiles (Fig. 3d). Sequencing of the ITS inserts corresponding to these two different melt curve profiles matched to either ITS short or ITS long sequence in a 3:1 distribution ratio respectively. Melt profile of the more dominant shorter ITS clone alone revealed the 3 melt peaks similar to the original E. coli ITS melt curve. Only 2 melt peaks were observed in the longer ITS clones. Mixing amplicons from each group in various DNA concentration ratios followed by HRM assay did not contribute to additional melt peaks.

(a) Clustal Omega Multiple Sequence Alignment of E. coli ATCC 25922 (GenBank Accession number CP009072) ITS short (361 bp) and ITS long (453 bp) sequences. (b) ITS homoduplex-heteroduplex profiles obtained after 20 (1), 25 (2), 30 (3), 35 (4) and 40 (5) number of PCR cycles. Slow migrating bands (at 800 and 1000 bp) were visible starting from cycle number 25, suggesting the heteroduplex nature of the bands. Expected homoduplex bands were at 540 bp (361 bp ITS short + 179 bp of 3′ of 16S and 38 bp of 5′ 23S), and 632 bp (453 bp + 179 bp). Lane M contains 100-bp DNA marker. (c) Agarose gel electrophoresis showing E. coli ITS PCR products treated (2) and untreated (1) with mung bean nuclease, an enzyme that recognizes and cleaves single stranded DNA, even when it is located in double-stranded DNA products. The loss of the higher molecular weight bands confirms the heteroduplex nature of the bands, leaving the true homoduplexes. Lane M contains 100-bp DNA marker. (d) ITS HRM analysis on 20 colonies resulted in 2 distinct melt curve groups (ITS short and ITS long). Combinations of the amplicons from each group did not recreate the original E. coli melt curve.
Development of classification algorithm
Our ITS melt curve library was generated from the 89 aforementioned bacterial species and an additional 10 randomly selected clinical positive blood culture samples that represented 10 different species that we had multiple samples of. Using this library, we performed a supervised classification analysis by training the proposed adaptive Naïve Bayes algorithm on a reference panel and testing the classifier on a separate panel of unknown samples. The goal is two-fold: 1) to predict whether the curve for the unknown sample belongs to those in the reference panel; if so, 2) to determine the species identity of the unknown sample. Our algorithm adaptively introduces an auxiliary label indicating the possibility of the tested sample to be unrepresented in the reference panel. This mechanism enables us to reliably identify novel samples, which have not been identified before. Our algorithm first calculates curve similarities based on both curve shapes and curve peak positions. Hilbert transform is used to enhance the similarity. With reliable similarity metric, our algorithm seeks nearest samples in the reference panel and constructs a probabilistic decision boundary based on the similarities to the test sample. Leave-one-out cross validation (LOOCV) analysis was performed on the reference panel with this algorithm by testing one replicate of a strain against the rest of the panel including the other replicates. This analysis revealed that 95% of species curves were reliably identified. Reference data and training results are available upon request.
ITS HRM assay on positive blood culture samples
To measure the accuracy of our ITS HRM assay (ITS PCR HRM analysis coupled with classification algorithm), we tested our assay against a convenience sample of 87 de-identified residual positive-blood-culture specimens prospectively collected from the clinical microbiology laboratory. Ten of the 87 samples were used for training our algorithm as mentioned in the previous section. Seventeen of the remaining 77 samples were omitted due to poly-bacterial or yeast culture results. We then performed 16S sequencing on 60 samples. The culture and sequencing results matched for 59 out of 60 samples. We omitted the sample with the discordant culture and 16S sequencing results, leaving us with 59 test samples representing 21 different bacterial species of which 5 species were not in our database.
We performed the ITS HRM assay on these 59 samples in duplicate, and utilized the 16S sequencing results as the reference for correct melt curve classification. Our classification algorithm incorporated both duplicate melt curves of a sample for analysis and determined the top 2 curve matches accompanied by their respective posterior probability values. Considering only the top 1 match, the algorithm correctly classified 53 samples and misclassified 6 samples with 90% classification accuracy. When we include the top 2 matches, the algorithm correctly classified 54 samples resulting in 92% classification accuracy (Table 1).
