
Abstract
Chediak–Higashi syndrome (CHS) is a rare autosomal recessive disease characterized by varying degrees of oculocutaneous albinism, recurrent infections, and a mild bleeding tendency, with late neurologic dysfunction. This syndrome is molecularly characterized by pathognomonic mutations in the LYST (lysosomal trafficking regulator). Using whole genome sequencing (WGS) we attempted to identify novel mutations of CHS based on a family of CHS with atypical symptoms. The two patients demonstrated a phenotypic constellation including partial oculocutaneous albinism, frequency upper respiratory infection or a marginal intelligence, without bleeding tendency and severe immunodeficiency. WGS revealed two compound LYST mutations including a maternally inherited chr1:235969126G > A (rs80338652) and a novel paternally inherited chr1: 235915327A > AT, associated with autosomal recessive CHS. These two variants fall in the coding regions of LYST, resulting in premature truncation of LYST due to R1104X/N2535KfsX2 induced incomplete translation. Notably, the heterozygous carriers (i.e. parents) were unaffected. Our finding also reveals decreased plasma serotonin levels in patients with CHS compared with unaffected individuals for the first time. The present study contributes to improved understanding of the causes of this disease and provides new ideas for possible treatments.
Similar content being viewed by others

Introduction
Chediak-Higashi Syndrome (CHS) is a rare autosomal recessive disease, characterized by partial oculocutaneous albinism (OCA), increased susceptibility to infection, a mild bleeding tendency, and/or late-onset progressive neurological impairment1. Bone marrow transplantation (BMT) is an acceptable curative treatment for CHS2. Defective structure or function of melanosomes in melanocytes is causative of OCA in CHS3. Large eosinophilic, peroxidase-positive inclusion bodies in circulating granulocytes cytotoxic T lymphocytes (CTLs), NK cells, and macrophages increase susceptibility to infection of patients with CHS4. The absence of dense bodies in CHS result in mild bleeding tendencies due to the inability to form platelet aggregation and hemostatic plugs1,3,5. Late-onset progressive neurologic symptoms observed were identical to those previously described in adults with CHS with a mild clinical course of the disease and who did not undergo BMT2.
In addition to the clinical manifestations and laboratory examination, molecular studies are also essential for the diagnosis of this disease. Pathologic mutations in the lysosomal trafficking regulator gene (LYST, also known as Chediak-Higashi Syndrome1, CHS1) at 1q42.1–2 are responsible for this defect6,7. LYST is a member of the BEACH (beige and Chediak-Higashi) family, defined by the presence of a BEACH motif in the C-terminal. The BEACH motif consists of a WIDL-enriched sequence followed by several WD-40 domains known to serve as a platform for protein-protein interaction8,9,10. The N-terminal of LYST has several ARM/HEAT repeats known to play a role in membrane interaction10. To date, 41 mutations of LYST associated with Chediak-Higashi syndrome have been reported (Table 1)11. These mutations are located from nucleotide 118 to 11,173, predominantly after nucleotide 2,000 and largely (75.6%) leading to truncated mutants of LYST (Table 1).
In the present study, we report a new autosomal recessive form of Chediak-Higashi syndrome with atypical symptom, including partial oculocutaneous albinism, frequency upper respiratory infection or a marginal intelligence. Furthermore, WGS results showed chr1:235969126G > A (rs80338652)/chr1: 235915327A > AT compound heterozygote (R1104X/N2535KfsX2) in LYST. While LYST R1104X has been previously reported7, a novel LYST mutation N2535KfsX2 identified is expected to result in a deletion of the protein. These provide new laboratory evidence for the clinical and prenatal diagnosis of CHS.
Results
Clinical features
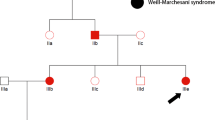
The proband was a 6-year-old girl who presented to a dermatologist because of the appearance of darkening skin on her face (Fig. 1B left) and limbs (Supplementary Figure 1), noted “silvery” colored hair with an abnormal pigment clumping under light microscopy (Fig. 2A). A peripheral blood smear identified an abnormal WBC granules compared with unaffected members of the family (Fig. 2B). Left temporal skin biopsy of the proband (HE × 400) identified edema and an increased number of intracellular pigment particles in basal epithelial cells (Fig. 2C). The parents reported a normal pregnancy and delivery; however, in the first six months of life progressive hyperpigmentation of the cheeks and limbs extensor side, gray hair and mild photophobia appearance was noted. History revealed frequent upper respiratory infection of the proband, no family history of CHS, no history of severe re-infection and mild bleeding tendency. Physical examination revealed a height of 105 cm, 3% below the normal height of Chinese children. Fundus examination demonstrated abnormal retinal vascular and pigments (data not shown). Intriguingly, Wechsler Intelligence Scale test noted that the proband had a marginal intelligence, however cerebral MRI showed no obvious abnormality (Supplementary Figure 2).

