
Abstract
Diamond is an evidence for carbon existing in the deep Earth. Some diamonds are considered to have originated at various depth ranges from the mantle transition zone to the lower mantle. These diamonds are expected to carry significant information about the deep Earth. Here, we determined the phase relations in the MgCO3-SiO2 system up to 152 GPa and 3,100 K using a double sided laser-heated diamond anvil cell combined with in situ synchrotron X-ray diffraction. MgCO3 transforms from magnesite to the high-pressure polymorph of MgCO3, phase II, above 80 GPa. A reaction between MgCO3 phase II and SiO2 (CaCl2-type SiO2 or seifertite) to form diamond and MgSiO3 (bridgmanite or post-perovsktite) was identified in the deep lower mantle conditions. These observations suggested that the reaction of the MgCO3 phase II with SiO2 causes formation of super-deep diamond in cold slabs descending into the deep lower mantle.
Similar content being viewed by others
Introduction
Carbon is circulated around the surface and interior of the Earth with subducting slabs and volcanic eruptions; subduction carries carbon-bearing rocks to the Earth’s interior and volcanic eruption expels carbon-bearing gas, lavas and rocks from the interior of the Earth1. The flux of subducted carbon within oceanic plates is estimated to be more than 5 Tmol/yr, almost twice as large as the expelled-carbon flux, 2–3 Tmol/yr, through arc magmatism2. This difference suggests the existence of carbon reservoirs in the deep Earth1,2.
One source of direct evidence for deep carbon is carbon-bearing samples originating from the Earth’s interior. Diamond is evidence of quite a deeper-origin carbon. In particular, some diamonds, called ‘super-deep diamond’, are thought to arise from the mantle transition zone or the lower mantle3,4,5,6,7,8. The inclusions in super-deep diamond may supply information on the lithology, water content, and/or elemental distribution in deep parts of the Earth3,4,5,6,7,8.
Subducting slabs play a key role for carrying carbon-bearing phases into the deep Earth and forming super-deep diamond3,4,5. Altered rocks in the oceanic crust contains the large amount of carbon such as organic carbon or carbonate minerals, which can be the deep reservoir of subducted carbon9,10. These carbonate minerals or melts may change to diamond if they become unstable in the Earth during the subduction process3,5,11,12,13. The stability of carbonates has been investigated using high-pressure experiments and ab initio calculations, and MgCO3 magnesite is determined to be stable under the high-pressure and high-temperature conditions expected during subduction11,12,13,14,15,16,17,18,19,20. The existence of carbonate (or carbonatite melt) in the deep mantle is supported by the discovery of carbonate as inclusions in diamonds originating from the mantle transition zone and/or the lower mantle3,5.
Since SiO2 is one of the abundant components and also is an important phase in deeply subducted slabs21,22,23, the MgCO3-SiO2 system may be applied to the slabs descending into the lower mantle. SiO2 phases may change to its high-pressure polymorph, such as coesite, stishovite, CaCl2-type phase and seifertite24,25. Magnesite is expected to break down to CO2 or diamond by reacting with silica minerals in the MgCO3-SiO2 system in subducting process12,13,26,27 although the detail of its phase relation has not yet been clarified. Knowledge of the reactions in this system at high pressure and high temperature may provide important insights into the carbon-related processes in the deep mantle, such as the origin of super-deep diamond and melting or oxidation by release of volatile components13,28. We used a laser-heated diamond anvil cell (LHDAC) combined with a high-pressure and high-temperature in situ synchrotron X-ray diffraction (XRD) technique to quantify the phase relations of the MgCO3-SiO2 system down to the lowermost-mantle conditions. Our objective is to clarify the behavior of carbon in the lower mantle and to model the origin of super-deep diamond.