Discussion
Our single-plex ITS HRM assay can be easily implemented to identify unknown bacterial species by observing their highly variable melt curve shapes with multiple peaks and wide Tm range (15 °C). In this study, we successfully differentiated and generated a training library of unique melt profiles for 89 different clinically relevant bacterial species, some from closely related species of the same genera. Based on this library, our ITS HRM assay correctly identified 59 positive blood culture bottles with 6 discrepancies for a total combined accuracy of 95% at the genus-level and 90% at the species-level. This included correct classification of 5 unanticipated species not included in our library. Of note, our algorithm identified two of the discrepant samples as Staphylococcus epidermis and Staphylococcus caprae whereas 16S sequencing identified them as Staphylococcus hominis. All three species fall in the coagulase-negative Staphylococcus group, among which the clinical need to identify them to species level remains controversial22. Two other discrepant samples were misclassified as unknowns, which in a clinical setting would have required additional analysis such as sequencing or await culture for identification. The overall classification accuracy could likely be improved with further expansion of our training samples.
The ITS sequence between the 16S-23S rRNA is less evolutionarily constrained than its flanking genes23, making it better suited as a single species-level phylogenetic locus to simplify assay format for routine clinical adoption. Although our library of available reference species is not exhaustively comprehensive but still reasonably broad, our preliminary results suggest greater resolving power of ITS over 16S HRM analysis in species-level discrimination for multiple genera. Heteroduplex formation of ITS amplicons due to intragenomic sequence variants was demonstrated; however, contrary to our hypothesis, it is unlikely the main contributor to curve shape complexity. We postulate that multiple melt domains harbored within the short (361 bp) ITS sequences are more likely the major contributing factor to multiple melt peaks observed5. Future work is needed to assign melt domains to sequence domains. One potential advantage of whole amplicon HRM for microbial typing over probe-based or other sequence fingerprinting approaches capable of resolving single nucleotide polymorphisms is its tolerance for small intraspecies sequence differences24. We did observe identical intraspecies melt profiles based on very limited number of strains for several species in our test panel. However, previous studies had targeted ITS for strain differentiation within one bacterial species25,26. Although curve difference is more prominent between species, ITS was shown to have enough intraspecies sequence and copy number variation to produce strain specific melt curves27. This may limit our assay capability to identify a certain type of strain to its correct species if the strain variation is not in our database. Further testing with expanded number of species and their many strains is required to better assess inclusivity. Resulting data sets will also train the algorithm by incorporating subtle strain variation for a robust species-level identification.
Varying template concentrations did not significantly affect overall curve integrity as long as amplicon accumulation reaches plateau phase. Despite improvement in detection limit of our ITS PCR by increasing PCR cycle number, we chose the standard 40-cycle PCR format given considerations for reduced assay time. Subtle variations in peak height due to concentration differences can still be learned and correctly classified by the algorithm.
Our current platform is more reliable for diagnosing mono-bacterial infection. In the case of a poly-bacterial sample, the contribution of individual bacterial populations to the ensemble melt curve is difficult to decouple and identify. We previously developed Universal digital High Resolution Melt (U-dHRM) to overcome this challenge by diluting the starting material such that each genotype is distributed into its own digital PCR reaction and generates a pure melt curve7. Targeting the 16S, this approach successfully identified bacterial species at a single molecule level. The ability to quantitatively resolve sample heterogeneity may aid discrimination between true infection versus colonization or contamination, but U-dHRM’s reliance on melt curve Tm limits extent of analysis. For future assessment, replacing 16S rDNA with ITS as the target sequence may considerably increase U-dHRM’s specificity to resolve all species in a mixed sample. The simplicity of our single-plex assay also facilitates scaling to the single cell/molecule format with advanced microfluidic digital droplet platforms28, making it highly sensitive to low level targets.
We have developed a new adaptive Naïve Bayes algorithm, which we demonstrate to be capable of differentiating between closely related species, a task previously challenging with 16S rRNA-based methods. Our algorithm consists of three steps, as described in Supplementary Methods. First, we align each curve according to a preset temperature to ensure improved accuracy in the subsequent curve similarity calculation. Second, our algorithm employs a Hilbert transformation of the melt curves, and constructs an enhanced similarity metric between curves. The resulting similarity metric is robust to noise in the curve measurements and contributes to the accuracy of the classifier. Finally, our algorithm performs Naïve Bayes-based classification and reports a final classification results by integrating posterior distributions for all melt curves from one species. Our algorithm is also extended with the function of identifying unseen samples outside the reference panel and reporting them as such. It is important to note that the classification capability of the proposed classifier depends heavily on the amount of training data, and should improve as more data become available29.
