
Abstract
In this study, the mechanistic and kinetic analysis for reactions of CF3OCH(CF3)2 and CF3OCF2CF2H with OH radicals and Cl atoms have been performed at the CCSD(T)//B3LYP/6-311++G(d,p) level. Kinetic isotope effects for reactions CF3OCH(CF3)2/CF3OCD(CF3)2 and CF3OCF2CF2H/CF3OCF2CF2D with OH and Cl were estimated so as to provide the theoretical estimation for future laboratory investigation. All rate constants, computed by canonical variational transition state theory (CVT) with the small-curvature tunneling correction (SCT), are in reasonable agreement with the limited experimental data. Standard enthalpies of formation for the species were also calculated. Atmospheric lifetime and global warming potentials (GWPs) of the reaction species were estimated, the large lifetimes and GWPs show that the environmental impact of them cannot be ignored. The organic nitrates can be produced by the further oxidation of CF3OC(•)(CF3)2 and CF3OCF2CF2• in the presence of O2 and NO. The subsequent decomposition pathways of CF3OC(O•)(CF3)2 and CF3OCF2CF2O• radicals were studied in detail. The derived Arrhenius expressions for the rate coefficients over 230–350 K are: k T(1) = 5.00 × 10−24T3.57 exp(−849.73/T), k T(2) = 1.79 × 10−24T4.84 exp(−4262.65/T), kT(3) = 1.94 × 10−24 T4.18 exp(−884.26/T), and k T(4) = 9.44 × 10−28T5.25 exp(−913.45/T) cm3 molecule−1 s−1.
Similar content being viewed by others
Introduction
With people’s increasing awareness of environment protection, the issue of fine global warming potential (GWP) should be given more attention. Global warming will affect the viability and health of ecosystems, influencing shifts in the distribution of plants, animals, and even human settlements and health. Hydrofluorinated ethers (HFEs) have been considered as potential replacements to chlorofluorocarbons (CFCs) in many industrial applications1 for having no contribution to the stratospheric ozone depletion2,3. However, HFEs may have large GWP due to the presence of C–F bonds4. The HFEs can react with OH radicals and possibly with Cl atoms in the atomosphere5,6,7, so that they have a reduced effect on global warming. The oxidation of HFEs initiated by OH radicals and Cl atoms can lead to the formation of hydrofluorinated esters (FESs). Then, the FESs’ degradation can contribute to the environmental burden of trifluoroacetic acid (TFA) in troposphere8. In order to evaluate the environmental acceptability of HFEs and the possible contribution of the photo-oxidation of FESs to TFA formation or derivates in the environment, it is necessary to know the rate coefficients for the reactions of HFEs. The kinetics of reactions of HFEs with free radicals can aid our understanding of their environmental impacts and estimate their atmospheric lifetimes.
During these years, many theoretical9,10,11,12,13,14,15,16,17 and experimental18,19,20,21,22,23 investigations about the reactions of some volatile organic compounds (VOCs) with free radicals have been carried out in kinetic properties, reaction mechanisms, temperature dependence and more. Previous studies have generally evaluated the fates of HFEs at 298 ± 2 K only. First high-temperature calculations for the rate constants of the reactions of HFEs with OH radicals and Cl atoms are estimated to improve our knowledge of the combustion and atmospheric chemistry of HFEs. In this work, we have carried out a kinetic study of the CF3OCH(CF3)2 and CF3OCF2CF2H with OH radicals and Cl atoms, using density functional theory (DFT) and the dual-level direct dynamic method. As reported by Stevens et al.24. using the B3LYP/6-311 G(2d,2p) level, CF3OCF2CF2H has two stable conformers (defined as conformer a and b), within energy difference of 0.016 kcal/mol. Based on our calculation, the energy difference between the two conformers is only 0.059 kcal/mol at the B3LYP/6-311++G(d,p) level. The energy difference is so small that both of them may have contribution to the title reaction. In experiment, Andersen et al.20. reported reactions of CF3OCH(CF3)2 and CF3OCF2CF2H with OH radicals and Cl atoms through Fourier transform infrared (FTIR) smog chamber techniques at 296 ± 2 K. Subsequently, Wilson et al.25. investigated the kinetics of reaction CF3OCF2CF2H with OH by relative rate experiments. For reactions of CF3OCF2CF2H (b) + OH/Cl, Singh et al.26. have studied the mechanism and rate constants at only 296 K by conventional transition state theory (CTST). The calculated rate constants are close to the previous experimental work. Even so, little is known about the temperature dependencies and contributions ratios of two conformers of CF3OCF2CF2H from theory and experiment. In addition, the tunneling effects are found to be important for the HFEs reactions with free radicals, and canonical variational transition state theory (CVT) involving small curvature tunneling correction (SCT) can provide the relevant analysis for the tunneling and variational effects. Therefore, in the present work, high-level ab initio calculations and the dual-level direct dynamics method are performed on the mechanisms and kinetics of CF3OCH(CF3)2 + OH/Cl reactions and the dynamic properties of CF3OCF2CF2H + OH/Cl reactions.
Isotopic analysis allows us to understand the significant advances of atmospheric chemistry and global change. For instance, the air pollution, global warming, and climate change have been expounded by the study of the distribution of stable isotopes27. Kinetic isotope effects (KIEs) values can also provide useful information for explaining the stable isotope composition of atmospheric organic compounds28. In order to establish the isotopic effects in CF3OCH(CF3)2 and CF3OCF2CF2H and lay a direction for future experiments, KIEs for CF3OCH(CF3)2/CF3OCF2CF2H + OH/OD and CF3OCD(CF3)2/CF3OCF2CF2D + OH/Cl reactions are estimated firstly in theory. The temperature dependence of the H and D-abstraction rate constants of the title reactions in a wide temperature range are also discussed firstly.
In the atmosphere, the complex peroxy radicals can be produced by alkyl radical (CF3OC(•)(CF3)2 and CF3OCF2CF2•) reaction with O2. Then, the peroxy radicals are converted to the corresponding alkoxy radicals through reaction with NO, peroxy radicals, and other ways. For the reactions of peroxy radicals with NO, organic nitrates may also be formed in this cycle. Organic nitrates have been observed in ambient atmospheric aerosols and in secondary organic aerosols (SOA) formed from the oxidation of various volatile organic compounds in the presence of NO. The organic nitrates are relatively stable and hygroscopic and thus will play an important role in affecting the climate by acting as the components of aerosol organic matter. The above processes are recommended in experiment and theory as follows:




The alkoxy radical plays a significant role in the destruction of a variety of VOCs. Andersen et al.20. experimentally predicted the degradation mechanism and some products including CF3OC(O•)(CF3)2 and CF3OCF2CF2O• radicals. However, the information about the barrier heights for their subsequent degradation channels is not provided. Theoretical calculations can not only predict kinetic data in term of reaction barriers but also give the thermodynamic content of the products such as reaction energies and enthalpies. Our aim is to gain a further insight into the mechanism of the subsequent behaviors for CF3OC(O•)(CF3)2 and CF3OCF2CF2O• radicals. This is the first study to reveal the decomposition mechanisms of CF3OC(O•)(CF3)2 and CF3OCF2CF2O• in theory.
