
Abstract
The neurologic manifestations of neonatal hyperbilirubinemia in the central nervous system (CNS) exhibit high variations in the severity and appearance of motor, auditory and cognitive symptoms, which is suggestive of a still unexplained selective topography of bilirubin-induced damage. By applying the organotypic brain culture (OBC: preserving in vitro the cellular complexity, connection and architecture of the in vivo brain) technique to study hyperbilirubinemia, we mapped the regional target of bilirubin-induced damage, demonstrated a multifactorial toxic action of bilirubin, and used this information to evaluate the efficacy of drugs applicable to newborns to protect the brain. OBCs from 8-day-old rat pups showed a 2–13 fold higher sensitivity to bilirubin damage than 2-day-old preparations. The hippocampus, inferior colliculus and cerebral cortex were the only brain regions affected, presenting a mixed inflammatory-oxidative mechanism. Glutamate excitotoxicity was appreciable in only the hippocampus and inferior colliculus. Single drug treatment (indomethacin, curcumin, MgCl2) significantly improved cell viability in all regions, while the combined (cocktail) administration of the three drugs almost completely prevented damage in the most affected area (hippocampus). Our data may supports an innovative (complementary to phototherapy) approach for directly protecting the newborn brain from bilirubin neurotoxicity.
Similar content being viewed by others

Introduction
Neonatal hyperbilirubinemia is a common and benign event in newborns, characterized by an increased level of unconjugated bilirubin (UCB), which has antioxidant effects1. The vast majority of UCB exists in the blood bound to its carrier protein albumin. However, a small fraction of UCB remains unbound as free bilirubin (Bf), which is responsible for the pathological effects on the central nervous system (CNS)2,3. When hyperbilirubinemia is left untreated, both bound and unbound forms of bilirubin are elevated, with the fraction of Bf increasing as the amount of available albumin decreases4,5. Presently, deaths due to hyperbilirubinemia are rare in Western countries thanks to the feasibility and efficacy of phototherapy. However, in past years, there has been a resurgence of kernicterus (the most severe and permanent form of bilirubin brain toxicity, RC0180; RP0060)6. If added to the still occurring severe damage and death in low and mild-incoming countries6, as well as the lifelong risk of developing kernicterus experienced by Crigler-Najjar Type I patients (OMIM218800; ORPHA79234; ICD-10: E80.5), the consequences of hyperbilirubinemia continue to merit attention, and it is crucial to improve the risk assessment and the therapies for this condition.
It is well accepted that the clinical symptoms of bilirubin toxicity in the brain reflect the selective topography of bilirubin-induced damage: motor disorders and athetosis (basal ganglia and cerebellum), auditory dysfunction (inferior colliculus), and learning impairments (hippocampus and cerebellum)7. Nevertheless, this pathological condition still has unexplained variability in the severity and occurrence of the above reported symptoms8. A possible reason for this variability has been attributed to the level and duration of hyperbilirubinemia9. As learned from other neonatal neurological diseases, alternative explanations exist. As described in Rice and Barone, windows of CNS vulnerability to stimuli have been documented to strongly depend upon the developmental events occurring at the time of exposure to a toxicant, rather than before or after, and might influence the outcome10.
To map bilirubin targets in the post-natal brain during development and to elucidate the mechanisms as a basis for possible therapeutic intervention, we used the organotypic brain culture (OBC) technique11 to study bilirubin neurotocicity. OBCs are slices of a specific region of the brain that conserve cellular heterogeneity and connections12 and exhibit synaptic plasticity and can reveal mechanisms of pathological insults comparable to what is obtained in vivo. OBCs are easily grown in vitro thus allowing for direct exposure to outside agents. They, therefore, represent an ideal tool to assess ex vivo the effect of a compound such as Bf on a specific CNS region13. In addition, OBCs can be prepared from animals at different postnatal ages, thus allowing one to mimic the various stages of CNS maturation. We also evaluated the use of various drugs aimed at directly protecting the brain pharmacologically, as an innovative treatment to be used as a complement to traditional phototherapy.
Results
Recovery of OBCs in Bf medium
To compare the viability of OBCs in standard medium vs. OBC media (see “Methods”) LDH release assay was performed. As shown in Fig. 1, the OBCs showed a significant LDH release immediately after slicing (which reflects procedural stress). This initial increase in LDH release decreased to normal levels in approximately 5 days (recovery time). No differences in LDH release were observed between OBCs cultured in standard vs. OBC media indicating that the modified media does not affect this assay. No differences were detected for any brain region or at any post-natal age (data not shown). Based on these results, Bf treatment was initiated at day 6 in vitro to allow for the complete recovery of the OBCs.

Comparison of membrane damages (LDH release) in usual OBC medium (solid line) and in Bf-adapted medium (dotted line) after cutting (refer to culture medium paragraph in Material and Methods section). The results are expressed as the percentage vs. maximum release (day 1). Data are expressed as the mean ± SD of three repetitions.
Regional sensitivity to bilirubin toxicity
Three viability tests were used to extensively follow the damage induced by Bf treatment. Each monitored a different typology of damage: membrane leakage (LDH release assay), apoptosis induction (Hoechst staining) and impairment of mitochondrial activity (MTT test).
LDH release
OBCs obtained from 2-day-old rat pups (Fig. 2A) did not show statistically significant increases in LDH activity at 70-nM Bf (sensitivity threshold, see “Methods” for details). Conversely, 140-nM Bf (toxic concentration) resulted in a toxic effect on the hippocampus (Hip: 3.5-fold vs. DMSO, p < 0.01), inferior collicula (IC: 3.2-fold vs. DMSO, p < 0.01) and cerebral cortex (Ctx: 4.4-fold vs. DMSO, p < 0.001). When Bf was increased to 300-nM (higher level to exacerbate the damage), a dose-dependent effect was observed in the IC and Ctx (2.0- and 2.5-fold, respectively, vs. OBCs treated with 140-nM Bf, p < 0.05; and 5.9-fold and 10.8-fold, respectively, vs OBCs treated with DMSO, p < 0.001), with the latter showing the maximum damage. No dose effect was observed in the Hip (3.8-fold vs. DMSO, p < 0.001). Only at 300-nM Bf concentration was a loss of membrane integrity observed in the cerebellum (Cll: 3.3-fold, p < 0.01) and superior collicula (SC: 1.8-fold, p < 0.05).