Results
We observed the phase relation of MgCO3 and SiO2 system up to 152 GPa and 3,100 K (see Supplementary Tables S1 and S3). Diamond and bridgmanite may be formed through the following reactions12,13,28:


Or, the following reaction is also possible:

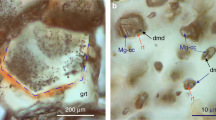
Bridgmanite and diamond were observed using XRD, in a run product recovered from 83 GPa and 1,780 K (Fig. 1a). The X-ray pattern was taken at an ambient condition without DAC, of which surface showed no damage after the experiments. The high-pressure phase of CO2 was not detected in most runs, matching the previous studies on the MgCO3-SiO2 system12,13. We successfully detected the high-pressure phase of CO2, CO2-VI29 in one run made by in situ X-ray diffraction at 83 GPa and 1,780 K (see Supplementary Table S1). Thus, occurrence of reactions (1) and (2) or (3) were thus confirmed.

(a) XRD patterns obtained under the ambient condition for the samples recovered from 83.1(5) GPa and 1,780(210) K. The X-ray pattern was taken at ambient condition without DAC, of which surface showed no damage after the experiment. The abbreviations represent as follow: St: stishovite, Brd: bridgmanite, Dia: diamond, Pt: platinum, W: tungsten (gasket), NaCl: sodium chloride (pressure medium). (b) In situ XRD patterns obtained at 145.5 (31) GPa and 2,700 (360) K. (c) 2D XRD images of a sample recovered from 145.5 (31) GPa and 2,700 (360) K obtained at ambient condition without DAC. The abbreviations represent as follow: Se: seifertite, Mgs: magnesite, MgsII: MgCO3 phase II, PPv: MgSiO3 post-perovskite phase, Dia: diamond, Pt: platinum, W: tungsten (gasket).
The structures of the MgCO3 high-pressure phases and their phase transition boundary are controversial and the data on their compression behaviors are very limited. A recent ab initio study reported a monoclinic post-magnesite phase, MgCO3 phase II (C2/m) at 300 K and pressures from 82 to 138 GPa16. On the other hand, the latest study reported the stabilization of another post-magnesite phase, having space group P-1, at 300 K and 85–101 GPa, which transforms to MgCO3 phase II (C2/m) at 101 GPa and 300 K19. We conducted an in situ XRD study of MgCO3 using a double sided laser-heated diamond anvil cell in the pressure range from 85 GPa to 132 GPa at about 2,500–3,000 K. Diffraction peaks after heating corresponded to the MgCO3 phase II16 in the same pressure range (see Supplementary Fig. S1 and Supplementary Table S2). The volumes of MgCO3 phase II in each run were estimated by fitting XRD patterns (see Supplementary Table S2). We then fitted the estimated volumes using the second order Birch-Murnaghan equations of state (BMEOS). As a result, the unit-cell volume at ambient condition (V0) and isothermal bulk modulus (K300K, 0) of magnesite phase II were estimated to be 498.9(5) Å3 and 154.9(7) GPa, respectively (K’ = 4; fixed). These values are consistent with the V0 and KT0 calculated by ref. 16, V0 = 503.36 Å3 (Z = 12) and K0 = 156.76 GPa using the third order BMEOS (K’ = 4.12).
MgCO3 phase II was identified in MgCO3-SiO2 system at pressures above 100 GPa by comparing the XRD patterns of the runs in the MgCO3-SiO2 system with the results of MgCO3 compression (see Supplementary Information Table S2). This enabled us to distinguish the MgSiO3 bridgmanite/post-perovskite phase from MgCO3 phase II in the XRD patterns (Fig. 1b). The MgSiO3 post-perovskite phase is thought to be formed by either reaction (1) or (3), where MgCO3 and SiO2 are considered to be MgCO3 phase II and seifertite, respectively. XRD spots (111) indicating diamond were also observed in some runs conducted at pressures greater than 100 GPa (see Supplementary Fig. S2). The number of diamond spots was limited and their intensities were weak. They did not appear in all 1D XRD patterns at high pressure. However, diamond created in high-pressure and high-temperature conditions was confirmed in the recovered run products (Fig. 1a and c).