(A) Pedigree of the two-generation, Chinese family with two affected individuals. Squares indicate males, and circles represent females. Black and white symbols represent affected and unaffected individuals, respectively. The proband is indicated by an arrow. (B) Partial oculocutaneous albinism. Face of the proband(II-2) and younger brother(II-3). The two patients were noted that hyperpigmentation with bleaching spots of the cheeks and “silvery” colored hair.

(A) Hair shafts. Irregular coarse melanin granules distributed in the patient’s hair (II-2 and II-3), evenly distributed pigment in the normal hair (II-1, I-1 and 1–2). (B) Wright staining of peripheral smear. Peripheral smear showed a polymorphonuclear leukocyte with abundant giant intracytoplasmic granules (100 ×) of the patients (II-2 and II-3 arrow), and normal polymorphonuclear leukocyte inclusion of unaffected members (I-1, I-2). (C) The proband left temporal skin biopsy (HE × 400). Skin projections becomes flat, basal cells are edematous, intracellular pigment particles are increasing, a small amount of lymphoid tissue cells exists in superficial dermal perivascular, occasionally pigment cells infiltration is visible (narrow).
The second case was the younger brother (1 year old) of the proband, with essentially the same clinical presentations. In this case, brown pigmentations are visible on the patient’s cheeks (Fig. 1B right). His hair was a“silvery” color (Fig. 1B right), and light microscopy examination revealed the presence of small, homogeneously distributed pigment clumps. These findings were in contrast to the normal pattern of distribution of unaffected family members (Fig. 2A). Peripheral blood smear showed giant inclusions in the cytoplasm of polymorphonuclear neutrophils (PMNs) not observed in the parents (Fig. 2B).
Whole genome sequencing data analysis
Since the two affected siblings have a unique phenotype and no history of re-infection and bleeding, we performed whole genome sequencing of all family members to identify the causative variants. Average coverage for the affected and unaffected members was >60× and >30×, respectively (Table 2). To improve the reliability of variant calls, we retained variants identified by VarScan and GATK pipelines12,13 (see Methods). In total, we observed 5,246,911 positions at which at least one family member had an allele that varied from the reference genome. On average, each family member had approximately four million positions with variant, identified and annotated using ANNOVAR14 (MAF < 0.01 in Asian cohort by 1000 Genomes Project (2012 April) annotations, see Methods).
To search for possible causative variants in coding and potential regulatory regions, we hypothesized that the causative variants would be recessive, loss of heterozygosity (LOH) or compound heterozygous. 28 and 11 variants were identified by recessive and LOH model analyses. These variants distribute in coding, upstream (<1000 bp to TSS) or 5′ UTR regions as shown in Supplementary Table 1. Of the recessive variants, we observed only one missense variant of a chimeric POTE-actin protein (POTEF, chr1:130877752T > C), while the altered amino acid falls within nonfunctional region of POTEF protein. Although 25 recessive and 11 LOH variants located within upstream or 5′ UTR regions, there were no conserved transcriptional factor binding sites annotated by ANNOVAR. Thus, there were no obvious recessive and LOH variants in potential regulatory and coding regions that could be responsible for this disease. Besides, we identified four genes (LYST, ALMS1, TAS2R46, and TAS2R43) in which mutations identified are consistent with a compound heterozygous mode of inheritance (Supplementary Table 2). Two variants, chr1:235969126G > A (rs80338652) and chr1: 235915327A > AT, fall in the coding regions of LYST (Lysosomal Trafficking Regulator), and were predicted to be stop-gain and a frameshift insertion (Table 3), respectively. These two variants together comprise multiple alleles formed compound heterozygotes.