Overall, our results suggest that ITS HRM assay is a simple, rapid, and practical molecular approach for broad-scale microbial species identification. It is the first to readily identify 89 bacterial species using a single-plex approach, without the need for microbial culture, hybridization probes, mass spectrometry, or sequencing. Our expandable reference database and trainable curve-matching algorithm with statistical interpretation enable discovery of unanticipated organisms with which our database can be updated after sequence verification. Similar use of ITS region may be extended to phylogenetic identification of other organism classes30,31. We recognize that a larger scale clinical study with a more comprehensive training library of species and strains would be needed to validate our preliminary findings. Nonetheless, as a simple add-on to most commonplace qPCR with HRM capabilities or emerging molecular point-of-care platforms designed for direct sample testing, our ITS HRM assay holds promise for clinical adoption as an early adjunctive test to traditional microbiological methods for etiologic diagnosis of acute febrile illnesses.
Methods
Bacterial Genomic DNA for Library Generation
Archived from previous studies, eighty-nine bacterial species were clinically isolated from the clinical microbiology laboratory at Johns Hopkins Hospital or purchased from American Type Culture Collection (ATCC). Bacterial genomic DNA from these species were extracted as previously described13.
PCR HRM Analysis
Real-time PCR amplification and HRM analysis of ITS and 16S rRNA gene was performed on the Rotor-Gene Q thermal cycler (Qiagen, Venlo, Netherlands) with ITS primers pairs ITS1F (5′-TTGTACACACCGCCCG-3′) and ITS2R (5′-YGCCAAGGCATCCACC-3′) and 16S primers pairs V1F (5′-GYGGCGNACGGGTGAGTAA-3′) and V6R (5′-AGCTGACGACANCCATGCA-3′). All standard measures to prevent sample contamination were taken including the use of a designated PCR workstation. PCR reactions were performed in a 20 μL volume using Type-It HRM kit (Qiagen, Venlo, Netherlands) with the addition of 200 nM low temperature calibrator32 and 2 μL genomic DNA. Before adding the low temperature calibrator and genomic DNA, each reaction was treated with 0.5 μL of dsDNase and 0.5 μL DTT (ArcticZymes PCR Decontamination Kit, ArcticZymes, Tromsø, Norway) followed by incubation at 37 °C for 20 minutes and 60 °C for 20 minutes to eliminate possible contaminating DNA. Reactions were covered with a 20 μL overlay of PCR grade mineral oil (Fisher Scientific, Hanover Park, IL) and cycling conditions were 95 °C for 5 min, followed by 40 amplification cycles of 95 °C for 15 s, 55 °C for 30 s, and 72 °C for 60 s for ITS or 90 s for 16S, 95 °C for 30 s and 28 °C for 30 s. HRM acquisition at every 0.1 °C immediately followed from 50 °C to 95 °C. The entire run was completed within 3 hours. HRM analysis was performed utilizing the RotorGene-Q software. Raw melt data files were exported and sent for further analysis utilizing the classification algorithm.
Heteroduplex Analysis
One μg of E. coli ATCC 25922 ITS PCR product was added to 25 μL final reaction volume containing 2.5 μL 10x reaction buffer and 1 μL of 10 U/μL mung bean nuclease. The reaction was incubated at 30 °C for 45 minutes. 25 μL of 0.02% SDS was added to stop the nuclease activity. The product was purified using MinElute Reaction Clean Up kit (Qiagen, Venlo, Netherlands) and eluted in 10 μL elution buffer. The purified nuclease treated samples were run in a 1% agarose gel, and imaged. For HRM analysis, ITS PCR reactions were performed as mentioned above and taken out after cycle number 20, 25, 30, 35, and 40. We put the reactions back into the thermal cycler and amplicons were melted from 65 °C–95 °C. Melt profiles were evaluated utilizing the RotorGene-Q software.