Results and Discussion
Electronic structures of stationary points
As for the prototypical species of HFEs, CF3OCH(CF3)2 and CF3OCF2CF2H can be attacked by OH radicals and Cl atoms by the following reaction channels:






At the B3LYP/6-311++G(d,p) and M06-2X/6-311++G(d,p) levels, the optimized structural parameters of the reactants, products, complexes, and transition states for reactions of CF3OCH(CF3)2 + OH/Cl and CF3OCF2CF2H (a, b) + OH/Cl are shown in Figure S1, along with the limited experimental bond distance and angle values29. It is seen that the optimized parameters of the stationary points are reasonable consistent with each other at the two levels. The theoretical bond distance of OH, H2O, HCl and bond angle in H2O are in good agreement with the corresponding experimental values. The vibrational frequencies of the stable points computed at the B3LYP/6-311++G(d,p) level are listed in Table S2, together with the experimental data of OH, H2O, and HCl30,31 The theoretical and experimental frequencies are in good agreement with each other within the largest deviation of 4.2%. The values of imaginary frequencies for TS1-TS6 are 1412i, 1101i, 958i, 848i, 949i, and 856i cm−1, respectively. Figure S1 demonstrates that the TS1 is reactant-like, and the reaction 5 may proceed via early transition state. However, the TS2 is product-like, and reaction 6 may proceed via late transition state. This phenomenon also appears in some similar species (CHF2OCF2CH2F, (CF3)2CHOCH3, (CF3)2CFOCH3, CF3CHFOCH3, CF3CHFOCF3, CHF2OCHFCF3, CF3OCH2CHF2, CF3OCHFCHF2)8,9,12−16 reaction with OH radicals or Cl atoms.
Reactions mechanism and energetics
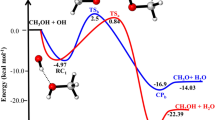
The schematic potential energy diagrams of CF3OCH(CF3)2 + OH/Cl and CF3OCF2CF2H (a, b) + OH/Cl, obtained at the CCSD(T)//B3LYP/6-311++G(d,p) level, are plotted in Fig. 1 and Figure S2, respectively. For CF3OCF2CF2H (b) + OH/Cl, the calculated results are in line with the theoretical values reported by Singh et al.26. Therefore, we just discuss the reactions of CF3OCH(CF3)2 + OH/Cl (reactions 5 and 6). For reaction 5, the relative energy of ER1 is lower than those of the reactants by 4.26 kcal/mol. Then, the corresponding product complex EP1 is produced with relative energies of 9.59 kcal/mol. The relative energy of transition state is calculated to be 5.89 kcal/mol. For reaction 6, product complex (EP2), with −0.09 kcal/mol energy, is formed via TS2 with the 10.17 kcal/mol potential barrier. Comparing the potential barriers of CF3OCH(CF3)2 + OH radicals with Cl atoms, we find that the barrier height of reaction with OH radical is lower than that with Cl atom. Thus, the hydrogen abstraction reaction rate constants of OH radical will be larger than that of Cl atom. Same conclusion can be found in similar reactions14,15,26.

The relative energies (in kcal/mol) are calculated at the CCSD(T)//B3LYP/6-311++G(d,p) + ZPE level.
Table 1 lists the reaction enthalpies ( ), and reaction Gibbs free energies (
), and reaction Gibbs free energies ( ) for reactions 5 and 6 at the B3LYP/6-311++G(d,p), and CCSD(T)//B3LYP/6-311++G(d,p) levels (kcal/mol) with the ZPE or TZPE corrections and the calculated bonds dissociation (
) for reactions 5 and 6 at the B3LYP/6-311++G(d,p), and CCSD(T)//B3LYP/6-311++G(d,p) levels (kcal/mol) with the ZPE or TZPE corrections and the calculated bonds dissociation ( ) of the C–H in molecules CF3OCH(CF3)2 and CF3OCF2CF2H (a, b). The relative energies (Er) of main species for all reactions with ZPE corrections and T1 diagnostic values at CCSD(T)/6-311++G(d,p) level are listed in Table S3. The reaction enthalpies (
) of the C–H in molecules CF3OCH(CF3)2 and CF3OCF2CF2H (a, b). The relative energies (Er) of main species for all reactions with ZPE corrections and T1 diagnostic values at CCSD(T)/6-311++G(d,p) level are listed in Table S3. The reaction enthalpies ( ), reaction Gibbs free energies (
), reaction Gibbs free energies ( ) for reactions 7–10 are displayed in Table S4. Standard enthalpies of formation (
) for reactions 7–10 are displayed in Table S4. Standard enthalpies of formation ( ) for the species at the B3LYP/6-311++G(d,p) and CCSD(T)//B3LYP/6-311++G(d,p) levels are listed in Table 2. As shown in Table S3, the T1 values of all species are within 0.006–0.017, indicating the multi-reference character in CCSD(T) wave functions may be ignored. Unless otherwise specified, energies used in the following discussion are at CCSD(T)//B3LYP/6-311++G(d,p) level. From Table 1, we can see that reaction of CF3OCH(CF3)2 with OH radical is exothermic with reaction enthalpy of −5.49 kcal/mol. However, reaction of CF3OCH(CF3)2 + Cl is endothermic (
) for the species at the B3LYP/6-311++G(d,p) and CCSD(T)//B3LYP/6-311++G(d,p) levels are listed in Table 2. As shown in Table S3, the T1 values of all species are within 0.006–0.017, indicating the multi-reference character in CCSD(T) wave functions may be ignored. Unless otherwise specified, energies used in the following discussion are at CCSD(T)//B3LYP/6-311++G(d,p) level. From Table 1, we can see that reaction of CF3OCH(CF3)2 with OH radical is exothermic with reaction enthalpy of −5.49 kcal/mol. However, reaction of CF3OCH(CF3)2 + Cl is endothermic ( = 9.85 kcal/mol). For reaction of CF3OCH(CF3)2 with OH and Cl, the value of
= 9.85 kcal/mol). For reaction of CF3OCH(CF3)2 with OH and Cl, the value of  is −7.93 and 6.18 kcal/mol, respectively. The
is −7.93 and 6.18 kcal/mol, respectively. The  of C–H in molecule CF3OCH(CF3)2 is 113.96 kcal/mol, which is apparently higher than that of CF3OCF2CF2H. This can explain that the reactions of CF3OCH(CF3)2 + OH/Cl is hard to happen than CF3OCF2CF2H + OH/Cl. The following kinetic results will test this view. In summary, for CF3OCH(CF3)2, the reaction with OH radical is more favorable than its reaction with Cl atom in thermodynamics according to the reaction barrier heights and bonds dissociation energy. We also calculated enthalpies of formation (
of C–H in molecule CF3OCH(CF3)2 is 113.96 kcal/mol, which is apparently higher than that of CF3OCF2CF2H. This can explain that the reactions of CF3OCH(CF3)2 + OH/Cl is hard to happen than CF3OCF2CF2H + OH/Cl. The following kinetic results will test this view. In summary, for CF3OCH(CF3)2, the reaction with OH radical is more favorable than its reaction with Cl atom in thermodynamics according to the reaction barrier heights and bonds dissociation energy. We also calculated enthalpies of formation ( ) for the species CF3OCH(CF3)2, CF3OC(CF3)2, CF3OCF2CF2H (a, b), and CF3OCF2CF2 (a, b) by the reactions 16–27. The experimental enthalpies of formation (
) for the species CF3OCH(CF3)2, CF3OC(CF3)2, CF3OCF2CF2H (a, b), and CF3OCF2CF2 (a, b) by the reactions 16–27. The experimental enthalpies of formation ( ) values9,11,12,14 involved in the reactions 16–27 are listed in Table S5. The enthalpies of formation (
) values9,11,12,14 involved in the reactions 16–27 are listed in Table S5. The enthalpies of formation ( ) for the species CF3OCH(CF3)2, CF3OC(CF3)2, CF3OCF2CF2H (a), CF3OCF2CF2 (a), CF3OCF2CF2H (b), and CF3OCF2CF2 (b) are −525.61, −467.24, −412.89, −361.86, −412.88, and −361.70, respectively. Above content can enrich thermal chemical databases of CF3OCH(CF3)2 and CF3OCF2CF2H.
) for the species CF3OCH(CF3)2, CF3OC(CF3)2, CF3OCF2CF2H (a), CF3OCF2CF2 (a), CF3OCF2CF2H (b), and CF3OCF2CF2 (b) are −525.61, −467.24, −412.89, −361.86, −412.88, and −361.70, respectively. Above content can enrich thermal chemical databases of CF3OCH(CF3)2 and CF3OCF2CF2H.