Validation of mutations by Sanger sequencing
We validated the two variants by Sanger sequencing (Fig. 3A). The screening sample included genomic DNA from frozen samples. Our results demonstrated that the father carries a chr1:235969126G > A (rs80338652) mutant, while mother carries a chr1: 235915327A > AT mutant. The affected patients carry both variants. The older sister (II-1), who is unaffected, carries a single LYST mutant (chr1: 235915327A > AT).

(A) Sanger sequencing validation studies. Sanger sequencing confirmed the compound heterozygotes mutations chr1:235969126G > A (rs80338652) (R1104Ter) and chr1: 235915327A > AT (N2535KfsX2) of the LYST gene identified. The two patients (II-2 and II-3) carry both of these two variants. The elder sister, who is unaffected, carries chr1: 235915327A > AT mutant. The father carries a chr1:235969126G > A (rs80338652) mutant, while mother carries chr1: 235915327A > AT mutant. (B) Human LYST protein and mutants. Domains and motifs responsible diagram of the mutations affected LYST protein. ARM/HEAT, the ARM/HEAT repeats region; PH, PH domain-like superfamily; BEACH, a BEACH motif in the C-terminal; WD, WD-40 domains. Recurrent R1104Ter mutationlocates in the ARM/HEAT repeats region. Mutation N2535KfsX2 leads to truncated of LYST protein at 2536 amino acid leading to deletion of PH-BEACH-WD domain.
Serotonin in the plasma of patients with CHS
Absent or reduced number and irregular morphology of platelet-dense bodies is one of the characteristics in CHS patients. Due to serotonin is the main component of dense bodies, we assumed that CHS patients may have lower level of serotonin in the plasma. ELISA was performed to detect the plasma level of serotonin. Although unaffected members of the family are healthy, each individual carries chr1: 235969126G > A (rs80338652) or chr1: 235915327A > AT mutant. We thus add two unrelated healthy volunteers as control. The result shows that patients (II-2 and II-3, Serotonin 28.08 (27.83–28.32) ng/mL) with CHS exhibited significantly decreased plasma serotonin levels compared with unaffected family controls (II-1, I-1, I-2); plus two unrelated healthy controls (63.72 (35.66–90.17) ng/mL, p < 0.05* see Fig. 4).

Data are presented as medians. Plasma serotonin level is lower in CHS patients (II-2 and II-3) compared with healthy controls (Unaffects; II-1, I-1, I-2 and two healthy controls).
Discussion
The dermatologist diagnosed two patients in the family as atypical CHS with reiterative upper respiratory tract infection, without bleeding tendency, neurodegeneration and severe immunodeficiency. Previously these patients had been diagnosed with griscelli syndrome. We performed WGS analysis on five samples derived from the two patients, their sibling and parents, to characterize the underlying genetic mechanisms resulting in the atypical presentation. We identified a novel compound heterozygous mutations generating truncated LYST and leading to autosomal recessive CHS. Intriguingly, Wechsler Intelligence Scale Test unveiled the proband appears a marginal intelligence phenotype, thus excluding intelligence related mutations. The R1104X mutation was reported to lead to truncated LYST, causative of autosomal recessive CHS7. The novel mutation N2535KfsX2, results in a frameshift and consequent translational termination at codon 2536, disrupting important domains such as PH-BEACH-WD (Fig. 3B). Thus, this compound heterozygous mutations R1104X/N2535KfsX2 lead to autosomal recessive CHS. The present study is the first time to report the novel mutation chr1: 235915327A > AT, along with chr1:235969126G > A (rs80338652) together, forming a compound heterozygous in LYST. These provide new laboratory evidence for the clinical and prenatal diagnosis of CHS. Besides, our findings also reveal, for the first time, decreased plasma serotonin levels in patients with CHS compared with unaffected individuals.