(A) LDH release in P2 OBCs; (B) LDH release in P8 OBCs; (C) Hoechst staining of P2 OBCs; (D) Hoechst staining of P8 OBCs; (E) MTT assay of P2 OBCs; (F) MTT assay of P8 OBCs. LDH release and Hoechst are expressed as fold change of control (DMSO = 1). MTT is expressed as % change of control (DMSO = 100%). Data are expressed as the mean ± SD of three-five repetitions. Statistical significance: (a) p < 0.05, (b) p < 0.01, (c) p < 0.001 vs. DMSO; §p < 0.05, §§p < 0.01, §§§p < 0.001 70-nM vs. 140-nM; #p < 0.05, ##p < 0.01, ###p < 0.001 140-nM vs. 300-nM. Hip: hippocampus; Cll: cerebellum; IC: inferior colliculus; SC: superior colliculus; Ctx: cortex.
In OBCs from 8-day-old animals (Fig. 2B), irrespective of the bilirubin concentration, the Hip displayed the maximum toxicity, followed by the IC and Ctx. In these three regions, LDH activity was already significantly increased after 24 hrs at 70-nM Bf (6.4-, 3.6- and 2.8-fold, respectively, p < 0.001). Increasing bilirubin concentration (140-nM) increased the damage in the Hip, IC and Ctx (to 19.0-, 10.6- and 8.9-fold vs. DMSO, p < 0.001). On the contrary, no damage was detected in the Cll and SC at 140-nM Bf. At 300-nM Bf, we did not observe a further increase in LDH release in the Hip, IC and Ctx. As in the case of OBCs from 2-day-old animals, statistically significant changes were present in the Cll (3.6-fold, p < 0.001) and SC (1.8-fold, p < 0.01) after only 24 hrs of exposure at 300-nM Bf.
Hoechst staining
The numerical data and a representative picture of the chromatin condensation are shown in Fig. 2C and D. In OBCs from 2-day-old rats (Fig. 2C), 70-nM Bf treatment resulted in statistically significant apoptosis in only the Hip (2.2-fold vs. control, p < 0.001), with a clear dose effect when increasing Bf to 140-nM (2.3-fold vs. OBCs treated with 70, p < 0.05; p < 0.01 vs. OBCs treated with DMSO) and 300-nM Bf (12.0-fold vs. OBCs treated with DMSO, p < 0.001). At 140-nM Bf, apoptosis was significant in the Ctx (2.2-fold, p < 0.05), without any further increase at 300-nM, and in the IC (10.4-fold, p < 0.01 vs. control, 3.1-fold vs. OBCs treated with 70-nM Bf, p < 0.05), again with no change when increasing concentration to 300-nM Bf. Moreover, 300-nM Bf was needed to induce apoptosis in the Cll (2.0-fold vs. control, p < 0.05) and SC (4.5-fold vs. control, p < 0.05).
Hippocampal slices from more developed animals (P8 – Fig. 2D) confirmed a high sensitivity to the low bilirubin dose of 70-nM (p < 0.001), with further increases at both 140 and 300-nM (61-fold vs. control, p < 0.001, and 3.7-fold vs. OBCs treated with 70-nM Bf, p < 0.01). The IC was also damaged after each treatment, but with a clear dose-dependent effect (2.9-fold at 70-nM, p < 0.01; 17-fold at 140-nM, p < 0.01; and 43-fold vs. OBCs treated with DMSO, p < 0.001). No significant bilirubin-induced apoptosis was observed in the Cll or Ctx.
Mitochondrial activity
Despite some change in MTT activity (Fig. 2E), no significant changes were found in P2 OBCs exposed to all Bf concentrations, except for a 25% decrease (p < 0.05) in the SC exposed to 140-nM Bf.
In P8 OBCs (Fig. 2F), a significant reduction in mitochondrial metabolism was detected in the Hip (p < 0.001) and IC (p < 0.01) exposed to Bf concentrations of 140 and 300-nM. The Ctx and SC showed a significant reduction in mitochondrial activity only when exposed to the maximal dose (300-nM Bf, both p < 0.05). The Cll was insensitive to all Bf concentrations.
Developmental sensitivity to bilirubin toxicity
Because brain exhibits significant post-natal development10, two different post-natal ages were compared. To highlight the developmental sensitivity to bilirubin toxicity, viability was expressed as a ratio of P8/P2 results (Table 1). A ratio higher than 1-fold indicates an increased developmental sensitivity in the LDH assay and Hoechst staining results; the opposite is true for MTT activity.
Developmental sensitivity to membrane leakage
LDH revealed a clear age-related increase in the sensitivity to bilirubin in the Hip when exposed to 140 and 300-nM Bf (P8 higher than P2) (both p < 0.001) (Table 1). A similar response was observed in the IC (at 140-nM Bf concentration, p < 0.01). No developmental sensitivity to bilirubin toxicity was observed in the Cll, SC or Ctx.
Developmental sensitivity to apoptosis
As shown in Table 1, at all bilirubin concentrations, Hip sensitivity was increased in P8 vs. P2 OBCs (p < 0.01 at 70-nM Bf and p < 0.001 at 140 and 300-nM Bf), while in the IC, an increase in apoptosis was observed at only the highest concentration (p < 0.001). No developmental difference was observed in the Cll, SC or Ctx.
Developmental sensitivity to in mitochondrial activity
Despite a trend of decrease in the Hip, IC and Ctx, no significant differences in the mitochondrial activity in these regions were observed (Table 1, lower panel).
Screening of pathways of damage
Different analyses (histological, genomic, and enzymatic) were performed in OBCs treated with bilirubin to screen for biomolecular mechanisms of damage. Screening was performed at the post-natal age showing the maximal sensitivity (P8) and at the toxic Bf concentration of 140 nM for 24 hrs.
Hematoxylin and Eosin
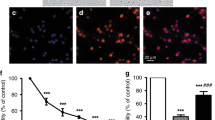
The damage to the Hip observed in the previously described viability tests was confirmed by histological evaluation. Tissue damage was evidenced by the appearance of large intracellular spaces, neuronal cell loss, apoptosis and some degree of necrosis. Edema, fibrosis, the presence of foam cells and microgliosis were also observed (see Fig. 3B = bilirubin treatment vs. A = control). Similar to the Hip, but to a lesser extent, the IC showed large intracellular spaces, apoptotic bodies and foam cells (Fig. 3H). Cellular death was also present in the Ctx (decreased cellular density, with a generalized weak nuclear staining – Fig. 3N). Some degree of degeneration was also present in the Cll, with peri-cellular edema, necrosis, and a slight detectable increase in apoptosis (Fig. 3E). The SC was only marginally affected by the exposure to bilirubin, with a negligible increase of cells with vacuoles and fibrillary matrix components with cellular debris, with respect to DMSO-exposed organotypic SC cultures (Fig. 3K).