 ) and reaction Gibbs free energies (
) and reaction Gibbs free energies ( ) for reactions 5 and 6 at the B3LYP/6-311++G(d,p), and CCSD(T)//B3LYP/6-311++G(d,p) levels (kcal/mol) and the calculated bonds dissociation energy (
) for reactions 5 and 6 at the B3LYP/6-311++G(d,p), and CCSD(T)//B3LYP/6-311++G(d,p) levels (kcal/mol) and the calculated bonds dissociation energy ( ) of the C–H in molecules CF3OCH(CF3)2 and CF3OCF2CF2H (a,b).
) of the C–H in molecules CF3OCH(CF3)2 and CF3OCF2CF2H (a,b). ) (in kcal/mol) for the species CF3OCH(CF3)2, CF3OC(CF3)2 CF3OCF2CF2H(a,b), and CF3OCF2CF2(a,b) at the B3LYP/6-311++G(d,p) and CCSD(T)//B3LYP/6-311++G(d,p) levels.
) (in kcal/mol) for the species CF3OCH(CF3)2, CF3OC(CF3)2 CF3OCF2CF2H(a,b), and CF3OCF2CF2(a,b) at the B3LYP/6-311++G(d,p) and CCSD(T)//B3LYP/6-311++G(d,p) levels.Reaction path properties
Figure S3a–d show the changes of the bond lengths along the MEP as functions of s (amu)1/2bohr. For reactions 5, 6, 7, and 8, the changes are very similar, that is, the breaking bond (C–H) and the forming bond (O–H or Cl–H) change strongly, while the other bonds are few changes. Figure S4a–d depict the classical potential energy curve (VMEP), ground-state vibrational adiabatic energy curve ( ), and zero-point energy curve (ZPE) as functions of s (amu)1/2bohr for the reactions 5, 6, 7, and 8, respectively. It can be seen that in Figure S4a–d, the plots of the
), and zero-point energy curve (ZPE) as functions of s (amu)1/2bohr for the reactions 5, 6, 7, and 8, respectively. It can be seen that in Figure S4a–d, the plots of the  and VMEP are similar in shape and locations, the curves of
and VMEP are similar in shape and locations, the curves of  and VMEP have the same zenith, indicating that the variational effect will be small in the calculation of the rate constant for the reactions 5, 6, 7, and 8. This will be borne out in the following study. The above conclusion about potential energy curves is similar with the previous theoretical work about the reactions of HFEs with OH radicals or Cl atoms8,10,12,13,14,15,16. The following study of the rate constants will also support this view deeply.
and VMEP have the same zenith, indicating that the variational effect will be small in the calculation of the rate constant for the reactions 5, 6, 7, and 8. This will be borne out in the following study. The above conclusion about potential energy curves is similar with the previous theoretical work about the reactions of HFEs with OH radicals or Cl atoms8,10,12,13,14,15,16. The following study of the rate constants will also support this view deeply.
The variations of the generalized normal-mode vibrational frequencies along with the MEPs of reactions 5, 6, 7, and 8 are shown in Figure S5a–d, respectively. Figure S5a shows that in the reactant region at about s = −2.0 (amu)1/2bohr, the frequencies are associated with those of reactant complex; And in the product region at about 1.5 (amu)1/2bohr, the frequencies are associated with the product complex. It is obvious that the mode 1 in Figure S5a–d, to connect the C–H stretching vibration mode of CF3OCH(CF3)2 and CF3OCF2CF2H with the O–H (Cl–H) stretching vibration mode of H2O (HCl), drops significantly near about the saddle point. The dramatical changes are used to call “reaction mode”, which is known to be the typical behavior of H-transfer reactions.
Kinetic rate constant calculations
Dual-level dynamics calculations are performed at the CCSD(T)//B3LYP/6-311++G(d,p) level to compute the reaction rate constants via the following reactions of 5–10. The CVT rate computations with the small-curvature tunneling (SCT) contributions are carried out for the six H-abstraction reactions in a wide temperature range from 230 to 1500 K. The TST, CVT, CVT/SCT rate constants of the reactions 5 (k1), 6 (k2), 7 (k3), and 8 (k4) are plotted in Figure S6a–d. The CVT/SCT rate constants of k1–k6 (for reactions 5–10), k1T–k4T, and the corresponding total rate constant kT(1)–kT(4), along with the experimental values are depicted in Fig. 2a–d. Because of only one H-abstraction reaction channel for reaction of (CF3)2CHOCF3 + OH/Cl, the total rate constants kT(1) = k1, kT(2) = k2. The total rate constants kT(3) (for the reaction of CF3OCF2CF2H with OH radical) and kT(4) (for the reaction of CF3OCF2CF2H with Cl atom) can be obtained from the following expressions:



The ω1 and ω2 are the temperature dependent weight factors calculated from the Boltzmann population distribution for each conformer of CF3OCF2CF2H. The knT (n = 1–4) is corrected rate constant by weight factors for each reaction of CF3OCF2CF2H conformer with OH radicals or Cl atoms. From the Figure S6a–d, we can find that the variational effects, defined as the ratios of the TST and CVT rate constants, can be ignored over the whole temperature range of 230–1500 K for reactions 5, 6, 7, and 8. On the other hand, it is found that the tunneling effects, defined as the ratios of CVT/SCT and CVT rate constants, play an important role at the low temperatures for reactions 5, 6, 7, and 8. The tunneling effects should be taken into account at the temperature of 230–375 K for reaction 5, 230–375 K for reaction 7, and 230–1000 K for reaction 8. Taking reaction 8 as an example, the specific values of CVT/SCT and CVT rate constants are 152.29, 32.35, 4.77, and 1.61 at 230, 296, 500, and 1000 K, respectively. In the theoretical study on reactions of (CF3)2CFOCH3, CF3CHFOCF3, CHF2OCHFCF312,14,15 with OH radicals and Cl atoms, similar tunneling effects can be found. Figure S6b shows that the CVT/SCT and CVT rate constants almost superposed on each other, implying that the tunneling contribution can be ignored for reaction 6.
Seen from Fig. 2a–d, for reactions of CF3OCH(CF3)2 + OH/Cl, the theoretical rate constants 1.88 × 10−16 and 9.17 × 10−19 cm3 molecule−1 s−1 are in good line with the corresponding experimental data 3.26 × 10−16 and 1.27 × 10−18 cm3 molecule−1 s−1 at 296 K20, respectively. It is pleasing to observe that the calculated rate constant, 2.11 × 10−15 cm3 molecule−1 s−1 of reaction CF3OCF2CF2H with OH radical at 296 ± 2 K, is in commendable accord with the experimental values of (2.26 ± 0.18) × 10−15 by Andersen et al.20. and 2.08 × 10−15 cm3 molecule−1 s−1 by Wilson et al.25. For reaction of CF3OCF2CF2H + Cl, theoretical overall rate constant (4.02 × 10−16 cm3 molecule−1 s−1) is in good agreement with the experimental value (2.70 × 10−16 cm3 molecule−1 s−1). The above good accordance between the experimental and theoretical result predicts the dynamic information is very credible. The positive temperature effect about the rate constants can be found for the reactions 5–10. This is not only the feature of the present reaction systems but also has been found in other similar reactions with OH radicals and Cl atoms by Devi et al.9. and other authors8,10,11,12,13,14,15,16.
The contributions ratios
Figure 3a and b show the contributions of CF3OCF2CF2H (a) and CF3OCF2CF2H (b) to the reactions CF3OCF2CF2H + OH and CF3OCF2CF2H + Cl versus 1000/T between 230 and 1500 K, respectively. It can be seen from Fig. 3a that the contribution of CF3OCF2CF2H (a) to the overall reaction is less important than that of CF3OCF2CF2H (b) in the whole temperature range, and the contribution of CF3OCF2CF2H (b) increases with the increasing of the temperature. Figure 3b suggests that below 365 K, the contribution of CF3OCF2CF2H (b) to the overall reaction is more important than that of CF3OCF2CF2H (a). For example, the values of k4T/kT(4) are 0.81 at 230 K and 0.55 at 350 K, respectively. However, as the temperature increases, CF3OCF2CF2H (a) prevails over CF3OCF2CF2H (b) and then becomes the major one, and the ratios of k3T/kT(4) are 0.54 at 400 K and k3T/kT(4) = 0.91 at 1500 K.