In addition to the present study, previous study has reported another case carrying compound heterozygous for two LYST gene mutations, both of which are predicted to result in truncated proteins1. The two mutations are a nonsense mutation (c.1540 C > T, CGA > TGA, R514X) and a one base pair deletion (del c.9893 T, F3298fsX3304), coding for part of the LYST protein’s BEACH domain1. To date, including our study, two pairs of compound heterozygous LYST gene mutations have been found. Although CHS is known as a monogenic disease, next generation sequencing technology provides us a perfect tool to investigate potential gene mutations that may have effects on this rare disease. We performed whole genome sequencing to search for possible causative variants. We used recessive, loss of heterozygosity (LOH) or compound heterozygous modes to call rare mutations which have potential regulatory functions. The 28 recessive and 11 LOH variants were identified felling in coding, upstream or 5′ UTR regions. However, there were no obvious recessive and LOH variants in potential regulatory and coding regions that could be responsible for this disease. In compound heterozygous mode of inheritance, besides chr1:235969126G > A (rs80338652) and chr1: 235915327A > AT in LYST, one pair of compound heterozygous mutations in ALMS1, three pairs of compound heterozygous mutations in TAS2R43, and three pairs of compound heterozygous mutations in TAS2R46 were identified (see Supplementary Table 2). In ALMS1, F926S and P2960L compound heterozygous mutations falls within nonfunctional region, indicating these mutations don’t play key role CHS disease. In TAS2R43, S11I and R268G mutations fall in transmembrane-region 1 and transmembrane-region 7, respectively. Similarly, in TAS2R46, E265Q and T232I mutations fall in transmembrane-region 7 and transmembrane-region 6. TAS2R43 and TAS2R46 belong to the TAS2R family, and expressed on the surface of taste receptor cells, mediating the perception of bitterness through a G protein-coupled second messenger pathway15. Since the expression and function of TAS2R43 and TAS2R46 are specific related to taste, we considered these compound heterozygous mutations not related to CHS disease.
The diagnosis of CHS should be considered in individuals with pigment dilution defects of the hair, skin, or eyes; congenital or transient neutropenia; immunodeficiency; and otherwise unexplained neurologic abnormalities or neurodegeneration. The differential diagnosis of CHS with several diseases should be noted, such as oculocutaneous albinism (OCA), hermanskypudlak syndrome (HPS), griscelli syndrome (GS). Each of these diseases can be distinguished with CHS by the presence of specific molecular findings. OCA can be caused by TYR, TYRP1 or MATP gene mutation; GS may be caused by pathogenic variants in MYO5A, RAB27A or MLPH; at least nine subtypes of hermansky-pudlak syndrome (HPS), HPS2 caused by variants in AP3B1 which most closely resembles CHS. GS is characterized by mild skin hypopigmentation and silvery gray hair, but normal platelet function; these clinical features are highly similar to the phenotypic presentation of our patients. Although symptoms are similar among these diseases and CHS, the peripheral blood smear is sufficient to distinguish CHS. Moreover, LYST is the only gene known to be associated with CHS, thus genetic testing represents an important component to confirm the diagnosis16,17. In this study, the compound heterozygous mutations in LYST were shared by both patients. R1104X in LYST has been reported in CHS patients with decreased cognitive abilities2. However, in the present study, the R1104X heterozygote mutation carrier (mother of the patients) appears to have normal intelligence. Deletion of the PH-BEACH-WD domain of LYST can be caused by both R1104X and N2535KfsX2 mutations. We thus considered the compound heterozygous mutation in LYST as causative of the marginal intelligence in the proband. Interestingly, previous studies have shown that cerebral MRI findings consisted of supratentorial and cerebellar volume loss2. In the proband, however, cerebral MRI observed no obvious abnormalities. Several studies have been focused on intellectual limitation. Xue et al. found several genes (WNT1, BMP4, POU3F4, TFAP2C, PAX3, KCNA1, KCNC3, KCNG4, KCNJ3, KCNK9 and KCNT1) associated with neuronal differentiation of fragile X syndrome, which is a common hereditary disorder associated with intellectual limitations18. Besides, Posthuma et al. found ATXN1 and TRIM31 genes are related to intelligence in an attention deficit/hyperactivity disorder background19. However, we didn’t found any mutation fall in these genes. In order to expand our study, long-term follow-up observation of patients’ nervous system is necessary. Pigment dilution defects of the hair, skin, or eyes and large granules in leucocytes could support the diagnosis of CHS. The nature of CHS is due to various LYST gene mutations that produce different phenotypes3,20. Presently, clinicians and researches have been trying to further describe the relationship between genetic mutations and clinical symptoms. Oversized melanosomes accumulate in the perinuclear area are observed in epidermal melanocytes of individuals with CHS3. They are not transferred to surrounding keratinocytes, which would explain the skin hypomelanosis. Enlarged mature lytic granules in CHS cytotoxic T-lymphocytes are unable to mediate targeted cell killing21. Lack of platelet dense body can cause coagulation disorders22. Thus the specific role of LYST mutants requires further studies.