Images of H&E staining of OBCs treated with DMSO (A,D,G,J,M), 140 nM Bf (B,E,H,K,N) or cocktail (C,F,I,L,O) of Hip (A,B,C), Cll (D,E,F), IC (G,H,I), SC (J,K,L) and Ctx (M,N,O). Red arrow: apoptosis. Green arrow: microgliosis. Red circle: foam cells. Red square: fibrosis. Green circle: inflammation. Blue square: edema. Hip: hippocampus; Cll: cerebellum; IC: inferior colliculus; SC: superior colliculus; Ctx: cortex. Scale Bar 50 μm.
Quantitative real-time PCR analysis of markers of damage
To evaluate the possible pathway of damage triggered by bilirubin in each region, we assessed the expression of selected genes as markers of specific biomolecular processes of damage (Table 2).
Oxidative imbalance was revealed in all regions with the exception of the Ctx. A 2.5- to 4-fold increase in expression of the inducible heme oxygenase-1 (HO-1) (a marker of oxidative stress) was detected in the Hip, IC (both p < 0.001), Cll (p < 0.01) and SC (p < 0.05). Sulfiredoxin 1 (Srnx1, involved in antioxidant metabolism, and induced in response to an oxidant milieu) was maximally modulated in the Hip and IC (p < 0.001 and p < 0.05, respectively), followed by the SC and Cll (p < 0.01 and p < 0.05, respectively).
The activation of the inflammatory pathway was assessed by the expression of multiple cytokines. Tumor necrosis factor alfa (Tnfα) mRNA was significantly induced in only the Hip and SC (p < 0.05 and p < 0.001, respectively). Interleukin 1β (Il1β) was maximally altered in the IC (p < 0.001), followed by the Hip (p < 0.01), Cll and SC (both p < 0.05).
The most striking alteration involved the interleukin 6 (Il6) and the cyclooxygenase (Cox2) genes, which were up-regulated approximately 300- and 50-fold, respectively, in the Hip and 100- and 60-fold, respectively, in the IC (all p < 0.001). In the third most damaged area (Ctx), Il6 expression was up-regulated approximately 12.4-fold (p < 0.001), with Cox2 expression up-regulated approximately 3.5-fold (p < 0.05). Both genes were modulated in the Cll (both p < 0.001) and in the SC (both p < 0.05).
Glutamate release in culture medium
The involvement of the glutamate excitotoxicity pathway was assessed by the quantification of glutamate release into the culture media (Table 2). Significant release was detected in the Hip, 2.4-fold (p < 0.001), and IC, 2.0-fold (p < 0.01), after bilirubin treatment.
Evaluation of possible therapeutic approaches
Finally, we used the OBCs as a screening platform to identify neuroprotective therapeutics. The LDH test was chosen to evaluate the protective effect of each treatment because of the level of sensitivity to Bf-induced damage that this test exhibited (Fig. 4). The main rationale for the drugs selected in this work was based on their potential to be used safely as new therapeutic treatments in severely jaundiced newborns14; therefore, only drugs already in clinical use, and known to cross the blood brain barrier were considered. The maximal dose not inducing adverse effects was experimentally identified, starting from the information available in literature for each compound. The current gold-standard drug treatment for bilirubin-based neurotoxicity, minocycline15,16, has been shown to suppress cerebellar hypoplasia in the Gunn rat17, the animal model for Crigler-Najjar type I and kernicterus. Minocycline was also effective in restoring Bf-induced damage in OBCs (Hip −35%, IC −80% and Ctx −57%, all p < 0.001) (Fig. 4). Curcumin, an antioxidant nutraceutical18,19, improved the viability of the challenged OBCs by approximately 69% (p < 0.001) in the IC, 43% (p < 0.01) in the Ctx and 18% (p < 0.05) in the Hip. MgCl2, a glutamate receptor blocker20,21, effectively improved viability by approximately 47% (p < 0.01), 44% (p < 0.01) and 37% (p < 0.001) in the Ctx, IC and Hip, respectively. Indomethacin, an anti-inflammatory drug22,23, increased the viability of OBCs by 63% (p < 0.01), 60% (p < 0.001) and 42% (p < 0.001) in the Ctx, IC and Hip, respectively. The cocktail of the 3 compounds greatly improved viability in the Hip (78%, p < 0.001 vs. Bf), while no additional difference in respect to single drugs was observed in the Ctx or IC (62%, 68%; both p < 0.001 vs. Bf). Exposure to a single drug or to the cocktail for 24 hrs did not increase LDH release in the OBCs compared to the DMSO control, indicating the safety of the dosage used in our system.

Recovery from damage (LDH release) in challenged OBCs is expressed as fold change of control (DMSO = 1). Data are expressed as the mean ± SD of three-five repetitions. Statistical significance: (a) p < 0.05; (b) p < 0.01; (c) p < 0.001. @: p < 0.05 cocktail vs. indomethacin. *p < 0.01 cocktail vs. minocycline; #,$p < 0.001 cocktail vs. curcumin and Mg, respectively. OBCs exposed to drugs alone as well as undamaged regions (Cll, SC) exposed to cocktail did not display signs of toxicity. Hip: hippocampus; Cll: cerebellum; IC: inferior colliculus; SC: superior colliculus; Ctx: cortex.
Assessment of the damage improvements after treatment
As a final demonstration of drug efficacy, both the histological examination (Fig. 3C,F,I,L,O) and quantification of the markers of biomolecular pathways of damage were performed using the cocktail as stated above (Table 3).
In agreement with the viability improvement evidenced by the LDH release results, the cocktail approach coincided with a significant recovery in all the 3 damaged regions (Hip, IC and Ctx; Fig. 3C,I and O, respectively). However, the treatment did not completely reverse the damage due to Bf, as there were still marginal signs of damage present. Histological evaluation of Bf-induced damage in OBCs receiving the cocktail revealed a significant reduction in apoptosis and a moderate reduction of cellular edema in the Hip. The IC showed a reduction of edema with a conservation of cellular elements, while the Ctx had a reduction of necrosis and of the fibrotic scenario. The SC and Cll images showed slight histologic improvement (Fig. 3).