The contribution ratios as a function of 1000/T for reactions of CF3OCF2CF2H (a,b) + OH (a) and CF3OCF2CF2H (a,b) + Cl (b).
Kinetic isotope effects (KIEs)
The kinetic isotope effects (KIEs), defined as the ratios of between kH and kD for the reactions of CF3OCH(CF3)2/CF3OCF2CF2H + OD (R*) and CF3OCD(CF3)2/CF3OCF2CF2D + OH/Cl (R’) have been investigated over the whole temperature range. The calculated KIEs are displayed in the Fig. 4a and b. Figure 4a shows that the KIEs are smaller than 1 for reaction of (CF3)2CHOCF3 + OD, this is, in this reaction a noticeable inverse (kH/kD < 1) exists. And the kT(1)/kT(1)* values increase with the increasing of temperature and the KIEs become negligible when the temperature is high. However, the KIEs of kT(3)/kT(3)* is larger than 1 (kH/kD > 1) at the whole temperature range. The reaction of CF3OCD(CF3)2 + OH (k1’ = kT(1)’) comes to normal KIEs and the KIEs values decrease with increasing of temperature. The KIEs for k2’ (kT(2)’) are smaller than 1, and a noticeable inverse can be found. For the reaction of CF3OCF2CF2D + OH (kT(3)’), the values of KIEs are within 1.05–1.31 (around 1.0) in the whole temperature, indicating that the KIEs can be ignored. Note the KIEs of reaction of CF3OCF2CF2D + Cl (kT(4)’) need to consider because the values of KIEs are around to be 12. Comparing the KIEs of k’ with k*, we find that the results of KIEs for reactions of CF3OCH(CF3)2/CF3OCF2CF2H + OD (R*) and CF3OCD(CF3)2/CF3OCF2CF2D + OH (R’) are different. Taking a comparison between the reactions CF3OCH(CF3)2 + OD and CF3OCD(CF3)2 + OH, it is seen from Fig. 4a and b that the KIEs are different in temperature dependencies and a noticeable inverse or normal. Above isotope effects have also been found in many reactions17,27,28,32,33,34. Up to now, there is no corresponding experimental value, thus, our theoretical prediction can provide helpful information for further experimental study.

The ratios of kT(1)/k T(1)*, kT(3)/k T(3)* in (a), and kT(1)/k T(1)’ kT(2)/kT(2)’, kT(3)/k T(3)’, kT(4)/kT(4) ’ in (b).
Subsequent oxidation and decomposition pathways
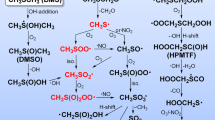
The mechanism and oxidation pathways of CF3OCH(CF3)2 and CF3OCF2CF2H to form the COF2 and CF3C(O)OCF3 is displayed in Fig. 5. From Fig. 5, the formed peroxy radicals (CF3OC(OO•)(CF3)2 and CF3OCF2CF2OO•) react with NO to form the organic nitrate and alkoxy radicals35. These processes are further refined and the geometries optimized at the B3LYP/6-311++G(d,p) level are listed in Figure S7. It is shown that the peroxynitrites were produced firstly and then it isomerizes to form organic nitrate or degrade to alkoxy radical and NO2. It can be seen from Figure S7a, CF3OC(OO•)(CF3)2 reacts with NO molecule to form IM1 (t-CF3OC(OONO)(CF3)2) and IM2 (c-CF3OC(OONO)(CF3)2) barrierlessly firstly. The IM1 can isomerize to IM2 via TSIM1-2 by overcoming 7.13 kcal/mol energy barrier. Then, IM1 can decompose into PIM1 (CF3OC(O•)(CF3)2 and NO2) via TSIM1-dec. The energy barrier for IM1 → CF3OC(O•)(CF3)2 + NO2 is 21.22 kcal/mol. Meanwhile, the IM2 can isomerize to IM3 (CF3OC(ONO2)(CF3)2) via TSIM2-iso by conquering a barrier of 7.01 kcal/mol. For the reaction of CF3OCF2CF2OO• with NO, we can find that the IM4 (t-CF3OCF2CF2OONO) and IM5 (c-CF3OCF2CF2OONO) are firstly formed with the relative energies of −23.07 and −24.25 kcal/mol, respectively. IM4 can also transform into IM5 through TSIM4–5 with an energy barrier of 9.26 kcal/mol. Afterwards, IM4 can dissociate into PIM4 (CF3OCF2CF2O• and NO2) through TSIM4-dec. The energy barrier for this dissociation process is 30.47 kcal/mol. However, the energy barrier for the process IM5 → TSIM5-iso → IM6 (CF3OCF2CF2ONO2) is 15.09 kcal/mol. Furthermore, the rate constant of IM1 decomposition is 2.75 × 10−4 s−1 at 298 K and ~10 order of magnitudes times smaller than that of isomerization pathway of IM2. In addition, the rate constant for IM4 dissociation is calculated to be 1.06 × 10−9 s−1 at 298 K, which is ~11 order of magnitudes times smaller than that of isomerization pathway of IM5. Therefore, we can get that the organic nitrate is more favorable to be produced than alkoxy radical. This result is in agreement with the investigation of RO2 reaction with NO (R=H, CH3, C2H5, n-C3H7, i-C3H7, 2-C5H11, and C6H5CHCH2OH) by Donahue et al.36 and An et al.37. The formed organic nitrates are relatively stable and thus the subsequent reactions from them will not be continued to discuss. We will pay our attention to the atmospheric behaviors of the alkoxy radicals. Atmospheric implications of organic nitrates will be discussed later.

Atmospheric oxidation mechanism for CF3OCH(CF3)2 and CF3OCF2CF2H.
The nature of the reaction mechanism for the unimolecular decomposition of CF3OC(O•)(CF3)2 and CF3OCF2CF2O• is carried out at the CCSD(T)//B3LYP/6-311++G(d,p) level of theory. For CF3OC(O•)(CF3)2, two possible channels are found in the subsequent reactions involved bond dissociation (C–C and C–O). Moreover, three possible processes included C–C/C–F bond dissociation and oxidation processes are listed as follows:





The optimized geometries and structural parameters of reactant and transition states for reactions 11–15 at the B3LYP level are depicted in Figure S8 in the Supporting Information. The thermodynamic data calculated at the CCSD(T)/6-311++G(d,p) and B3LYP/6-311++G(d,p) levels are summarized in Table 3 and the corresponding schematic potential energy surfaces are shown in Fig. 6. Transition vectors for all the transition states (TS1-CC, TS1-CO, TS2-CC, TS2-F, and TS2-O2) are obtained at 395i, 265i, 573i, 329i, and 414i cm−1, respectively, as recorded in Table S6 in the Supporting Information.
 ), enthalpies (
), enthalpies ( ), and Gibbs free energies (
), and Gibbs free energies ( ) for reactions involved in thermal decomposition of the alkoxy radicals at B3LYP/6-311++G(d,p) and CCSD(T)//B3LYP/6-311++G(d,p) levels of theory.
) for reactions involved in thermal decomposition of the alkoxy radicals at B3LYP/6-311++G(d,p) and CCSD(T)//B3LYP/6-311++G(d,p) levels of theory.
Potential energy profile (in kcal/mol) for the decomposition and oxidation pathways of CF3OC(O•)(CF3)2 and CF3OCF2CF2O• radicals at the CCSD(T)//B3LYP/6-311++G(d,p) level.