Figure 2 shows the phase diagram of the MgCO3-SiO2 system based on the present and previous studies. The temperature of magnesite decarbonation is consistent with ref. 13 but higher than ref. 12 up to 70 GPa. We discovered the phase transformations from CaCl2-type SiO2 + MgCO3 phase II to bridgmanite + diamond + O2 and from seifertite + MgCO3 phase II to bridgmanite/post-perovskite + diamond + O2 for the first time in this experiment. The decomposition boundary of CO2 has a steep gradient in the present phase diagram of MgCO3-SiO2 system which is not consistent with the results of any previous studies on CO2, such as decomposition28 or phase transition of CO230.

The closed and open symbols represent the results of the present study and ref. 13, respectively. The colors of the symbols show the observed phases as follow: blue: MgCO3 + SiO2, green: MgSiO3 ± MgCO3 ± SiO2 (+CO2), red: MgSiO3 + C ± MgCO3 ± SiO2. The present phase boundaries in the MgCO3-SiO2 system are shown by black solid and dotted lines. The non-equilibrium phases are shown as a small font in the index column above the phase diagram. The referred phase boundaries between CO2-V and ionic CO230 and between CO2-V and C + O228 are gray dotted lines, shown by ‘Y11’ and ‘L11’, respectively. The phase boundaries of SiO2 stishovite (St)-to-CaCl2-type SiO2 (CS)24 and CaCl2-type SiO2 (CS)-to-seifertite (Se)25, the phase boundary of MgSiO3 bridgmanite (Br)-to-post-perovskite phase (PPv)37, and those of MgCO3 magnesite-to-MgCO3 liquid and MgCO3 liquid-to-MgO + CO220 are shown by gray solid lines.
Discussion
Since the reactions between MgCO3 and SiO2 may be expected in deeply subducted slabs13,26,27, we should consider the phase relations in the MgCO3-SiO2 system on subducting depth-temperature paths. The phase boundaries of the MgCO3-SiO2 system are shown with several slab geotherms31 in Fig. 3. The slab geotherms in Fig. 3a are models based on the geological observations32 and mineralogical hypothesis31 in the present Earth. The super-deep diamonds form Juina kimberlite were considered to be related to Gondwana subduction and formed at 150 Ma5. Therefore, the colder geotherms like the modern Earth may be useful to be compared with the MgCO3-SiO2 reactions. The reactions in this system provide important information for understanding the stability limits of MgCO3 in deep subduction.

(a) The phase boundaries in the MgCO3-SiO2 system are denoted with the Earth’s geotherms. The gray solid zone shows the mantle adiabat33. The blue and red solid arrows are the depth-temperature paths of Tonga and Mexico subduction zones, where the coldest and hottest slabs were modeled in ref. 32, respectively. The hypothetical models of the “hot”, “cold” and “very cold” slabs31 are shown by the orange, light blue and blue arrows, respectively, and the zone of each color is extrapolation of each path. The series of paths of cold-slab geotherm below the mantle transition zone represent the case of the fast subduction, stagnant and the slow subduction31. (b) The schematic model of the super-deep diamond’s formation. The orange, light blue and blue lines represent the “hot”, “cold” and “very cold” subducting slabs31, respectively. The green circles and diamond-shaped symbols indicate the possible depth of CO2 and diamond formation, respectively.
The reaction between MgCO3 and SiO2 may not occur down to the top of the lower mantle but can occur at depths from 1,000 to 2,000 km in various subducting slabs. In “hot” and “cold” slabs31, which are comparable to Mexico and the various Pacific subduction paths respectively32, magnesite can react with stishovite to form bridgmanite and CO2 at depths of around 1,700 km, and subsequently CO2 can decompose to diamond and O2 at the greater depths. Therefore, super-deep diamond could be formed at depths greater than 1,700 km in cold slabs.
The path of “very cold” slabs31 such as Tonga subduction32 may pass through the ‘bottleneck’ region in the phase diagram at ~80 GPa corresponding to the depth near the MgCO3 magnesite-phase II boundary (~1,900 km). MgCO3 in such slabs may descend to the base of the lower mantle because the stability field of MgSiO3 + diamond + O2 is relatively narrow or absent in the pressure-temperature path of very cold slabs (Fig. 3a). If MgCO3 is able to survive beyond 1,900-km depth, MgCO3 phase II will be formed and subsequently decompose to MgSiO3 post-perovskite phase + diamond + O2 by a reaction with seifertite at the base of the lower mantle due to heating from the outer core.