The observed improvement in the viability was confirmed at the molecular level by real-time PCR. The mRNA expression of markers for oxidative imbalance was slightly increased for HO-1 in the Hip (increase of approximately 164% vs. bilirubin-treated OBCs, p < 0.01) (Table 3). The other regions did not show a significant change in HO-1 expression after treatment with Bf and cocktail.
Treatment with the Bf + cocktail also showed a reduction of interleukin 6 (Il6) and cyclooxygenase 2 (Cox2) levels compared to slices exposed to Bf alone, thus indicating that the neuro-inflammatory markers were modulated by bilirubin treatment (Table 3) and the effect attenuated by the drugs. Cox2 levels decreased by approximately 97% in the Hip, IC and Cll (p < 0.05, p < 0.01 and p < 0.01, respectively). Il6 mRNA was reduced by approximately 80–90% in the Hip, IC, Ctx and Cll (all p < 0.01). Similar down-regulation (about of 95% vs. bilirubin-treated OBCs) compared to normal expression was also observed for Il1β in the Hip (p < 0.05), IC and Cll (both p < 0.01). Less impressive was the reduction of Tnfα expression, which was statistically significant in only the SC (p < 0.001).
In accordance with our previous results showing significant glutamate-mediated damage in only the Hip and IC, the cocktail significantly reduced glutamate release in these same regions (Hip p < 0.01 and IC p < 0.05).
Discussion
Our data highlight a clear region-specific and developmental regulated in the brain’s sensitivity to bilirubin toxicity. The Hip emerged as the most sensitive brain region, followed by the IC. These data are in agreement with the most common clinical manifestations of bilirubin neurotoxicity: alterations in the auditory-evoked potentials24,25 and memory/learning deficits26,27. Additionally, our findings are in agreement with clinical findings8 that indicate damage to the IC and Hip8.
Despite being considered as a region resistant to bilirubin toxicity28, some evidence of bilirubin toxicity in the cerebral Ctx has been reported. Newborn electroencephalographic measurements have demonstrated that hyperbilirubinemia affects cerebro-cortical electrical activity in a time-limited manner29, and autopsy findings have shown apoptosis and necrosis in this area30. Moreover, nearly all in vitro evidence of the mechanisms of bilirubin-induced cytotoxicity have been obtained using primary cultures of neurons and astrocytes isolated from the cerebral Ctx31,32. In animal models, cortical sensitivity to bilirubin has also been shown by inhibiting the phosphorylation of synapsin I26, by observing histological abnormalities in the occipital cortex, and altering the balance of excitatory and inhibitory neurotransmitters33. Collectively, these findings suggest a possible cortical sensitivity to bilirubin. Though this sensitivity is probably less severe than that observed in other regions, potential damage to the Ctx likely needs to be considered in infants where bilirubin neurotoxicity is suspected.
Only P8 OBCs were sensitive to a bilirubin concentration of 70-nM (Bf) (Fig. 2), which is considered to be either not toxic or at least at the threshold of potentially causing damage3. This observation suggests an “age-dependent” sensitivity to bilirubin toxicity. This is in line with what has been reported in animal models for hyperbilirubinemia (Gunn rat), where the window of vulnerability to bilirubin toxicity has been observed at P6–10, a period preceded and followed by a much lower CNS vulnerability34,35,36. Unfortunately, no detailed biomolecular data are available on postnatal brain development in humans. Conversely, in rodents, the presence of a “dormant period” in the first 3 postnatal days has been described37. Thereafter, a sustained increase of NMDA receptor subunits38 and sensitivity to inflammation39 are observed in the second postnatal week. Along with these changes, the activation of neurogenesis, proliferation of non-neuronal cells, and increased apoptosis and remodeling in physiological tissues37 are all potential mechanisms for bilirubin toxicity10, contributing to the final toxic phenotype. This is in full agreement with the concept of the vulnerability of the developing brain to a toxic substance: “if exposure occurs before or after an organ develops, it is less vulnerable to perturbation than if exposure occurs during development of that organ”10, with respect to different brain insults and with our hypothesis that CNS sensitivity to UCB might be developmentally regulated.
Also of interest is the insensitivity of the Cll OBCs to acute (24 hrs) bilirubin insult. Cerebellar hypoplasia due to bilirubin toxicity is a landmark finding of animal models of hyperbilirubinemia34,36,40,41,42,43,44 and is reported in pre-term babies45,46. In our model, we found regions of the brain susceptible to an acute exposure (24 hrs) to UCB (Hip, IC) and others that are not. Specifically, for the Cll, our results suggest that a longer (chronic) exposure to toxic levels of UCB may be required to initiate the damage in this CNS region. This finding is in line with what has been reported in animal models. In both Gunn rats and UGT−/− mice, cerebellar damage took at least 7–9 days to become significant and was not detectable at early post-natal ages, despite the presence of hyperbilirubinemia early after birth34,36,40,41,42,43,44. Also in line with this hypothesis is the observation that 4 days of exposure to 140-nM Bf increased LDH release (>2.0-fold) in our Cll OBCs (data not shown).
In this study, we discovered a multifactorial etiology of bilirubin toxicity by cross-validating the data obtained from the analysis of mechanisms of damage (Table 3 and Fig. 3) and treatments (Fig. 4). Neuroinflammation (revealed by cytokine modulation – Table 3 and microgliosis – Fig. 3) and oxidative stress (Ho1/Srnx1 modulation – Table 3, and foam cells observed in Fig. 3) were observed in all three damaged areas (Hip, IC and Ctx), with glutamate release present in only the Hip and IC OBCs (Fig. 3). Additionally, our findings regarding the neurological damage in Hip, IC and Ctx are in agreement with the histological findings at autopsy30,45. A part being in agreement with the literature based on cell lines and the animal model that we used as reference (see M&M), the pro-inflammatory action of bilirubin we observed well agreed with the two unique previous studies using cerebellar and hippocampal OBCs40,47. The discrepancy in the glutamate finding (not investigated in the Hip47, showing a temporary significant release in Cll OBCs exposed to UCB40, but never significant in the Cll in our acute scheme) might be explained by the different experimental set-ups, and with the amount of Bf used (20-nM in Barateiro et al.40 vs. 140-nM in. our work) being the major difference.