For reaction 11, the CF3OC(O•)(CF3)2 can proceed via TS1-CC to produce the hydrofluorinated esters of CF3C(O)OCF3 and CF3• radical by the C-C bond breakage. This process is exothermic ( = −0.74 kcal/mol). From Figure S7, we can see that the C–O bond in TS1-CO will be broken. Table 3 shows that the process of bond breaking of C–O (reaction 12) bond is endothermic (
= −0.74 kcal/mol). From Figure S7, we can see that the C–O bond in TS1-CO will be broken. Table 3 shows that the process of bond breaking of C–O (reaction 12) bond is endothermic ( = 15.22 kcal/mol). Figure 6 shows the barrier heights of reactions 11 and 12 are 9.41 and 36.45 kcal/mol, respectively. This shows that the C–C bond fracture is more favorable than C–O bond fracture. These results are in good line with the experimental research20.
= 15.22 kcal/mol). Figure 6 shows the barrier heights of reactions 11 and 12 are 9.41 and 36.45 kcal/mol, respectively. This shows that the C–C bond fracture is more favorable than C–O bond fracture. These results are in good line with the experimental research20.
The CF3OCF2CF2O• can be dissociated into CF3OCF2• radical and COF2 by TS2-CC (reaction 13) with the C–C bond fracture. It is easy from Table 3 the C–C bond-cleavage pathway is significantly exothermic thus thermodynamically more facile. This process is accompanied by the energy barrier of 8.25 kcal/mol at the CCSD(T)//B3LYP/6-311++G(d,p) level. The transition state TS2-F corresponding to the C–F bond scission of CF3OCF2CF2O• (reaction 14) leading to F atom and CF3OCF2CFO is accompanied by an absorption of heat of 21.36 kcal/mol and thus is thermodynamically less probable. Table 3 and Fig. 6 suggest that the barriers of TS2-O2 are significantly higher than those of TS2-CC and TS2-F. Based on barrier heights, it may be concluded that thermal decomposition pathway of C–F bond (reaction 14) and oxidative pathway (reaction 15) not seem to vie with thermal decomposition pathway of C–C bond (reaction 13). In the end, ester CF3C(O)OCF3 may be the primary product for the subsequent decomposition pathways of CF3OC(O•)(CF3)2 radical. It can form CF3C(O)OH by hydrolysis38 and thus may contribute to the environmental burden of TFA. We can also summarize that the subsequent decomposition pathways of CF3OCF2CF2O• radical to produce CF3OCF2• radical and COF2. COF2 play an important role in producing CO2 and HF in the atmosphere and have potential to cause health or environmental problems. The above results are in good line with the experimental findings by Andersen et al.20.
As reported by Finlayson-Pitts et al.39, the reaction of alkoxy radical with NO2 is slow in the gas phase. Even at 100 ppb of NO2, the reaction rate constant for 3-alkoxy hexane with NO2 (75 s−1) cannot vie with its decomposition (~3 × 104 s−1). Furthermore, the rate constants for the major decomposition pathways CF3OC(O•)(CF3)2 (reaction 11) and CF3OCF2CF2O• (reaction 13) are computed to be 3.0 × 109 s−1 and 1.1 × 1012 s−1 at 298 K, respectively. The large decomposition rates may show that the decomposition reactions will precede their reactions with NO2 to form the corresponding nitrates. Thus, we predict that the alkoxy radicals of CF3OC(O•)(CF3)2 and CF3OCF2CF2O• may be unlikely reaction with NO2 to form CF3OC(ONO2)(CF3)2 and CF3OCF2CF2ONO2, rather than dissociate of themselves.
Atmospheric Implications of CF3OCH(CF3)2 and CF3OCF2CF2H
The tropospheric lifetimes (τ) of CF3OCH(CF3)2 and CF3OCF2CF2H can be estimated by their removal reactions with OH radicals or Cl atoms. The atmospheric lifetimes can be shown as:



In the consideration of good agreement between the theoretically calculated and experimentally measured rate constants, we adopted the computed rate constants to estimate the lifetimes. Using the global atmospheric OH and Cl concentrations of 1 × 106 and 1 × 103 molecule cm−3 5,7, the lifetimes of CF3OCH(CF3)2 and CF3OCF2CF2H are estimated to be 168 and 15 years at 296 K. The estimated lifetimes are respectively in good agreement with the experimental values (estimated based on the experimental rate constants) 117.0 and 16.9 years (18.8 years)40,41. These relatively long times suggest that in the atmosphere CF3OCH(CF3)2 and CF3OCF2CF2H are environmentally unfriendly.
In order to further assess their atmospheric implications, the global warming potentials (GWPs)42,43 to carbon dioxide of CF3OCH(CF3)2 and CF3OCF2CF2H are computed at different time horizons of 20, 100, and 500 years. We calculated the GWPs for CF3OCH(CF3)2 and CF3OCF2CF2H by the following formula:

Where (Ak) is the intensities of the corresponding vibrational mode k.  is the radiative forcing function per unit cross section per wave number (W m−2 (cm−1)−1 (cm2 molecule−1)−1) evaluated at the scaled band center frequency (
is the radiative forcing function per unit cross section per wave number (W m−2 (cm−1)−1 (cm2 molecule−1)−1) evaluated at the scaled band center frequency ( )43 TH is the time horizon.
)43 TH is the time horizon.  is the absolute global warming potential for carbon dioxide44. The radiative efficiencies (
is the absolute global warming potential for carbon dioxide44. The radiative efficiencies ( ) are 0.632 and 0.635 W m−2 ppb−1 for CF3OCH(CF3)2 and CF3OCF2CF2H, respectively, by using the method proposed by Pinnock et al.43. The underlying data of frequencies, IR intensities, and radiative forcing used in calculation of GWP of CF3OCH(CF3)2 and CF3OCF2CF2H are presented in Table S7 and S8, respectively. For compounds that are not well-mixed in the atmosphere, the correction factors, f(τ), based on the lifetime of the compounds are needed. The corresponding expression can be shown as in the Hodnebrog et al.’s work45. The f(τ) values for CF3OCH(CF3)2 and CF3OCF2CF2H are calculated to be 0.991 and 0.966, respectively. Herein, the lifetimes corrected radiative efficiencies are 0.626 W m−2 ppb−1 CF3OCH(CF3)2 and 0.613 W m−2 ppb−1 for CF3OCF2CF2H. The corresponding GWPs values are displayed in Table 4. We can see for Table 4 that the GWPs of CF3OCH(CF3)2 and CF3OCF2CF2H at 20 years of time horizon are 9371 and 6877, respectively. These large values show that the contribution of two HFEs to GWP will be significant.
) are 0.632 and 0.635 W m−2 ppb−1 for CF3OCH(CF3)2 and CF3OCF2CF2H, respectively, by using the method proposed by Pinnock et al.43. The underlying data of frequencies, IR intensities, and radiative forcing used in calculation of GWP of CF3OCH(CF3)2 and CF3OCF2CF2H are presented in Table S7 and S8, respectively. For compounds that are not well-mixed in the atmosphere, the correction factors, f(τ), based on the lifetime of the compounds are needed. The corresponding expression can be shown as in the Hodnebrog et al.’s work45. The f(τ) values for CF3OCH(CF3)2 and CF3OCF2CF2H are calculated to be 0.991 and 0.966, respectively. Herein, the lifetimes corrected radiative efficiencies are 0.626 W m−2 ppb−1 CF3OCH(CF3)2 and 0.613 W m−2 ppb−1 for CF3OCF2CF2H. The corresponding GWPs values are displayed in Table 4. We can see for Table 4 that the GWPs of CF3OCH(CF3)2 and CF3OCF2CF2H at 20 years of time horizon are 9371 and 6877, respectively. These large values show that the contribution of two HFEs to GWP will be significant.