For cloning of E. coli ITS, the ITS PCR product was purified using MinElute Reaction Clean Up kit (Qiagen, Venlo, Netherlands) and cloned using NEB® PCR Cloning Kit (New England Biolabs, Ipswich, MA) following manufacturer’s protocol. Twenty colonies were picked, screened for insert, and purified for plasmids. Each plasmid was subjected to ITS HRM analysis and the ITS PCR product of representative plasmids of ITS short and ITS long were sent for Sanger sequencing.
Blood Culture Sample Collection and Preparation
Eighty-seven waste positive blood culture bottles were consecutively collected between July and August 2015 in the clinical microbiology laboratory at Johns Hopkins Hospital according to a protocol approved by the Institutional Review Board of The Johns Hopkins University (IRB NA_00013251). Briefly, blood was drawn from patients as part of routine clinical care for suspected bloodstream infections as recommended by the central hospital laboratory. The clinical blood culture bottles (Bactec Lytic/10 anaerobic medium, the Bactec plus aerobic/F media and the Bactec Standard/10 aerobic medium) were processed using the Bactec FX blood culture continuous monitoring system (bioMérieux, Inc., Durham, NC). None of the bottles tested contained charcoal. Further identification of bacterial isolates was performed using the Phoenix microbial identification system (Becton, Dickinson, Sparks, MD) or Matrix Assisted Laser Desorption Ionization Time of Flight Mass Spectrometry using the Bruker MicroFlex instrument (Bruker Daltonics, Lowell, MA). After reference testing was complete, the remaining waste sample was de-identified for research purposes and stored at 4 °C. Pathogens identified by standard microbiological testing were recorded. Explicit consent was not sought, since residual waste samples were retrospectively tested in a de-identified manner. Results from the PCR/HRM test was compared to reference testing results but not used to inform clinical treatment. Bacterial DNA was extracted from 500 μL aliquots of blood culture sample using a previously described protocol based on the Roche MagNA Pure extraction instrument (Roche Diagnostics)33. All blood culture samples were initially stored at 4 °C. Processing occurred as quickly as possible, with a maximum of 1 week of storage prior to batch DNA extraction.
16S Sequence Analysis
All blood culture samples were amplified for 16S using primers pairs V1F and V6R. The PCR product was purified for sequencing utilizing the Qiagen QIAquick PCR purification kit (Qiagen, Venlo, Netherlands) and sent for Sanger sequencing at MCLAB (San Francisco, CA). The sequencing results (typically 600–700 bp long) were used to choose the best matching database sequence in the NCBI database of 16S sequences using nucleotide BLAST.
Supervised Validation for Library Generation by Adaptive Naïve Bayes
We have utilized a Naïve Bayes algorithm in order to classify 89 species. The proposed adaptive Naïve Bayes (See Supplementary Methods) is able to predict whether the unknown sample is among the reference panel and subsequently distinguish the species of the sample. We incorporated a low temperature calibrator DNA duplex to correct for tube-to-tube or run-to-run experimental variations for curve alignment and normalization.
Additional Information
How to cite this article: Andini, N. et al. Microbial Typing by Machine Learned DNA Melt Signatures. Sci. Rep. 7, 42097; doi: 10.1038/srep42097 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Centers for Disease, C. & Prevention. Outbreak of acute febrile illness among athletes participating in triathlons-Wisconsin and Illinois, 1998. MMWR Morb Mortal Wkly Rep 47, 585–588 (1998).
Jennings, G. J. et al. Brucellosis as a cause of acute febrile illness in Egypt. Trans R Soc Trop Med Hyg 101, 707–713, doi: 10.1016/j.trstmh.2007.02.027 (2007).
Suttinont, C. et al. Causes of acute, undifferentiated, febrile illness in rural Thailand: results of a prospective observational study. Ann Trop Med Parasitol 100, 363–370, doi: 10.1179/136485906X112158 (2006).
Mwaigwisya, S., Assiri, R. A. & O′Grady, J. Emerging commercial molecular tests for the diagnosis of bloodstream infection. Expert Rev Mol Diagn 15, 681–692, doi: 10.1586/14737159.2015.1029459 (2015).
Wittwer, C. T., Reed, G. H., Gundry, C. N., Vandersteen, J. G. & Pryor, R. J. High-resolution genotyping by amplicon melting analysis using LCGreen. Clin Chem 49, 853–860 (2003).