In less damaged regions, single drugs were effective in reducing tissue damage. Curcumin and indomethacin emerged as the most effective drugs in ameliorating the damage in the IC with similar results for indomethacin in the Ctx. In these regions, all therapeutic schemes applied matched with the protection offered by minocycline (Fig. 4), a drug shown to be fully effective in in vivo models of kernicterus15,16. Notably, in addition to the antioxidant and anti-inflammatory mechanisms, minocycline most likely acts as a chelator for bivalent ions, such as calcium which is probably strongly relevant in bilirubin encephalopathy, as demonstrated by Daood et al.48. However, minocycline use is not allowed in neonates due to its side effects, and safe calcium chelators are still not available in clinics. In agreement with the multifactorial pathway of damage, drugs applied as a cocktail seem to be to the most effective way to ameliorate the damage in the Hip, the most damaged region both in our experimental model and in newborns. Indeed, the additive effect of the cocktail vs. single drug administration (Fig. 4) indicates that bilirubin acts simultaneously on different, but not necessarily connected, pathways. Importantly, we observed no evidence of drug toxicity in damaged (Hip, IC and Ctx) or undamaged (Cll, SC) brain regions after exposure to either the single or cocktail drug treatments, indicating that the cocktail is potentially a more effective treatment to confer the maximum and widespread protection to the brain in severe hyperbilirubinemia.
In conclusion, the application of OBCs to study bilirubin neurotoxicity proved well-suited for the reproduction of findings observed in vivo and clinically. We have demonstrated a multifactorial toxic action of bilirubin, with some regional specificity, and we have shown ex vivo the efficacy of a new theoretical approach aimed at protecting the brain with drugs that are currently used in the clinical setting.
Methods
Organotypic brain culture preparation
Wistar HanTM Rats, at 2 (P2) and 8 (P8) days after birth, were obtained from the animal facility of the University of Trieste. Animal experiments were performed according to the Italian Law (decree 87–848) and European Community directive (86-606-ECC). The study was approved by the animal care and use committee of the University of Trieste, along with regular communication with the Italian Ministry. Maximal effort was used to minimize the number of the animals used and their suffering, with the respect to the 3R rule. Immediately after sacrifice, the Hip, Cll, IC (all pathologically involved in kernicterus)28, SC and Ctx (considered resistant) were dissected and maintained in dissection medium (ice cold Gey’s Balanced Salt Solution plus D-Glucose 10 mg/mL) until use. A McIIwain tissue chopper (Gomshall Surrey, U.K.) was used to cut transversely 300 (SC, IC), 350 (Hip, Ctx) and 425 (Cll) μm slices. Healthy slices, selected for structural integrity under stereomicroscope inspection, were maintained in dissection medium for 60 min to allow clearing of cutting surfaces from preparation procedure stress. Slices were then transferred to sterile, semi-porous Millicell-CM inserts (PICM03050, Millipore, Darmstadt, Germany), fed by 1 mL of media and maintained at 37 °C, 5% CO2, 95% humidity in a humidified incubator11.
Cultures medium
Standard culture medium (50% Basal Medium Eagle – BME - medium, 25% Hank’s Balanced Salt Solution - HBSS, 25% Heat-Inactivated Horse Serum, 1% L-Glutamine, 2% Penicillin/Streptomycin, 10 mg/mL glucose)11 was modified to allow for Bf quantification. Bf adapted medium (OBCs medium) was composed of 65% Basal Medium Eagle (Life Technologies Corporation, Grand Island, NY), 10% heat-inactivated Fetal Bovine Serum (Euroclone, Milan, Italy), 25% Hank’s Balanced Salt Solution (Sigma Aldrich, St. Louis, MO, USA), 1% L-Glutamine (Life Technologies Corporation, Grand Island, NY), 2% Penicillin/Streptomycin (Life Technologies Corporation, Grand Island, NY), 10 mg/mL D-Glucose (Sigma Aldrich, St. Louis, MO, USA). The media was changed the day after cutting and every two days thereafter. Slices were maintained in culture 5 days before starting Bf exposure, this amount of time was required to allow recovery from the stress of slicing (Fig. 1)49.
Bf treatment
After 6 days in vitro, slices were challenged for 24 hrs with different bilirubin concentrations based on our experience in vitro31,43,47 and in the range of the in vivo brain Bf 50. The concentration of UCB dissolved in DMSO required to reach the desired Bf concentrations in the OBC medium was quantified according to Roca et al.51. Three concentrations of Bf were used: 70-nM (sensitivity threshold), 140-nM (toxic concentration), 300-nM (higher level to exacerbate the damage). Control slices were exposed to the same final concentration of DMSO used to dissolve the UCB.
Viability tests
Lactate dehydrogenase release
The amount of total extracellular LDH in media, indicative of membrane leakage, was determined using a CytoTox-ONE™ Homogeneous Membrane Integrity Assay (G7891, Promega, Madison, WI, USA). After Bf exposure (24 hrs), the supernatant was collected, and the reaction was carried out according to the manufacturer’s instructions. The fluorescence (560Ex/590Em) was determined using an EnSpire Multimode Plate Reader (PerkinElmer, Waltham, MA, USA), and the background fluorescence subtracted. The amount of LDH released in treated slices was expressed as the fold change compared to the control slices.
Hoechst staining
Evidence of chromatin condensation, a marker of cell death by apoptosis, was obtained by administration of 1 μg/mL Hoechst 33258 (Sigma Aldrich, St. Louis, MO, USA). Slices treated with Bf or DMSO alone were fixed for 30 min at room temperature in 3% paraformaldehyde (PFA)13. Apoptotic cells were counted at 40X magnification, by fluorescence microscopy using a Leica DM2000 (Leica Mycrosystems Srl, Solms, Germany) by three separate individuals. At least three different fields were analyzed in each repetition. The results were expressed as the percentage of apoptotic cells relative to the total number (apoptotic plus unaffected) of cells in the control (100%).
Mitochondrial activity
Mitochondrial metabolic activity was assessed using a 1-(4,5-dimethylthiazol-2-yl)-3, 5-diphenylformazan (MTT) assay (Sigma-Aldrich, St. Louis, MO, USA). Post-challenge experiments, slices for each repetition were incubated with 0.5 mg/mL of MTT in media at 37 °C for 1 hr, harvested, and the precipitated salt dissolved in DMSO. Absorbance was detected at 562 nm using a LD 400C Luminescence Detector (Beckman Coulter, Milan, Italy). The results were expressed as the percentage of activity relative to controls (100%).