With regard to the atmospheric implications of CF3OCH(CF3)2 and CF3OCF2CF2H, the following statements can be taken into account: (1) CF3OCH(CF3)2 and CF3OCF2CF2H will not contribute to stratospheric ozone depletion. (2) It is noteworthy that the atmospheric lifetimes of CF3OCH(CF3)2 and CF3OCF2CF2H are approximately 168 and 15 years, respectively, and consequently they have remarkable GWP (see above). (3) The atmospheric oxidation of CF3OCH(CF3)2 and CF3OCF2CF2H gives organic nitrates (CF3ONO2, CF3OCF2NO2, CF3OCF2CF2NO2, (CF3)2C(ONO2)OCF3), CF3OCOF, CF3COOCF3, COF2, and CF3COCF3. The organic nitrates are hygroscopic, and thus they influence the climate by serving as cloud condensation nuclei and components of aerosol organic matter. COF2 is removed from the troposphere via contact with water surfaces and hydrolysis to HF and CO2 in atmosphere. The CF3COF, CF3COOCF3, and CF3COCF3 can be hydrolysis to produce TFA and HF. All in all, the contributing oxidation products may increase the acidity of precipitation and consequently they have negative impact on the air quality.
The three-parameter Arrhennius fits based on the calculated rate constants (in units of cm3 molecule−1 s−1) at the CCSD(T)//B3LYP/6-311++G(d,p) level within 230–350 K for the main emphasis is on atmospheric applications give expressions as follows:




Discussion and Summary
DFT methods are adopted to investigate kinetic contents of reactions of CF3OCH(CF3)2 and CF3OCF2CF2H with OH radical and Cl atom. The high-level energies for the stationary points are refined at the CCSD(T)//B3LYP/6-311++G(d,p) level. All the H-abstraction reactions rate constants are calculated by CVT combined with SCT correction. The agreement between theoretical and experiment result is very excellent. In the temperature range of 230–1500 K, the variational effect is almost not existing or almost negligible. The tunneling effect is significant in the whole lower temperatures for reactions 5, 7, and 8. All the rate constants have positive temperature effects. Calculated results show that two conformers of CF3OCF2CF2H are both important for total rate constant of reactions of CF3OCF2CF2H + OH/Cl.
In addition, kinetic isotope effects for the reactions CF3OCH(CF3)2/CF3OCF2CF2H + OD (R*) and CF3OCD(CF3)2/CF3OCF2CF2D + OH/Cl (R’) have been investigated. The atmospheric lifetimes of (CF3)2CHOCF3 and CF3OCF2CF2H are estimated to be 168 and 15 years, respectively. The GWP(20 years) values are calculated to be 9371 and 6877, respectively, which show that their contributions to the global warming are significant and should be given more concerns. The peroxy radicals of CF3OC(OO•)(CF3)2 and CF3OCF2CF2OO• formed by the alkyl radicals with O2 can produce the CF3OC(ONO2)(CF3)2 and CF3OCF2CF2ONO2 in the NO-rich environment. The CF3OC(ONO2)(CF3)2 and CF3OCF2CF2ONO2 are thermally stable nitrate and thus will play a very important role in SOA formation. The subsequent reaction pathways of CF3OC(O•)(CF3)2 and CF3OCF2CF2O• radicals in the gas phase are also investigated in detail. The primary degradation channels of CF3OC(O•)(CF3)2 and CF3OCF2CF2O• radicals are the C–C bond cleavage to form the COF2 and CF3C(O)OCF3, respectively, which is in consistent with the experimental findings. The high toxic excitant gas COF2 can be hydrolysis into CO2 and HF which can increase the acidity of precipitation. The CF3C(O)OCF3 can be transformed into highly soluble compounds TFA and HF which may be rapidly incorporated into cloud droplets, contributing to the acidity of precipitation. The standard enthalpies of formation for the species are also calculated. The three-parameter Arrhenius expressions for the title reactions are k T(1) = 5.00 × 10−24T3.57 exp(−849.73/T), k T(2) = 1.79 × 10−24T4.84 exp(−4262.65/T), kT(3) = 1.94 × 10−24 T4.18 exp(−884.26/T), and k T(4) = 9.44 × 10−28T5.25 exp(−913.45/T) cm3 molecule−1 s−1. We hope our theoretical studies may provide a good estimate for further laboratory understanding the relevant chemical reactions.
Methodology
Electronic structure calculations
The equilibrium geometries of the reactants, products, complexes, and transition states are optimized at the B3LYP46,47,48 of theory conjunct with the standard 6-311++G(d,p) basis set using the Gaussian 09 package49. The B3LYP method is more widely used for predicting the reliable geometries of the stationary points, and found to be accurate from the previous studies12,15,16,50,51,52,53,54,55. Moreover, B3LYP theory is able to suppress effectively the problem of spin contamination. All the transition states are verified by the intrinsic reaction coordinate (IRC) calculations to connect the corresponding reactants to products at the B3LYP/6-311++G(d,p) level. Geometries of stationary points are also optimized at the M06-2X/6-311++G(d,p)56 level to ensure the accuracy of data. In order to obtain more reliable relative energy, single points energy calculations have been performed by the method of CCSD(T)/6-311++G(d,p)57,58. The differences of energies for these transition states between the CCSD(T)//B3LYP/6-311++G(d,p) and CCSD(T)//M06-2X/6-311++G(d,p) levels are within 0.07–0.49 kcal/mol (See Table S1), suggesting that the B3LYP level is reasonable and applicable in the current system. As also reported in the previous work for the HFEs with radicals11, the single points energies obtained at the CCSD(T) level based on the M06-2X geometries are very close to the results based on the B3LYP geometries (within the largest difference of 0.40 kcal/mol), thus, we just discuss the single points energy at the CCSD(T)//B3LYP/6-311++G(d,p) level in this paper. The T1 values of all species are checked to guarantee that CCSD(T) wave function is not affected by multi-reference character.
Thermodynamics and Kinetics
The enthalpies of formation ( ) are the important thermodynamic parameters which can be estimated by using isodesmic reactions. Herein, we predict the values of
) are the important thermodynamic parameters which can be estimated by using isodesmic reactions. Herein, we predict the values of  for CF3OCH(CF3)2, CF3OC(CF3)2, CF3OCF2CF2H (a, b), and CF3OCF2CF2 (a, b) by using the following reactions:
for CF3OCH(CF3)2, CF3OC(CF3)2, CF3OCF2CF2H (a, b), and CF3OCF2CF2 (a, b) by using the following reactions:












The kinetic rate constants, over the temperature range of 230–1500 K, are computed using the canonical variational transition state theory (CVT)59,60 involving small curvature tunneling correction (SCT)61,62 through the POLYRATE-Version 9.7 computer program63. Dynamic computations are performed using the values obtained from the CCSD(T)//B3LYP/6-311++G(d,p) level to make a direct comparison to the previous investigations9,11,12,15,16. The relevant expressions are given as:


In above expressions, s is the location of the generalized transition state on the IRC; σ is the symmetry factor; β equals (kBT)−1 where kB is Boltzmann’s constant; h is Planck’s constant; The κ is the tunneling factor; and QGT and QR are partition functions for the generalized transition state and reactants, respectively. In the calculations of the electronic partition functions, two electronic states for OH radical, with a 140 cm−1 splitting in the 2Π1/2 and 2Π3/2 ground states, are included. The two low-lying electronic states, 2P1/2 and 2P3/2, with a splitting of 881 cm−1 of Cl atom, are also included. For the title reactions of 5 and 7–10, there are weak hydrogen-bonded complexes formed at the entrance channels. Hence, the stepwise mechanism occurs, that is, the first reversible step leads to the formation of reactant complex, and the second irreversible step yields the corresponding products. For this case, we use “well” option to think about the contributions of reactant complex in the Polyrate suite. This method has been applied successfully to describe the reactions of the free radical with several volatile organic chemicals14,15,16,64,65,66. Detailed discussions can be shown in Alvarez-Idaboy et al.’s works64,65,66.