Tong, S. Y. & Giffard, P. M. Microbiological applications of high-resolution melting analysis. Journal of clinical microbiology 50, 3418–3421, doi: 10.1128/JCM.01709-12 (2012).
Fraley, S. I. et al. Universal digital high-resolution melt: a novel approach to broad-based profiling of heterogeneous biological samples. Nucleic Acids Res 41, e175, doi: 10.1093/nar/gkt684 (2013).
Athamanolap, P. et al. Trainable high resolution melt curve machine learning classifier for large-scale reliable genotyping of sequence variants. PloS one 9, e109094, doi: 10.1371/journal.pone.0109094 (2014).
Hardick, J. et al. Identification of bacterial pathogens in ascitic fluids from patients with suspected spontaneous bacterial peritonitis by use of broad-range PCR (16S PCR) coupled with high-resolution melt analysis. J Clin Microbiol 50, 2428–2432, doi: 10.1128/JCM.00345-12 (2012).
Jeng, K. et al. Application of a 16S rRNA PCR-high-resolution melt analysis assay for rapid detection of Salmonella Bacteremia. Journal of clinical microbiology 50, 1122–1124, doi: 10.1128/JCM.05121-11 (2012).
Masek, B. J. et al. Sensitive detection and serovar differentiation of typhoidal and nontyphoidal Salmonella enterica species using 16S rRNA Gene PCR coupled with high-resolution melt analysis. J Mol Diagn 16, 261–266, doi: 10.1016/j.jmoldx.2013.10.011 (2014).
Niimi, H. et al. Melting Temperature Mapping Method: A Novel Method for Rapid Identification of Unknown Pathogenic Microorganisms within Three Hours of Sample Collection. Scientific reports 5, 12543, doi: 10.1038/srep12543 (2015).
Won, H. et al. Rapid identification of bacterial pathogens in positive blood culture bottles by use of a broad-based PCR assay coupled with high-resolution melt analysis. Journal of clinical microbiology 48, 3410–3413, doi: 10.1128/JCM.00718-10 (2010).
Yang, S. et al. Rapid identification of biothreat and other clinically relevant bacterial species by use of universal PCR coupled with high-resolution melting analysis. J Clin Microbiol 47, 2252–2255, doi: 10.1128/JCM.00033-09 (2009).
Lilliebridge, R. A., Tong, S. Y., Giffard, P. M. & Holt, D. C. The utility of high-resolution melting analysis of SNP nucleated PCR amplicons-an MLST based Staphylococcus aureus typing scheme. PloS one 6, e19749, doi: 10.1371/journal.pone.0019749 (2011).
Fraley, S. I. et al. Nested Machine Learning Facilitates Increased Sequence Content for Large-Scale Automated High Resolution Melt Genotyping. Scientific reports 6, 19218, doi: 10.1038/srep19218 (2016).
Janda, J. M. & Abbott, S. L. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. J Clin Microbiol 45, 2761–2764, doi: 10.1128/JCM.01228-07 (2007).
Palais, R. A., Liew, M. A. & Wittwer, C. T. Quantitative heteroduplex analysis for single nucleotide polymorphism genotyping. Anal Biochem 346, 167–175, doi: 10.1016/j.ab.2005.08.010 (2005).
Reja, V. et al. ScreenClust: Advanced statistical software for supervised and unsupervised high resolution melting (HRM) analysis. Methods 50, S10–14, doi: 10.1016/j.ymeth.2010.02.006 (2010).
Gurtler, V. & Stanisich, V. A. New approaches to typing and identification of bacteria using the 16S-23S rDNA spacer region. Microbiology 142 (Pt 1), 3–16, doi: 10.1099/13500872-142-1-3 (1996).
Sievers, F. & Higgins, D. G. Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol Biol 1079, 105–116, doi: 10.1007/978-1-62703-646-7_6 (2014).
Becker, K., Heilmann, C. & Peters, G. Coagulase-negative staphylococci. Clin Microbiol Rev 27, 870–926, doi: 10.1128/CMR.00109-13 (2014).