Slabs in the early Earth, Archean/Proterozoic age, might descend into the hotter mantle than the modern adiabat33. The hotter subduction could restrict the MgCO3 subduction into the deep lower mantle because of the reaction (1) in the shallower mantle (Fig. 3). In this case, CO2 might be a significant carbon carrier in the subducted slabs although its stability has been still controversial under high pressure and high temperature conditions28,30. If early slab temperatures had been higher than the modern mantle adiabat33, the formation of CO2 fluid28 or melting in the MgCO3-SiO2 system26 would be expected down to the mantle transition zone. These phenomena might prevent the subduction of carbon-bearing phases into the lower mantle. This might be the reason why the reports of the old super-deep diamonds are absent.
The reaction in the “very cold” slab may play an important role in formation of super-deep diamond in the deep lower mantle. Reference 5 summarized the mineral inclusions in diamond, and estimated the formation depths of diamonds in the mantle. Some diamonds from São Luiz, Juina province, Brazil have inclusions of orthopyroxene which are a psuedomorph of bridgmanite and TAPP (tetragonal almandine-pyrope phase) suggesting the top of the lower mantle, 660–750 km depths. They also suggested that some diamonds with inclusions of aluminous bridgmanite pseudomorph from the same locality, Juina, Brazil, might be originated at the depths greater than 750 km, although the exact depth limit was not estimated. On the other hand, Diamonds containing the iron-rich (Mg,Fe)O inclusions6,8 were considered to be originated from the deep lower mantle (1,700–2,900 km) because of iron enrichment by the spin transition at depths greater than about 1,700 km34,35,36, or by the phase transition from bridgmanite to post perovskite at around 2,700-km depth37,38,39,40,41, although it is a debated matter whether iron-rich (Mg,Fe)O inclusions are the signature of the bottom of the lower mantle or not.
The mechanism of diamond formation discovered here may explain the origin of super-deep diamond from the deep lower mantle such as those reported from the limited locality in Brazil5,6,8. Although we did not consider the effects of fO242,43 in the reaction (3) because of the technical difficulty for controlling fO2 in the DAC experiments, we can expect the above reaction in the presence of iron in the average lower mantle with the fO2 condition below the iron-wüstite buffer44. When metallic iron exists in the system, iron oxide can be formed by the reaction with O2 in the run products. The role of iron is also important in formation of high-pressure polymorphs of carbonate18,45,46. The latest studies reported that the iron-magnesium carbonate transforms into the several new phases: Fe4(CO4)3 phase having unknown structure18, orthorhombic (Mg,Fe)CO3 phase II45, or the unquenchable phases with unexpected stoichiometry (Mg2Fe2(C4O13) + Fe13O19) that coexist under the lower mantle46. Therefore, we may need to modify the present phase relation further in the iron bearing system. However, we can consider that the reaction of diamond formation could also occur in the iron-bearing system since iron oxides and/or FeO-bearing bridgmanite and ferropericlase can be formed in deep subducted slabs.
Released oxygen will oxidize the ferrous iron in mantle minerals or the metallic iron penetrated from the outer core at the core-mantle boundary28,47. The oxidation of metallic iron can generate FeO. As a result, the iron-rich magnesiowüstite of (Mg,Fe)O6,8 would be formed as inclusions in the deepest diamonds, although it is not a single process for generation of FeO enrichment in magnesiowustite as inclusions in diamond and other processes of the redox change in the shallower depths could also have generated similar iron enrichment. Future studies are needed to elucidate the ultra-deep iron-carbon redox coupling processes and their influence on formation of super-deep diamond.