Screening of pathway of damage
Histology
After Bf treatment, OBCs were immediately fixed in neutral buffered formalin and embedded in 4% paraffin. Tissue were sectioned to a thickness of 3 μm by a microtome (Microm-hm 340e- BioOptica, Milan, It), and dried in oven a 60 °C for one hour. Sections were stained with hematoxylin & eosin (H&E) using a Leica ST5020 Multistainer (Leica Microsystem, Milano, It). Histology was read by two independent pathologists, blinded to experimental design and treatment groups, at the Department of Pathology of Academic Medical Center Hospital of Cattinara. Images were collected by a D-Sight plus image digital microscope & scanner (Menarini Diagnostics, Firenze, Italy).
Quantitative real-time PCR of selected markers of bilirubin toxicity
Genes reported to be involved in the biomolecular pathways of neuronal damage were selected as markers of these pathway47,49,52 (see Table 4). The mRNA expression of the genes of interest was analyzed by quantitative real-time PCR. Total RNA was extracted using TRI Reagent® RNA Isolation Reagent (Sigma-Aldrich, St. Louis, MO, USA), following the manufacturer’s instructions. Complementary DNA (cDNA) was synthesized with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Monza, Italy). For the quantitative real-time PCR, primers were designed using the Beacon designer 4.2 software (Premier Biosoft International, Palo Alto, CA, USA) on rat sequences available in GenBank (Table 1). The reaction was performed in a final volume of 15 μL in an iQ5 Bio-Rad Thermal cycler (BioRad Laboratories, Hercules, CA, USA). Briefly, 25 ng of cDNA and the corresponding gene-specific sense/antisense primers (250 nM each, with the exception of Cyclo-Oxygenase 2 -Cox2 and Interleukin 1β -Il1β, 500 and 750 nM, respectively) were diluted in the Sso Advance SYBER green supermix (Bio-Rad Laboratories, Hercules, CA, USA). Amplification of target genes was accomplished using the following protocol: 3 min at 95 °C, 40 cycles at 95 °C for 20 sec, 60 °C for 20 sec, and 72 °C for 30 sec. The specificity of the amplification was verified by a melting-curve analysis, and non-specific products of PCR were not found in any case. The relative quantification was made using the iCycleriQ software, version 3.1 (Bio-Rad Laboratories, Hercules, CA, USA) by the ΔΔCt method, taking into account the efficiencies of the individual genes and normalizing the results to the housekeeping genes (Hypoxanthine guanine phosphoribosyl transferase: Hprt, Glyceraldehyde 3-phosphate dehydrogenase: Gapdh)57,58.
Glutamate quantification in culture media
The amount of extracellular glutamate (glu), a marker of bilirubin-induced excitotoxicity31,59,60, was quantified using a Glutamate Assay Kit (MAK004, Sigma-Aldrich, St. Louis, MO, USA). Briefly, after the Bf exposure, the supernatant was collected, and the assay was performed according to the manufacturer’s instructions. The absorbance (450 nm), proportional to the glutamate present, was determined using an EnSpire Multimode Plate Reader (PerkinElmer, Waltham, MA, USA). Glutamate release in media was expressed as fold change compared to the control slices.
Screening of therapeutic drugs
Slices from P8 animals (the most sensible post-natal age), were exposed (for 24 hrs) to the following: (1) DMSO alone, as control; (2) 140 nM Bf–to induce the damage; (3) drugs alone–to assess possible toxic effects; (4) 140 nM Bf plus single drugs; and (5) 140 nM Bf plus the three principles simultaneously (cocktail). Drugs screened in this study included the following: curcumin, an antioxidant (50 μM, Sigma-Aldrich, St. Louis, MO, USA), magnesium chloride, a glutamate channel blocker, (10 mM, Sigma-Aldrich, St. Louis, MO, USA) as alternative to the bilirubin displacer magnesium sulfate14 already used in newborns, and indomethacin, an anti-inflammatory agent (50 μM, Liometacen, Promedica Srl. Parma, Italy). Minocycline was used at 60 μM (Sigma-Aldrich, St. Louis, MO, USA).
Statistical analysis
Data were analyzed with GraphPad Prism version 5.00 for Windows (GraphPad Software, La Jolla California USA). Statistical significance was evaluated by paired two-tailed test. ANOVA was applied to the results in Table 2, where different post-natal ages have been compared. A p value < 0.05 was considered as statistically significant. The results are expressed as the mean ± SD of at least three independent repetitions.
Additional Information
How to cite this article: Dal Ben, M. et al. Evaluation of region selective bilirubin-induced brain damage as a basis for a pharmacological treatment. Sci. Rep. 7, 41032; doi: 10.1038/srep41032 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
15 February 2017
The version of this Article previously published quoted an incorrect abbreviation for Matteo Dal Ben in the 'How to cite this article' section. This has now been corrected in the PDF and HTML versions of the paper.
References
Wagner, K.-H. et al. Looking to the horizon: the role of bilirubin in the development and prevention of age-related chronic diseases. Clin. Sci. Lond. Engl. 1979 129, 1–25 (2015).
Ostrow, J. D., Mukerjee, P. & Tiribelli, C. Structure and binding of unconjugated bilirubin: relevance for physiological and pathophysiological function. J. Lipid Res. 35, 1715–1737 (1994).
Ostrow, J. D., Pascolo, L. & Tiribelli, C. Reassessment of the unbound concentrations of unconjugated bilirubin in relation to neurotoxicity in vitro . Pediatr. Res. 54, 98–104 (2003).
American Academy of Pediatrics Subcommittee on Hyperbilirubinemia. Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation. Pediatrics 114, 297–316 (2004).
Bhutani, V. K., Johnson, L. H. & Shapiro, S. M. Kernicterus in sick and preterm infants (1999–2002): a need for an effective preventive approach. Semin. Perinatol. 28, 319–325 (2004).
Bhutani, V. K. et al. Neonatal hyperbilirubinemia and Rhesus disease of the newborn: incidence and impairment estimates for 2010 at regional and global levels. Pediatr. Res. 74 Suppl 1, 86–100 (2013).
Watchko, J. F. & Tiribelli, C. Bilirubin-induced neurologic damage–mechanisms and management approaches. N. Engl. J. Med. 369, 2021–2030 (2013).
Shapiro, S. M. Chronic bilirubin encephalopathy: diagnosis and outcome. Semin. Fetal. Neonatal Med. 15, 157–163 (2010).