Additional Information
How to cite this article: Bai, F.-Y. et al. Theoretical insight into OH- and Cl-initiated oxidation of CF3OCH(CF3)2 and CF3OCF2CF2H & fate of CF3OC(X•)(CF3)2 and CF3OCF2CF2X• radicals (X=O, O2). Sci. Rep. 7, 40264; doi: 10.1038/srep40264 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Tsai, W. T. Environmental Risk Assessment of Hydrofluoroethers (HFEs). J. Hazard. Mater. 119, 69–78 (2005).
Ravishankara, A. R. et al. Do Hydrofluorocarbons Destroy Stratospheric Ozone? Science 263, 71–75 (1994).
Hanmmitt, J. K. et al. Future Emission Scenarios for Chemicals that May Deplete Stratospheric Ozone. Nature 330, 711–716 (1987).
Bera, P. P., Francisco, J. S. & Lee, T. J. Identifying the Molecular Origin of Global Warming. J. Phys. Chem. A 113, 12694–12699 (2009).
Kurylo, M. J. & Orkin, V. L. Determination of Atmospheric Lifetimes via the Measurement of OH Radical Kinetics. Chem. Rev. 103, 5049–5076 (2003).
Spicer, C. W. et al. Unexpectedly High Concentrations of Molecular Chlorine in Coastal Air. Nature 394, 353–356 (1998).
Wingenter, O. W. et al. Tropospheric Hydroxyl and Atomic Chlorine Concentrations, and Mixing Timescales Determined from Hydrocarbon and Halocarbon Measurements Made over the Southern Ocean. J. Geophys. Res. 104, 21819–21828 (1999).
Blanco, M. B., Bejan, I., Barnes, I., Wiesen, P. & Teruel, M. A. Atmospheric Photooxidation of Fluoroacetates As a Source of Fluorocarboxylic Acids. Environ. Sci. Technol. 44, 2354–2359 (2010).
Song, G. C. et al. Theoretical Studies on the Mechanisms and Dynamics of OH Radicals with CH2FCF2OCHF2 and CH2FOCH2F. J. Phys. Chem. A 114, 9057–9068 (2010).
Jogeshwari Devi, K. & Chandra, A. K. Theoretical Investigation of the Gas-Phase Reactions of (CF3)2CHOCH3 with OH Radical. Chem. Phys. Lett. 502, 23–28 (2011).
Bai, F. Y. et al. Theoretical Investigation of the Mechanisms and Dynamics of the Reaction CHF2OCF2CHFCl + Cl. J. Mol. Model. 20, 2419 (2014).
Bai, F. Y. et al. Theoretical Study on the Reactions of (CF3)2CFOCH3 + OH/Cl and Reaction of (CF3)2CFOCHO with Cl Atom. J. Phys. Chem. A 119, 1256–1266 (2015).
Sun, H. et al. Theoretical Investigation of the Reaction of CF3CHFOCH3 with OH Radical. J. Phys. Chem. A 113, 5951–5957 (2009).
Jia, X. J. et al. Theoretical Investigation of the Reactions of CF3CHFOCF3 with the OH Radical and Cl Atom. J. Phys. Chem. A 114, 417–424 (2010).
Yang, L., Liu, J. Y., Wan, S. Q. & Li, Z. S. Theoretical Studies of the Reactions of CF3CHClOCHF2/CF3CHFOCHF2 with OH Radical and Cl Atom and Their Product Radicals with OH. J. Comput. Chem. 30, 565–580 (2009).
Yang, L., Liu, J. Y. & Li, Z. S. Computational Studies on the Mechanisms and Dynamics of OH Reactions with CHF2CHFOCF3 and CHF2CH2OCF3 . J. Chem. Theor. Comput. 4, 1073–1082 (2008).
Bai, F. Y., Wang X., Sun, Y. Q., Wang, R. S. & Pan, X. M. Atmospheric Chemistry of Ethers, Esters, and Alcohols on the Lifetimes, Temperature Dependence, Kinetic Isotope Effect: As Example of CF3CX2CX2CX2OX with OX Reactions (X=H, D). RSC Adv. 6, 36096–36108 (2016).
Orkin, V. L., Khamaganov, V. G. & Guschin, A. G. Photochemical Properties of Hydrofluoroethers CH3OCHF2, CH3OCF3, and CHF2OCH2CF3: Reactivity Toward OH, IR Absorption Cross Sections, Atmospheric Lifetimes, and Global Warming Potentials. J. Phys. Chem. A 118, 10770–10777 (2014).
Ninomiya, Y. et al. Reaction with OH Radicals and Cl Atoms and Atmospheric Fate of n-C3F7OCH2O(·) Radicals. Environ. Sci. Technol. 34, 2973–2978 (2000).
Andersen, M. P. S., Nielsen, O. J., Wallington, T. J., Hurley, M. D. & DeMore, W. B. J. Phys. Chem. A 109, 3926–3934 (2005).
Andersen, L. L., Østerstrøm, F. F., Nielsen, O. J., Andersen, M. P. S. & Wallington, T. J. Atmospheric chemistry of (CF3)2CFOCH3 . Chem. Phys. Lett. 607, 5–9 (2014).
Østerstrøm, F. F., Wallington, T. J., Sulbaek Andersen, M. P. & Nielsen, O. J. Atmospheric Chemistry of (CF3)2CHOCH3, (CF3)2CHOCHO, and CF3C(O)OCH3 . J. Phys. Chem. A 119, 10540–10552 (2015).
Sulbaek Andersen, M. P., Nielsen, O. J., Karpichev, B., Wallington, T. J. & Sander, S. P. Atmospheric Chemistry of Isoflurane, Desflurane, and Sevoflurane: Kinetics and Mechanisms of Reactions with Chlorine Atoms and OH Radicals and Global Warming Potentials. J. Phys. Chem. A 116, 5806–5820 (2012).
Stevens, J. E., Macomber, L. D. & Davis L. W. IR Spectra and Vibrational Modes of the Hydrofluoroethers CF3OCH3, CF3OCF2H, and CF3OCF2CF2H and Corresponding Alkanes CF3CH3, CF3CF2H, and CF3CF2CF2H. Open Phys. Chem. J. 4, 17–27 (2010).
Wilson, E. W. Jr, Hamilton, W. A., Mount, H. R. & DeMore, W. B. Rate Constants for the Reactions of Hydroxyl Radical with Several Fluoroethers by the Relative Rate Method. J. Phys. Chem. A 111, 1610–1617 (2007).
Singh, H. J., Rao, P. K. & Tiwari, L. Theoretical Studies on OH and Cl Initiated Hydrogen Atom Abstraction of HFE-227pc (CF3OCF2CHF2). J. Atmos. Chem. 70, 257–268 (2013).
Johnson, M. S., Feilberg, K. L., Hessberg, P. V. & Nielsen, O. J. Isotopic Processes in Atmospheric Chemistry. Chem. Soc. Rev. 31, 313–323 (2002).
Brenninkmeijer, C. A. et al. Isotope Effects in the Chemistry of Atmospheric Trace Compounds. Chem. Rev. 103, 5125–5161 (2003).
Lide, D. R. CRC Handbook of Chemistry and Physics, 95th ed. Ch. 9, 23–24, CRC Press, New York (1999).
Shimanouchi, T. Tables of Molecular Vibrational Frequencies Consolidated, Vol. 1, National Bureau of Standards: U. S. GPO, Washinton, D. C. (1972).
Chase, M. W. et al. JANAF, Vol. 14 (Supp. 1), American Chemical Society: Washington D.C. (1985).
Baeza-Romero, M. T. et al. A Combined Experimental and Theoretical Study of the Reaction Between Methylglyoxal and OH/OD Radical: OH Regeneration. Phys. Chem. Chem. Phys. 9, 4114–4128 (2007).
Wang, L., Zhao, Y., Wen, J. & Zhang, J. Mechanisms and Kinetics of Hydrogen Abstraction of Methylhydrazine and Deuterated Methylhydrazine with H/D Atoms. Theor. Chem. Acc. 132, 1–14 (2013).