LYST is a member of the BEACH (beige and Chediak-Higashi) family of proteins, which are defined by the presence of a C-terminal BEACH motif. The BEACH motif consists of a WIDL-enriched sequence followed by several WD-40 domains known to serve as a platform for protein-protein interaction8,9,10. The N-terminal of LYST has several ARM/HEAT repeats known to play a role in membrane interaction10. Mutation chr1:235969126G > A (rs80338652) leads to truncated ARM/HEAT domain in sister chromatid, while chr1: 235915327A > AT results in deletion of PH-BEACH-WD domain in the other one. Thus the two mutations implement sister chromatid truncated LYST protein. The R1104X mutation (caused by rs80338652 G > A) in LYST is located in the ARM/HEAT repeats region, and result in LYST translational termination at codon 1104, indicating that may affect the membrane interaction function and protein-protein interaction of LYST (Fig. 3B). The N2535KfsX2 (caused by chr1: 235915327A > AT mutant) mutation is located between ARM/HEAT repeats region and PH domain and leads translational termination of LYST at codon 2536. Thus, we assumed that N2535KfsX2 mutant may not affect membrane interaction but affect protein-protein interaction.
A previous study identified twenty-one proteins that interact with LYST using a yeast two-hybrid screens23. R1104X mutation in LYST leads to loss of interaction with 18 proteins, that likely play key roles in vesicle tethering, docking and fusion, phosphorylation and signal transduction (Such as 14-3-3 proteins, Hepatocyte growth factor–regulated tyrosine kinase substrate and Casein kinase II β-subunit). Only 14-3-3β/τ and estrogen receptor-related protein (ERR1), involved in protein kinase C (PKC) signaling transduction, exocytosis and transcriptional regulation, can interact with R1104X truncation. LYST N2535KfsX2 truncation results in loss of 9 interaction proteins, involved in neurodegenerative disorder like Atrophin-1, transcription regulation and signal transduction. Thus, N2535KfsX2 can alleviate the symptoms caused by R1104X, thereby explaining the phenotype of our patients.
Several studies have been focused on varies of CHS phenotype by using LYST mutation mice. Two mouse strains, C57BL/6 souris mice and beige mice, are commonly used in CHS mechanism studies. Souris mice have a truncated mutation which ending at 2482 amino acid within the ARM/HEAT domain of LYST. Beige mice have an Ile deletion at 3741 amino acid within the WD domain. These two strains of mice have similar phenotype: diluted coat color, typical inclusion bodies in granulocytes and NK-cell degranulation and cytotoxicity24. Although both strains lacked NK-cell cytotoxicity, only souris mice developed all clinical and histopathologic signs of hemophagocytic lymphohistiocytosis (HLH) after infection with lymphocytic choriomeningitis virus24. In addition, CTLs in beige strain eliminated virus infection, while souris CTLs failed to control them. This suggests CTL cytotoxicity may be predictive for the risk of CHS patients to develop HLH24. Thus, we will follow up this family, investigating how LYST mutations affect CTLs function accelerating HLH in CHS for further study.
Most CHS platelets show absent dense bodies; a few platelets harbor enlarged and irregularly shaped dense bodies)3. Due to serotonin is the main component of dense bodies, we assumed that CHS patients may have lower level of serotonin in the plasma. As a result, our finding reveals decreased plasma serotonin levels in patients with CHS compared with unaffected individuals for the first time. Absences of dense bodies in CHS result in mild bleeding tendencies due to the inability to form hemostatic plugs, however, lower levels of serotonin in our patients don’t lead to bleeding tendency.
In the present study, we report a novel autosomal recessive form of CHS–R1104X/N2535KfsX2 compound heterozygote in LYST. R1104X recessive mutation in LYST has been previously reported7. Herein, we report a novel LYST mutation N2535KfsX2 together with R1104X is expected to result in a deletion protein, which leads to truncated ARM/HEAT domain and deletion of PH-BEACH-WD domain of LYST of the two sister chromatid respectively, in a newly diagnosed child and her family and provide phenotypic correlations via a thorough description of the affected individual. Thus this study contributes to improved understanding of the causes of this disease and provides new ideas for possible treatments.
Methods
Ethics Statement
The present study was approved by the Ethics Committee of the Beijing Children’s Hospital Affiliated to the Capital Medical University, Beijing, China, and was conducted according to the principles expressed in the Declaration of Helsinki. Participants and/or their legal guardians involved in this study gave a written informed consent prior to inclusion in the study.