Ahlfors, C. E., Wennberg, R. P., Ostrow, J. D. & Tiribelli, C. Unbound (free) bilirubin: improving the paradigm for evaluating neonatal jaundice. Clin. Chem. 55, 1288–1299 (2009).
Rice, D. & Barone, S. Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ. Health Perspect. 108 Suppl 3, 511–533 (2000).
Stoppini, L., Buchs, P. A. & Muller, D. A simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods 37, 173–182 (1991).
Gähwiler, B. H., Capogna, M., Debanne, D., McKinney, R. A. & Thompson, S. M. Organotypic slice cultures: a technique has come of age. Trends Neurosci. 20, 471–477 (1997).
Lossi, L., Alasia, S., Salio, C. & Merighi, A. Cell death and proliferation in acute slices and organotypic cultures of mammalian CNS. Prog. Neurobiol. 88, 221–245 (2009).
Strauss, K. A. et al. Management of hyperbilirubinemia and prevention of kernicterus in 20 patients with Crigler-Najjar disease. Eur. J. Pediatr. 165, 306–319 (2006).
Geiger, A. S., Rice, A. C. & Shapiro, S. M. Minocycline blocks acute bilirubin-induced neurological dysfunction in jaundiced Gunn rats. Neonatology 92, 219–226 (2007).
Lin, S. et al. Minocycline blocks bilirubin neurotoxicity and prevents hyperbilirubinemia-induced cerebellar hypoplasia in the Gunn rat. Eur. J. Neurosci. 22, 21–27 (2005).
Gunn, C. H. Hereditary Acholuric Jaundice in a New Mutant Strain of Rats. J. Hered. 29, 137–139 (1938).
Ghoneim, A. I., Abdel-Naim, A. B., Khalifa, A. E. & El-Denshary, E. S. Protective effects of curcumin against ischaemia/reperfusion insult in rat forebrain. Pharmacol. Res. 46, 273–279 (2002).
Lim, G. P. et al. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J. Neurosci. Off. J. Soc. Neurosci. 21, 8370–8377 (2001).
Crowther, C. A., Hiller, J. E., Doyle, L. W. & Haslam, R. R. Australasian Collaborative Trial of Magnesium Sulphate (ACTOMg SO4) Collaborative Group. Effect of magnesium sulfate given for neuroprotection before preterm birth: a randomized controlled trial. JAMA 290, 2669–2676 (2003).
Westermaier, T. et al. Magnesium treatment for neuroprotection in ischemic diseases of the brain. Exp. Transl. Stroke Med. 5, 6 (2013).
Chechneva, O., Dinkel, K., Cavaliere, F., Martinez-Sanchez, M. & Reymann, K. G. Anti-inflammatory treatment in oxygen-glucose-deprived hippocampal slice cultures is neuroprotective and associated with reduced cell proliferation and intact neurogenesis. Neurobiol. Dis. 23, 247–259 (2006).
Miller, S. P. et al. Prolonged indomethacin exposure is associated with decreased white matter injury detected with magnetic resonance imaging in premature newborns at 24 to 28 weeks’ gestation at birth. Pediatrics 117, 1626–1631 (2006).
Agrawal, V. K., Shukla, R., Misra, P. K., Kapoor, R. K. & Malik, G. K. Brainstem auditory evoked response in newborns with hyperbilirubinemia. Indian Pediatr. 35, 513–518 (1998).
Sharma, P. et al. Brainstem evoked response audiometry (BAER) in neonates with hyperbilirubinemia. Indian J. Pediatr. 73, 413–416 (2006).
Hansen, T. W., Bratlid, D. & Walaas, S. I. Bilirubin decreases phosphorylation of synapsin I, a synaptic vesicle-associated neuronal phosphoprotein, in intact synaptosomes from rat cerebral cortex. Pediatr. Res. 23, 219–223 (1988).
Song, S., Hu, Y., Gu, X., Si, F. & Hua, Z. A novel newborn rat kernicterus model created by injecting a bilirubin solution into the cisterna magna. PloS One 9, e96171 (2014).
Shapiro, S. M. Definition of the clinical spectrum of kernicterus and bilirubin-induced neurologic dysfunction (BIND). J. Perinatol. Off. J. Calif. Perinat. Assoc. 25, 54–59 (2005).
Gürses, D., Kiliç, I. & Sahiner, T. Effects of hyperbilirubinemia on cerebrocortical electrical activity in newborns. Pediatr. Res. 52, 125–130 (2002).
Zangen, S. et al. Fatal kernicterus in a girl deficient in glucose-6-phosphate dehydrogenase: a paradigm of synergistic heterozygosity. J. Pediatr. 154, 616–619 (2009).
Falcão, A. S., Fernandes, A., Brito, M. A., Silva, R. F. M. & Brites, D. Bilirubin-induced inflammatory response, glutamate release, and cell death in rat cortical astrocytes are enhanced in younger cells. Neurobiol. Dis. 20, 199–206 (2005).
Gambaro, S. E., Robert, M. C., Tiribelli, C. & Gazzin, S. Role of brain cytochrome P450 mono-oxygenases in bilirubin oxidation-specific induction and activity. Arch. Toxicol., doi: 10.1007/s00204-014-1394-4 (2014).
Hu, W. et al. Ex vivo (1)H nuclear magnetic resonance spectroscopy reveals systematic alterations in cerebral metabolites as the key pathogenetic mechanism of bilirubin encephalopathy. Mol. Brain 7, 87 (2014).
Keino, H. & Kashiwamata, S. Critical period of bilirubin-induced cerebellar hypoplasia in a new Sprague-Dawley strain of jaundiced Gunn rats. Neurosci. Res. 6, 209–215 (1989).
Lee, Y.-K., Daito, Y., Katayama, Y., Minami, H. & Negishi, H. The significance of measurement of serum unbound bilirubin concentrations in high-risk infants. Pediatr. Int. Off. J. Jpn. Pediatr. Soc. 51, 795–799 (2009).
Conlee, J. W. & Shapiro, S. M. Development of cerebellar hypoplasia in jaundiced Gunn rats: a quantitative light microscopic analysis. Acta Neuropathol. (Berl.) 93, 450–460 (1997).
Bandeira, F., Lent, R. & Herculano-Houzel, S. Changing numbers of neuronal and non-neuronal cells underlie postnatal brain growth in the rat. Proc. Natl. Acad. Sci. USA 106, 14108–14113 (2009).