Methods
A natural single crystal of magnesite (Bahia, Brazil, Mg0.994Ca0.003Fe0.003CO3) and a reagent quartz (reagent grade, Woko) were ground to fine powders in an agate mortar for 1 hour. The powders were mixed 1:1 by mole fraction and ground in an agate mortar for 30 min to homogenize the mixture. High-pressure and high-temperature experiments were conducted using a double sided LHDAC. The culet diameters of the diamond anvils were between 100 and 350 μm. The sample chamber drilled in rhenium or tungsten gaskets ranges from 30 to 100 μm in diameter and from 40 to 80 μm in the thickness depending on the culet sizes. NaCl or SiO2 glass was used as the pressure medium and thermal insulator.
Platinum was included in the sample chamber as a laser absorber. We used three sample configurations to obtain stable temperatures at high pressures: Run Ch1-3-layer-chamber, Run Ch2-5-layer-chamber and Run Ch3-Pt/W-doughnut-chamber (Supplementary Table 1). In Run Ch1, 5 wt. % platinum powder (platinum black, purity 99.9%, Mitsuwa Chemicals) was mixed with magnesite and quartz starting material (Ch1) and this was sandwiched by a pressure medium. On the other hand, in Run Ch2, the magnesite and quartz mixture was sandwiched by platinum foils (purity 99.9%, Nilaco). Run Ch2 aimed to reduce the temperature gradient at the heating spot during double-sided laser heating.
The samples were heated by double-sided fiber lasers (SPI LASER) of wavelength 1.090 μm at Tohoku University in Sendai, Japan and 1.070 μm at BL10XU of SPring-8 in Hyogo, Japan. The heating duration was 10–120 minutes. Temperature was determined by fitting the emission spectra from the heated samples to the gray-body radiation formula.
In situ synchrotron X-ray diffraction (XRD) was conducted to identify the experimental products. We acquired the XRD patterns of the samples at high pressure up to 152 GPa in the temperature range from 300 K to 3,100 K at BL10XU of SPring-8. The collimator was 20 μm in diameter and the typical wavelength of X-ray was 0.41414(9) Å. An imaging plate (Rigaku, R-AXIS IV++) and a charge coupled device (Bruker, AXS SMART APEX) detectors were used for acquiring the XRD patterns. The pressures of in situ experiments were determined based on the equation of state of Pt48. Thermal pressure was estimated based on Mie–Grüneisen–Debye model49,50. In Runs Ch3-1 and Ch2-5, the sample was heated at Tohoku University and observed after temperature quenching at high pressure using XRD at BL10XU of SPring-8. In Ch3-1, the sample was recovered to the ambient condition at Tohoku University after the temperature quenching and pressure determination using ruby fluorescence method51. Since thermal pressure could not be estimated in Runs Ch3-1 and Ch2-5, we considered the pressure errors up to ±10 GPa for pressures at high temperature during laser heating. This is almost equivalent to the maximum pressure increase by laser heating in the in situ XRD experiments.
The high-pressure phase transition of MgCO3 was confirmed in two series of runs, Ms-1 and Ms-2 in addition to the MgCO3-SiO2 system (Supplementary Table 2). These runs were conducted using a membrane-type diamond anvil cell. The natural magnesite and Pt powder were sandwiched by the same magnesite in the sample chamber. Pt powder was used as a pressure maker and laser absorber. Magnesite was compressed to about 100 GPa, and then pre-heated to temperature of less than 1,500 K using CO2 laser at Tohoku University in order to crystallize the compressed sample in Run Ms-1. We conducted laser heating at 2,500–3,000 K for 5–20 min in order to obtain the obvious XRD patterns of the MgCO3 high-pressure phase using SPI fiber laser in two series of runs, Ms-1 and Ms-2. XRD patterns were acquired at high pressure and 300 K after quenching.
Temperatures in some experiments increased suddenly to above 3,000 K in the Runs Ch1-1, Ch1-2, Ch1-3 and Ch 2-1 using platinum as a laser absorber (Supplementary Table 1). This phenomenon may correspond to the ‘temperature jump’ reported by ref. 13. The sudden temperature increase was also observed at high pressures and above 2,000 K when a Pt powder (Run Ch1) and Pt foil (Run Ch2) was used as the laser absorber. These results indicate that large temperature fluctuation may be caused by the volume change following reactions, phase transitions and/or melting of the sample.