Patients
This study included two patients (II-2 and II-3) and three unaffected family members (elder sister (II-1), mother and father) as shown in Fig. 1A. Genomic DNA samples were obtained with written informed consent. TIAN amp Blood DNA Kit (Tiangen Biotech co., Ltd, Beijing) was used for extracting genomic DNA from blood samples.
Reads mapping & variants calling
Paired-end reads of 300 bp (150 bp at each end) were mapped to UCSC human reference genome (GRCh37/hg19 assembly) using BWA25 version 0.7.7-r441 ‘mem’ mode with default options followed by removal of PCR duplicates, low-quality reads (BaseQ < 20). After being sorted and indexed by samtools26, the bam files of each offspring and the parents were converted as three-sample (trio) mpileup format. Variant calls were performed by VarScan software using version 2.3.7 of Trios pipeline12 (http://varscan.sourceforge.net/). In addition, the Genome Analysis Toolkit (GATK)12 pipeline with version 2.6–5 was used to cross-validate the variants called by VarScan.
Variant annotation and prioritization
We used the ANNOVAR software for variant filtering and annotation13. Rare variants (minor allele frequency (MAF) <0.01 in Asian cohort) were filtered by 1000 Genomes Project data (2012 April) using ANNOVAR filter-based annotation14. By excluding common variants, the remaining rare variants were annotated by ANNOVAR RefSeq gene-based annotation13. In addition to removing synonymous, non-frameshift or noncoding variants, we also applied both recessive and compound heterozygous mode of inheritance to select candidate pathogenic variants.
Data Availability
All the clinical data and identified genetic variations have been deposited into the rare disease database - eRAM at http://www.unimd.org/eram/.
Amplification mutation sites and Sanger sequencing
We amplified the two mutations in LYST with a Veriti 96-well Thermal Cycler (Applied Biosystems, Thermo Fisher Scientific). The primer sequences used include: F: 5′-cag aagtcacaaagaggagca-3′, R: 5′-ttgtgtgatactgttggctgc-3′; F: 5′-tacatattacttgccatagaa-3′, R: 5′-agttggcaatacataagtga-3′. Mutations were confirmed by Sanger sequencing.
Enzyme-Linked Immunosorbent Assay (ELISA)
The plasma level of serotonin was measured by ELISA using a commercial serotonin antibody kit according to the manufacturer’s instructions (ab133053; Abcam, Cambridge, MA, USA). All ELISA measurements were made in duplicate and results were expressed as median values and interquartile ranges. Comparisons between two groups were made using a two-tailed Mann-Whitney U-test. A p-value of 0.05 was considered significant.
Additional Information
How to cite this article: Jin, Y. et al. Whole Genome Sequencing Identifies Novel Compound Heterozygous Lysosomal Trafficking Regulator Gene Mutations Associated with Autosomal Recessive Chediak-Higashi Syndrome. Sci. Rep. 7, 41308; doi: 10.1038/srep41308 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Zarzour, W. et al. Two novel CHS1 (LYST) mutations: clinical correlations in an infant with Chediak-Higashi syndrome. Molecular genetics and metabolism 85, 125–132, doi: 10.1016/j.ymgme.2005.02.011 (2005).
Tardieu, M. et al. Progressive neurologic dysfunctions 20 years after allogeneic bone marrow transplantation for Chediak-Higashi syndrome. Blood 106, 40–42, doi: 10.1182/blood-2005-01-0319 (2005).
Westbroek, W. et al. Cellular defects in Chediak-Higashi syndrome correlate with the molecular genotype and clinical phenotype. J Invest Dermatol 127, 2674–2677 (2007).
Faigle, W. et al. Deficient peptide loading and MHC class II endosomal sorting in a human genetic immunodeficiency disease: the Chediak-Higashi syndrome. J Cell Biol 141, 1121–1134 (1998).
Ambrosio, A. et al. Mechanism of platelet dense granule biogenesis: study of cargo transport and function of Rab32 and Rab38 in a model system. Blood 120, 4072–4081 (2012).
Barbosa, M. D. et al. Identification of the homologous beige and Chediak-Higashi syndrome genes. Nature 382, 262–265 (1996).
Nagle, D. L. et al. Identification and mutation analysis of the complete gene for Chediak-Higashi syndrome. Nat Genet 14, 307–311 (1996).