Haberny, K. A. et al. Ontogeny of the N-methyl-D-aspartate (NMDA) receptor system and susceptibility to neurotoxicity. Toxicol. Sci. Off. J. Soc. Toxicol. 68, 9–17 (2002).
Galic, M. A. et al. Postnatal inflammation increases seizure susceptibility in adult rats. J. Neurosci. Off. J. Soc. Neurosci. 28, 6904–6913 (2008).
Barateiro, A., Domingues, H. S., Fernandes, A., Relvas, J. B. & Brites, D. Rat cerebellar slice cultures exposed to bilirubin evidence reactive gliosis, excitotoxicity and impaired myelinogenesis that is prevented by AMPA and TNF-α inhibitors. Mol. Neurobiol. 49, 424–439 (2014).
Bortolussi, G. et al. Rescue of bilirubin-induced neonatal lethality in a mouse model of Crigler-Najjar syndrome type I by AAV9-mediated gene transfer. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 26, 1052–1063 (2012).
Bortolussi, G. et al. Age-dependent pattern of cerebellar susceptibility to bilirubin neurotoxicity in vivo in mice. Dis. Model. Mech. 7, 1057–1068 (2014).
Gazzin, S. et al. Bilirubin accumulation and Cyp mRNA expression in selected brain regions of jaundiced Gunn rat pups. Pediatr. Res. 71, 653–660 (2012).
Robert, M. C. et al. Alterations in the cell cycle in the cerebellum of hyperbilirubinemic Gunn rat: a possible link with apoptosis? PloS One 8, e79073 (2013).
Brito, M. A. et al. Cerebellar axon/myelin loss, angiogenic sprouting, and neuronal increase of vascular endothelial growth factor in a preterm infant with kernicterus. J. Child Neurol. 27, 615–624 (2012).
Tabarki, B., Khalifa, M., Yacoub, M., Tlili, K. & Essoussi, A. S. Cerebellar symptoms heralding bilirubin encephalopathy in Crigler-Najjar syndrome. Pediatr. Neurol. 27, 234–236 (2002).
Silva, S. L. et al. Neuritic growth impairment and cell death by unconjugated bilirubin is mediated by NO and glutamate, modulated by microglia, and prevented by glycoursodeoxycholic acid and interleukin-10. Neuropharmacology 62, 2398–2408 (2012).
Daood, M. J., Hoyson, M. & Watchko, J. F. Lipid peroxidation is not the primary mechanism of bilirubin-induced neurologic dysfunction in jaundiced Gunn rat pups. Pediatr. Res. 72, 455–459 (2012).
Daood, M. J. & Watchko, J. F. Calculated in vivo free bilirubin levels in the central nervous system of Gunn rat pups. Pediatr. Res. 60, 44–49 (2006).
Roca, L. et al. Factors affecting the binding of bilirubin to serum albumins: validation and application of the peroxidase method. Pediatr. Res. 60, 724–728 (2006).
Giraudi, P. J., Bellarosa, C., Coda-Zabetta, C. D., Peruzzo, P. & Tiribelli, C. Functional induction of the cystine-glutamate exchanger system Xc(-) activity in SH-SY5Y cells by unconjugated bilirubin. PloS One 6, e29078 (2011).
Falcão, A. S., Fernandes, A., Brito, M. A., Silva, R. F. M. & Brites, D. Bilirubin-induced immunostimulant effects and toxicity vary with neural cell type and maturation state. Acta Neuropathol. (Berl.) 112, 95–105 (2006).
Fernandes, A. et al. Inflammatory signalling pathways involved in astroglial activation by unconjugated bilirubin. J. Neurochem. 96, 1667–1679 (2006).
Liaury, K. et al. Morphological features of microglial cells in the hippocampal dentate gyrus of Gunn rat: a possible schizophrenia animal model. J. Neuroinflammation 9, 56 (2012).
Qaisiya, M., Coda Zabetta, C. D., Bellarosa, C. & Tiribelli, C. Bilirubin mediated oxidative stress involves antioxidant response activation via Nrf2 pathway. Cell. Signal. 26, 512–520 (2014).
Silva, S. L. et al. Features of bilirubin-induced reactive microglia: from phagocytosis to inflammation. Neurobiol. Dis. 40, 663–675 (2010).
Bustin, S. A. et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55, 611–622 (2009).
Vandesompele, J. et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, RESEARCH0034 (2002).
Grojean, S., Koziel, V., Vert, P. & Daval, J. L. Bilirubin induces apoptosis via activation of NMDA receptors in developing rat brain neurons. Exp. Neurol. 166, 334–341 (2000).
McDonald, J. W., Shapiro, S. M., Silverstein, F. S. & Johnston, M. V. Role of glutamate receptor-mediated excitotoxicity in bilirubin-induced brain injury in the Gunn rat model. Exp. Neurol. 150, 21–29 (1998).
Acknowledgements
We thank Paola Zarattini from the Local Animal Facility of the Università degli Studi di Trieste, and Andrea Lorenzon from the CBM scrl, AREA Science Park of Trieste, for their kind support and help with the procedures involving the animals. We thank Prof. Steven M. Shapiro and Dr. Sean M. Riordan for the critical review of the manuscript content.
Author information
Authors and Affiliations
Contributions
Designed and conceptualized the study (M.D.B. and S.G.). Performed the experiments (M.D.B.). Collected and analyzed and interpreted the data (M.D.B., C.B., F.Z. and S.G.). Wrote the manuscript (M.D.B., S.G. and C.T.). Drafted and revised the Ms. for intellectual content (M.D.B., S.G., C.T., C.B. and F.Z.). All the authors give final approval of the submitted Ms.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Dal Ben, M., Bottin, C., Zanconati, F. et al. Evaluation of region selective bilirubin-induced brain damage as a basis for a pharmacological treatment. Sci Rep 7, 41032 (2017). https://doi.org/10.1038/srep41032
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep41032
This article is cited by
-
An egg a day keeps kernicterus away
Pediatric Research (2024)
-
Molecular events in brain bilirubin toxicity revisited
Pediatric Research (2024)
-
Models of bilirubin neurological damage: lessons learned and new challenges
Pediatric Research (2023)
-
Long-Term Mental Health and Quality of Life Outcomes of Neonatal Insults in Kilifi, Kenya
Child Psychiatry & Human Development (2022)
-
Human liver stem cells express UGT1A1 and improve phenotype of immunocompromised Crigler Najjar syndrome type I mice
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