Additional Information
How to cite this article: Maeda, F. et al. Diamond formation in the deep lower mantle: a high-pressure reaction of MgCO3 and SiO2. Sci. Rep. 7, 40602; doi: 10.1038/srep40602 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Dasgupta, R. & Hirschmann, M. M. The deep carbon cycle and melting in the Earth’s interior. Earth Planet. Sci. Lett. 298, 1–13 (2010).
Kerrick, D. C. & Connolly, J. A. D. Metamorphic devolatilization of subducted marine sediments and the transport of volatiles into the Earth’s mantle. Nature 411, 293–296 (2001).
Brenker, F. E. et al. Carbonates from the lower part of transition zone or even the lower mantle. Earth Planet. Sci. Lett. 260, 1–9 (2007).
Harte, B. Diamond formation in the deep mantle: the record of mineral inclusions and their distribution in relation to mantle dehydration zones. Mineral. Mag. 74, 189–215 (2010).
Harte, B. & Richardson, S. Mineral inclusions in diamonds track the evolution of a Mesozoic subducted slab beneath West Gondwanaland. Gond. Res. 21, 236–245 (2012).
Hayman, P., Kopylova, M. & Kaminsky, F. Lower mantle diamonds from Rio Soriso (Juina area, Mato Grosso, Brazil). Contrib. Mneral. Petrol. 149, 430–445 (2005).
Pearson, D. G. et al. Hydrous mantle transition zone indicated by ringwoodite included within diamond. Nature 507, 221–224 (2014).
Wirth, R., Dobrzhineskaya, L., Harte, B., Schreiber, A. & Green, H. W. High-Fe (Mg, Fe)O inclusion in diamond apparently from the lowermost mantle. Earth Planet. Sci. Lett. 404, 365–375 (2014).
Plank, T. & Langmuir, C. H. The chemical composition of subducting sediment and its consequences for the crust and mantle. Chem. Geol. 145, 325–394 (1998).
Alt, J. C. & Teagle, D. A. H. The uptake of carbon during alteration of ocean crust. Geochem. Cosmochim. Acta 63, 1527–1535 (1999).
Biellmann, C., Gillet, P., Guyot, F., Peyronneau, J. & Reynard, B. Experimental evidence for carbonate stability in the Earth’s lower mantle. Earth Planet. Sci. Lett. 118, 31–41 (1993).
Takafuji, N., Fujino, K., Nagai, T., Seto, Y. & Hamane, D. Decarbonation reaction of magnesite in subducting slabs at the lower mantle. Phys. Chem. Miner. 33, 651–654 (2006).
Seto, Y., Hamane, D., Nagai, T. & Fujino, K. Fate of carbonates within oceanic plates subducted to the lower mantle, and a possible mechanism of diamond formation. Phys. Chem. Miner. 35, 223–229 (2008).
Fiquet, G. et al. Structural refinements of magnesite at very high pressure. Am. Mineral. 87, 1261–1265 (2002).
Isshiki, M. et al. Stability of magnesite and its high-pressure form in the lowermost mantle. Nature 427, 60–62 (2004).
Oganov, A. R., Ono, S., Ma, Y., Glass, C. W. & Garcia, A. Novel high-pressure structures of MgCO3, 176 CaCO3 and CO2 and their role in Earth’s lower mantle. Earth Planet. Sci. Lett. 273, 38–47 (2008).
Boulard, E. et al. New host for carbon in the deep Earth. Proc. Natl. Acad. Sci. USA 108, 5184–5187 (2011).
Boulard, E. et al. Experimental investigation of the stability of Fe-rich carbonates in the lower mantle. J. Geophys. Res. 117, B02208 (2012).
Pickard, C. J. & Needs, R. J. Structures and stability of calcium and magnesium carbonates at mantle pressures. Phys. Rev. B 91, 104101 (2015).