Jogl, G. et al. Crystal structure of the BEACH domain reveals an unusual fold and extensive association with a novel PH domain. EMBO J 21, 4785–4795 (2002).
De Lozanne, A. The role of BEACH proteins in Dictyostelium. Traffic 4, 6–12 (2003).
Kaplan, J., De Domenico, I. & Ward, D. M. Chediak-Higashi syndrome. Curr Opin Hematol 15, 22–29 (2008).
Karim, M. A. et al. Apparent genotype-phenotype correlation in childhood, adolescent, and adult Chediak-Higashi syndrome. Am J Med Genet 108, 16–22 (2002).
Koboldt, D. C. et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22, 568–576 (2012).
Van der Auwera, G. A. et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 11, 11.10.11–11.10.33 (2013).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38, e164 (2010).
Conte, C. et al. Identification and characterization of human taste receptor genes belonging to the TAS2R family. Cytogenet Genome Research 98(1), 45–53 (2002).
Introne, W. J. et al. Chediak-Higashi Syndrome in GeneReviews® (ed. Pagon, R. A. et al.) (Seattle, 1993–2016).
Huizing, M. et al. A. Disorders of lysosome-related organelle biogenesis: clinical and molecular genetics. Annu Rev Genomics Hum Genet 9, 359–386 (2008).
Lu P. et al. Integrated transcriptome analysis of human ips cells derived from a fragile × syndrome patient during neuronal differentiation. Science China Life sciences (2016).
Rizzi T. S. et al. The atxn1 and trim31 genes are related to intelligence in an adhd background: Evidence from a large collaborative study totaling 4,963 subjects. American journal of medical genetics Part B, Neuropsychiatric genetics: the official publication of the International Society of Psychiatric Genetics 156, 145–157 (2011).
Kaya, Z. et al. A novel single point mutation of the LYST gene in two siblings with different phenotypic features of Chediak Higashi syndrome. Pediatr Blood Cancer 56, 1136–1139 (2011).
Holland, P. et al. LYST affects lysosome size and quantity, but not trafficking or degradation through autophagy or endocytosis. Traffic 15, 1390–1405 (2014).
Golebiewska, E. M. & Poole, A. W. Platelet secretion: From haemostasis to wound healing and beyond. Blood Rev 29, 153–162 (2015).
Tchernev, V. T. et al. The Chediak-Higashi protein interacts with SNARE complex and signal transduction proteins. Molecular medicine 8, 56–64 (2002).
Jessen, B. et al. Subtle differences in CTL cytotoxicity determine susceptibility to hemophagocytic lymphohistiocytosis in mice and humans with Chediak-Higashi syndrome. Blood 118(17), 4620–9, doi: 10.1182/blood-2011-05-356113 (2011).
Li, H. Exploring single-sample SNP and INDEL calling with whole-genome de novo assembly. Bioinformatics 28, 1838–1844 (2012).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Acknowledgements
This work was supported in part by the National High Technology Research and Development Program of China (2015AA020108); the China Human Proteome Project (2014DFB30030); Beijing Municipal Science and Technology Project (D131100005313014); Capital’s Special Clinic Application Research Project (Z161100000516070); Beijing Municipal Administration of Hospitals Clinical Medicine Development of Special Funding Support (ZYLX201508). All authors reviewed the manuscript.
Author information
Authors and Affiliations
Contributions
Participated in Study Design: Zigang Xu, Tieliu Shi, and Yongli Guo. Conducted experiments: Yaqiong Jin, Li Zhang, Senfen Wang, Feng Chen, Yang Gu, Enyu Hong and Yongbo Yu. Performed data analysis: Li Zhang, Yaqiong Jin, Yongli Guo and Tieliu Shi. Wrote or contributed to the writing of the manuscript: Yaqiong Jin, Yongli Guo, Li Zhang, Senfen Wang, Feng Chen, Zigang Xu and Tieliu Shi. Project principal: Xin Ni, Tieliu Shi, and Zigang Xu.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Jin, Y., Zhang, L., Wang, S. et al. Whole Genome Sequencing Identifies Novel Compound Heterozygous Lysosomal Trafficking Regulator Gene Mutations Associated with Autosomal Recessive Chediak-Higashi Syndrome. Sci Rep 7, 41308 (2017). https://doi.org/10.1038/srep41308
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep41308
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