Lee, Z. M., Bussema, C., 3rd & Schmidt, T. M. rrnDB: documenting the number of rRNA and tRNA genes in bacteria and archaea. Nucleic Acids Res 37, D489–493, doi: 10.1093/nar/gkn689 (2009).
Rasmussen, J. P., Saint, C. P. & Monis, P. T. Use of DNA melting simulation software for in silico diagnostic assay design: targeting regions with complex melting curves and confirmation by real-time PCR using intercalating dyes. BMC Bioinformatics 8, 107, doi: 10.1186/1471-2105-8-107 (2007).
Jensen, M. A., Webster, J. A. & Straus, N. Rapid identification of bacteria on the basis of polymerase chain reaction-amplified ribosomal DNA spacer polymorphisms. Appl Environ Microbiol 59, 945–952 (1993).
Yasuda, M. & Shiaris, M. P. Differentiation of bacterial strains by thermal gradient gel electrophoresis using non-GC-clamped PCR primers for the 16S-23S rDNA intergenic spacer region. FEMS Microbiol Lett 243, 235–242, doi: 10.1016/j.femsle.2004.12.011 (2005).
Gurtler, V., Grando, D., Mayall, B. C., Wang, J. & Ghaly-Derias, S. A novel method for simultaneous Enterococcus species identification/typing and van genotyping by high resolution melt analysis. J Microbiol Methods 90, 167–181, doi: 10.1016/j.mimet.2012.05.002 (2012).
Rane, T. D., Zec, H. C., Puleo, C., Lee, A. P. & Wang, T. H. Droplet microfluidics for amplification-free genetic detection of single cells. Lab Chip 12, 3341–3347, doi: 10.1039/c2lc40537g (2012).
Zimic, M. J., Guerra, D. & Arevalo, J. DNA thermodynamic pressure: a potential contributor to genome evolution. Trans R Soc Trop Med Hyg 96 Suppl 1, S15–20 (2002).
Mandviwala, T., Shinde, R., Kalra, A., Sobel, J. D. & Akins, R. A. High-throughput identification and quantification of Candida species using high resolution derivative melt analysis of panfungal amplicons. J Mol Diagn 12, 91–101, doi: 10.2353/jmoldx.2010.090085 (2010).
Somogyvari, F. et al. Detection of invasive fungal pathogens by real-time PCR and high-resolution melting analysis. In Vivo 26, 979–983 (2012).
Fraley, S. I. et al. Universal digital high-resolution melt: a novel approach to broad-based profiling of heterogeneous biological samples. Nucleic Acids Research 41, e175, doi: 10.1093/nar/gkt684 (2013).
Jeng, K. et al. Comparative analysis of two broad-range PCR assays for pathogen detection in positive-blood-culture bottles: PCR-high-resolution melting analysis versus PCR-mass spectrometry. Journal of clinical microbiology 50, 3287–3292, doi: 10.1128/JCM.00677-12 (2012).
Acknowledgements
E.W. “Al” Thrasher Research Award, The Gerber Foundation, NIH R01AI117032, and Stanford Spark.
Author information
Authors and Affiliations
Contributions
N.A., S.T. and S.Y. conceived of experiments; N.A. and A.H. carried out experiments; N.A, B.W., P.A, T.W., S.B. and S.Y. conceived of algorithm, B.W. developed algorithm; J.H., B.M. and C.G. provided DNA samples for reference bacterial species and from positive blood culture samples; G.A, S.P., R.R. and K.C. provided characterized clinical isolates; S.C. performed statistical analysis; N.A., B.W., S.B. and S.Y. wrote manuscript; all authors reviewed manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Andini, N., Wang, B., Athamanolap, P. et al. Microbial Typing by Machine Learned DNA Melt Signatures. Sci Rep 7, 42097 (2017). https://doi.org/10.1038/srep42097
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep42097
This article is cited by
-
RAPD and ERIC-PCR coupled with HRM for species identification of non-dysenteriae Shigella species; as a potential alternative method
BMC Research Notes (2021)
-
Precision cancer monitoring using a novel, fully integrated, microfluidic array partitioning digital PCR platform
Scientific Reports (2019)
-
Diagnosis of Human Leptospirosis in a Clinical Setting: Real-Time PCR High Resolution Melting Analysis for Detection of Leptospira at the Onset of Disease
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