Solopova, N. A., Dubrovinsky, L., Spivak, A. V., Litvin, Y. A. & Dubrovinskaia, N. Melting and decomposition of MgCO3 at pressures up to 84 GPa. Phys. Chem. Miner. 42, 73–81 (2015).
Ishii, T., Kojitani, H. & Akaogi, M. High-pressure phase transitions and subduction behavior of continental crust at pressure–temperature conditions up to the upperpart of the lower mantle. Earth Planet. Sci. Lett. 357–358, 31–41 (2012).
Ono, S. Stability limits of hydrous minerals in sediment and mid-ocean ridge basalt compositions: Implications for water transport in subduction zones. J. Geophys. Res. 103, 18253–18267 (1998).
Ricolleau, A. et al. Phase relations and equation of state of a natural MORB: Imprication for the density profile of subducted oceanic crust in the Earth’s lower mantle. J. Geophys. Res. 115, B08202 (2010).
Murakami, M., Hirose, K., Ono, S. & Ohishi, Y. Stability of CaCl2-type and PbO2-type SiO2 at high pressure and temperature determined by in-situ X-ray measurements. Geophys. Res. Lett. 30, 1207 (2003).
Grocholski, B., Shim, S.-H. & Prakapenka, V. B. Stability, metastability, and elastic properties of a dense silica polymorph, seifertite. J. Geophys. Res.: Solid Earth 118, 4745–4757 (2013).
Litasov, K. D., Fei, Y., Ohtani, E., Kuribayashi, T. & Fumakoshi, K. Thermal equation of state of magnesite to 32 GPa and 2073 K. Phys. Earth Planet. Inter. 168, 191–203 (2008).
Kakizawa, S., Inoue, T., Suenami, H. & Kikegawa, T. Decarbonation and melting in MgCO3-SiO2 system at high temperature and high pressure. J. Mineral. Petrol. Sci. 110, 179–188 (2015).
Litasov, K. D., Goncharov, A. F. & Hemley, R. J. Crossover from melting to dissociation of CO2 under pressure: Implications for the lower mantle. Earth Planet. Sci. Lett. 309, 318–323 (2011).
Iota, V. et al. Six-fold coordinated carbon dioxide VI. Nature Matt. 6, 34–38 (2007).
Yoo, C.-S., Sengupta, A. & Kim, M. Carbon dioxide carbonates in the Earth’s mantle: implications to the deep carbon cycle. Angew. Chem. 125, 11415–11418 (2011).
Komabayashi, T., Omori, S. & Maruyama, S. Petrogenetic grid in the system MgO-SiO2-H2O up to 30 GPa, 1600 °C: applications to hydrous peridotite subducting into the Earth’s deep interior. J. Geophys. Res. 109, B03206 (2004).
Syracuse, E. M., van Keken, P. E. & Abers, G. A. The global range of subduction zone thermal models. Earth Planet. Sci. Lett. 183, 73–90 (2010).
Katsura, T., Yoneda, A., Yamazaki, D., Yoshino, T. & Ito, E. Adiabatic temperature profile in the mantle. Phys. Earth Planet. Inter. 183, 212–218 (2010).
Badro, J. et al. Iron partitioning in Earth’s mantle: toward a lower mantle discontinuity. Science 300, 789–791 (2003).
Speziale, S. et al. Iron spin transition in Earth’s mantle. Proc. Natl. Acad. Sci. 102, 17918–17922 (2005).
Lin, J.-F. et al. Pressure-induced electronic spin transition of iron in magnesiowustite-(Mg,Fe)O. Phys. Rev. B 73, 113107 (2006).
Komabayashi, T. et al. Simultaneous volume measurements of post-perovskite and perovskite in MgSiO3 and their thermal equations of state. Earth Planet. Sci. Lett. 265, 515–524 (2008).
Murakami, M., Hirose, K., Kawamura, K., Sata, N. & Ohishi, Y. Post-perovskite phase transition in MgSiO3 . Science 304, 855–858 (2004).