Jaramillo, V. I. & Smith, M. A. Temperature-Dependent Kinetic Isotope Effects in the Gas-Phase Reaction: OH + HBr. J. Phys. Chem. A 105, 5854–5859 (2001).
Renbaum, L. H. & Smith, G. D. Organic Nitrate Formation in the Radical-Initiated Oxidation of Model Aerosol Particles in the Presence of NOx. Phys. Chem. Chem. Phys. 11, 8040–8047 (2009).
Zhang, J. Y., Dransfield, T. & Donahue, N. M. On the Mechanism for Nitrate Formation via the Peroxy Radical + NO Reaction. J. Phys. Chem. A 108, 9082–9095, (2004).
Wang, H. H., Ji, Y. M., Gao, Y. P., Li, G. Y. & An, T. C. Theoretical Model on the Formation Possibility of Secondary Organic aerosol from •OH initialed oxidation reaction of styrene in the presence of O2/NO. Atmos. Environ. 101, 1–9 (2015).
Wallington, T. J. et al. A. The Environmental Impact of CFC Replacements-HFCs and HCFCs. Environ. Sci. Technol. 28, 320–326 (1994).
Finlayson-Pitts, B. J. & Pitts, J. N. Jr. Chemistry of the Upper and Lower Atmosphere: Theory, Experiments, and Applications, Vol. 1 Ch. 6, 188-189 (2000).
Blowers, P., Moline, D. M., Tetrault, K. F., Wheeler, R. R. & Tuchawena, S. L. Global Warming Potentials of Hydrofluoroethers. Environ. Sci. Technol. 42, 1301–1307 (2008).
Urata, S., Takada, A., Uchimaru, T. & Chandra, A. K. Rate Constants Estimation for the Reaction of Hydrofluorocarbons and Hydrofluo-roethers with OH Radicals. Chem. Phys. Lett. 368, 215–223 (2003).
Blowers, P., Tetrault, K. F. & Trujillo-Morehead, Y. Global Warming Potential Predictions for Hydrofluoroethers. Theor. Chem. Acc. 119, 369–381 (2008).
Pinnock, S., Hurley, M. D., Shine, K. P., Wallington, T. J. & Smyth, T. J. Radiative Forcing of Climate by Hydrochlorofluor-ocarbons and Hydrofiuorocarbons. J. Geophys. Res. Atmos. 100, 23227–23238 (1995).
Wuebbles D. J. Weighing Functions for Ozone Depletion and Greenhouse Gas Effects on Climate. Annu. Rev. Energy Environ. 20, 45–70 (1995).
Hodnebrog. O. et al. Global Warming Potentials and Radiative efficiencies of Halocarbons and Related compounds: A Comprehensive Review. Rev. Geophys. 51, 300–378 (2013).
Becke, A. D. Density-functional thermochemistry. II. The Effect of the Perdew-Wang Generalized-Gradient Correlation Correction. J. Chem. Phys. 97, 9173–9177 (1992).
Becke, A. D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 98, 5648–5652 (1993).
Lee, C., Yang, W. & Parr, R. G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 37, 785–789 (1988).
Frisch, M. J. et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT (2009)
Shenghur, A., Weber, K. H., Nguyen, N. D., Sontising, W. & Tao, F. M. Theoretical Study of the Hydrogen Abstraction of Substituted Phenols by Nitrogen Dioxide as a Source of HONO. J. Phys. Chem. A, 118, 11002–11014 (2014).
Wang, L., Li, Y. J., He, H. Q. & Zhang, J. L. Hydrogen Abstraction Reactions of OH Radicals with CH3CH2CH2Cl and CH3CHClCH3: A Mechanistic and Kinetic Study. J. Comput. Chem. 33, 66–75 (2014).
Bai, F. Y. et al. Theoretical Studies of the Reactions CFxH3-xCOOR + Cl and CF3COOCH3 + OH (X=1–3; R=CH3,CH2CH3). Chemphyschem 16, 1768–1776 (2015).
Prager, J., Najm, H. N. & Zador, J. Uncertainty quantification in the ab initio rate-coefficient calculation for the CH3CH(OH)CH3 + OH → CH3C(OH)CH3 + H2O reaction. P. Combust. Inst. 34, 583–590 (2013).
Gao, Y. P., Ji, Y. M., Li, G. Y. & An, T. C. Mechanism, Kinetics and Toxicity Assessment of OH-initiated Transformation of Triclosan in Aquatic Environments. Water Res. 49, 360–370 (2014).
Dalmasso, P. R. et al. Reactivity of Hydrohaloethers with OH Radicals and Chlorine Atoms: Correlation with Molecular Properties. Atmos. Environ. 91, 104–109 (2014).
Zhao, Y. & Truhlar, D. G. Density Functionals with Broad Applicability in Chemistry. Acc. Chem. Res. 41, 157–167 (2008).
Raghavachari, K., Trucks, G. W., Pople, J. A. & Head-Gordon, M. A Fifth-Order Perturbation Comparison of Electron Correlation Theories. Chem. Phys. Lett. 157, 479–483 (1989).
Pople, J. A., Head-Gordon, M. & Raghavachari, K. Quadratic Configuration Interaction. A General Technique for Determining Electron Correlation Energies. J. Chem. Phys. 87, 5968–5975 (1989).
Garrett, B. C. & Truhlar, D. G. Criterion of Minimum State Density in the Transition State Theory of Bimolecular Reactions. J. Chem. Phys. 70, 1593–1598 (1979).
Garrett, B. C. & Truhlar, D. G. Generalized Transition State Theory. Bond Energy-Bond Order Method for Canonical Variational Calculations with Application to Hydrogen Atom Transfer Reactions. J. Am. Chem. Soc. 101, 4534–4548 (1979).
Lu, D. H. et al. A New Version of A Computer Program for the Calculation of Chemical Reaction Rates for Polyatomics. Comput. Phys. Commun. 71, 235–262 (1992).
Liu, Y. P. et al. Molecular Modeling of the Kinetic Isotope Effect for the [1,5]-Sigmatropic Rearrangement of cis-1,3-Pentadiene. J. Am. Chem. Soc. 115, 2408–2415 (1993).
Corchado, J. C. et al. POLYRATE, version 9.7; University of Minnesota: Minneapolis, MN (http://comp.chem.umn.edu/polyrate) (2007).
Alvarez-Idaboy, J. R., Reyes, L. & Mora-Diez, N. The Mechanism of the Baeyer-Villiger Rearrangement: Quantum Chemistry and TST Study Supported by Experimental Kinetic Data. Org. Biomol. Chem. 5, 3682–3689 (2007).
Alvarez-Idaboy, J. R., Mora-Diez, N. & Vivier-Bunge, A. A Quantum Chemical and Classical Transition Sate Theory Explanation of Negative Activation Energies in OH Addition to Substituted Ethenes. J. Am. Chem. Soc. 122, 3715–3720 (2000).
Alvarez-Idaboy, J. R., Mora-Diez, N., Boyd, R. J. & Vivier-Bunge, A. On the Importance of Prereactive Complexes in Molecule-Radical Reactions: Hydrogen Abstraction from Aldehydes by OH. J. Am. Chem. Soc. 123, 2018–2024 (2001).
Acknowledgements
This work is supported by National Natural Science Foundation of China (No. 21377021). We thank Professor Donald G. Truhlar for providing the Polyrate, Version 9.7 program.
Author information
Authors and Affiliations
Contributions
The conception and design of computations: X. Pan. The execution and analysis of calculations: F. Bai., X. Pan., and X. Jia. Paper writing: F. Bai., Y. Ma., S. Lv., X. Pan. and X. Jia. All authors have reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Bai, FY., Ma, Y., Lv, S. et al. Theoretical insight into OH- and Cl-initiated oxidation of CF3OCH(CF3)2 and CF3OCF2CF2H & fate of CF3OC(X•)(CF3)2 and CF3OCF2CF2X• radicals (X=O, O2). Sci Rep 7, 40264 (2017). https://doi.org/10.1038/srep40264
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep40264
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