Murakami, M., Hirose, K., Sata, N. & Ohishi, Y. Post-perovskite phase transition and mineral chemistry in the pyrolytic lowermost mantle. Geophys. Res. Lett. 32, L03304 (2005).
Oganov, A. R. & Ono, S. Theoretical and experimental evidence for a post-perovskite phase of MgSiO3 in Earth’s D” layer. Nature 430, 445–448 (2004).
Sakai, T. et al. Fe–Mg partitioning between post-perovskite and ferropericlase in the lowermost mantle. Phys. Chem. Miner. 37, 487–496 (2010).
Rohrbach, A. & Schmidt, M. W. Redox freezing and melting in the Earth’s deep mantle resulting from carbon–iron redox coupling. Nature 472, 209–212 (2011).
Stagno, V. et al. The stability of magnesite in the transition zone and the lower mantle as function of oxygen fugacity. Geophys. Res. Lett. 38, L19309 (2011).
Frost, D. J. & McCammon, C. A. The redox state of Earth’s mantle. Annu. Rev. Earth Planet. Sci. 36, 389–420 (2008).
Liu, J., Lin, J.-F. & Prakapenka, V. B. High-pressure orthorhombic ferromagnesite as a potential deep-mantle carbon carrier. Sci. Rep. 5, 7640, doi: 10.1038/srep07640 (2015).
Merlini, M. et al. The crystal structures of Mg2Fe2C4O13, with tetrahedrally coordinated carbon, and Fe13O19, synthesized at deep mantle conditions. Am. Mineral. 100, 2001–2004 (2015).
Otsuka, K. & Karato, S. Deep penetration of molten iron into the mantle caused by a morphological instability. Nature 492, 243–246 (2012).
Fei, Y. et al. Toward an internally consistent pressure scale. Proc. Natl. Acad. Sci. 104 (2007).
Suzuki, I. Thermal expansion of periclase and olivine, and their anharmonic properties. J. Phys. Earth 23, 145–159 (1975).
Fei, Y., Mao, H.-K. & Hu, J. P–V–T equation of state of magnesiowüstite (Mg0.6Fe0.4)O. Phys. Chem. Miner. 18, 416–422 (1992).
Dewaele, A., Datchi, F., Loubeyre, P. & Mezouar, M. High pressure-high temperature equations of state of neon and diamond. Phys. Rev. B 77, 094106 (2008).
Acknowledgements
This work was supported by Grant-in-Aid awards for Scientific Research to E.O. (numbers 22000002 and 15H05748) from the Ministry of Education, Culture, Sports, Science, and Technology of Japanese Government. E.O. was also supported by Russian Science Foundation, Project 14B25.31.0032. The synchrotron radiation experiments were performed at SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (Proposal No. 2013B0104, 2014A0104, 2014B0104, 2015A0104, 2015B0104). F.M. was supported by the International Joint Graduate Program in Earth and Environmental Science (GP-EES), Tohoku Univirstiy.
Author information
Authors and Affiliations
Contributions
F.M., S.K., T.S., E.O., N.H. and Y.O. performed XRD measurements. F.M., S.K., E.O. and T.S. made arguments on geological applications of the experimental results and wrote the paper. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Maeda, F., Ohtani, E., Kamada, S. et al. Diamond formation in the deep lower mantle: a high-pressure reaction of MgCO3 and SiO2. Sci Rep 7, 40602 (2017). https://doi.org/10.1038/srep40602
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep40602
This article is cited by
-
How lowermost mantle viscosity controls the chemical structure of Earth’s deep interior
Communications Earth & Environment (2023)
-
Comparative study on high-pressure physical properties of monoclinic MgCO3 and Mg2CO4
Scientific Reports (2022)
-
First-principles calculations of high-pressure physical properties anisotropy for magnesite
Scientific Reports (2022)
-
Reversal of carbonate-silicate cation exchange in cold slabs in Earth’s lower mantle
Nature Communications (2021)
-
Redox-Induced Destabilization of Dolomite at Earth’s Mantle Transition Zone
Journal of Earth Science (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
