
Abstract
Cronobacter sakazakii is an important foodborne pathogens causing rare but life-threatening diseases in neonates and infants. CRISPR-Cas system is a new prokaryotic defense system that provides adaptive immunity against phages, latter play an vital role on the evolution and pathogenicity of host bacteria. In this study, we found that genome sizes of C. sakazakii strains had a significant positive correlation with total genome sizes of prophages. Prophages contributed to 16.57% of the genetic diversity (pan genome) of C. sakazakii, some of which maybe the potential virulence factors. Subtype I-E CRISPR-Cas system and five types of CRISPR arrays were found in the conserved site of C. sakazakii strains. CRISPR1 and CRISPR2 loci with high variable spacers were active and showed potential protection against phage attacks. The number of spacers from two active CRISPR loci in clinical strains was significant less than that of foodborne strains, it maybe a reason why clinical strains were found to have more prophages than foodborne strains. The frequently gain/loss of prophages and spacers in CRISPR loci is likely to drive the quick evolution of C. sakazakii. Our study provides a new insight into the co-evolution of phages and C. sakazakii.
Similar content being viewed by others


Introduction
Cronobacter sakazakii is an important foodborne pathogen associated with outbreaks of life-threatening necrotizing enterocolitis, meningitis, and sepsis in neonates and infants1,2. Although the incidence is low, the fatality rates of these diseases, caused by C. sakazakii infection, range between 40 to 80% and survivors are often left with severe neurological and developmental disorders3,4. Infections caused by Cronobacter spp. have been epidemiologically linked to the consumption of contaminated powdered infant form4. However, virulence genes and mechanism of pathogenicity of C. sakazakii remain unclear5.
As the most abundant biological form on the planet, phages constitute major players shaping bacterial communities in most ecosystems6, and they are closely associated with the virulence and evolution of several important bacterial pathogens7. External DNA acquired by horizontal gene transfer (HGT) is the main driving force in the evolution of bacterial genomes. Integrated phages (prophages), which represent a sizable fraction of bacterial chromosomes, are major contributors to differences among individuals within a bacterial species8. Although acquisition of phage DNA can increase the fitness of host bacteria under certain environmental conditions, the replication and maintenance of these nucleotide sequences could be a burden9. Overall, predation by phages presents a serious challenge to bacterial survival; hence, bacteria have evolved numerous mechanisms to resist phage infection9.
CRISPR-Cas system, comprised of clustered regularly interspaced short palindromic repeats (CRISPR) along with their associated (Cas) proteins, protects prokaryotic organisms from viral predation and invading nucleic acids10. A CRISPR array is composed of a cluster of identical short repeats separated by short variable DNA sequences of similar length (called ‘spacers’). Such sequences derived from phage or plasmid genomes that match the sequences of CRISPR spacers are called protospacers11. CRISPR–Cas system encompasses three distinct mechanistic stages: adaptation, expression and interference12,13,14,15,16. The adaptation stage involves the incorporation of new spacers deriving from foreign DNA (‘protospacers’) into the CRISPR array. These spacers preserve the sequence memory for a targeted defense against subsequent invasions by the corresponding phages or plasmid. The expression stage includes the transcription of CRISPR sequences and subsequent processing to produce CRISPR RNAs (crRNAs). During the interference stage, crRNAs, aided by Cas proteins, function as guides to specifically target and cleave the nucleic acids of cognate phages or plasmids10,11,15,16,17. CRISPR-Cas system is found in 48% of Eubacteria and 95% of Archaea and has been divided into two classes, five types, and 16 subtypes, based on the signature protein families and features of the architecture of cas loci10. Recent studies have reported that CRISPR-Cas systems could also be used for non-defense roles, such as regulation of collective behavior and pathogenicity11.
Previous studies have found that prophages and subtype I-E CRISPR-Cas system exist in the genomes of C. sakazakii strains18,19,20,21. However, the characteristic of integrated prophages and CRISPR-Cas system, and their impact on the evolution of C. sakazakii have not been reported. In this study, we combined 17 new whole genome sequences of C. sakazakii strains representing the major sequence types in China obtained from this study, with public whole genome sequences to explore the contribution of prophages and CRISPR-Cas system to the evolution of C. sakazakii. It is meaningful for further understanding of genetic diversity of C. sakazakii and provides a new insight to explore their pathogenicity in the future.
Results and Discussion
Impact of prophages on genetic diversity and pathogenicity of C. sakazakii
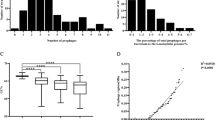
Prophages or prophage-derived elements were detected in all C. sakazakii strains. The average GC content of prophages (52.71%; ranging from 46.76% to 58.30%) was lower than that of C. sakazakii strains (57.04%; ranging from 56.62% to 57.70%). As shown in Fig. 1a, the number of integrated prophages in C. sakazakii strains ranged from 1 to 9, 83.78% of strains have less than 6 prophages. For there were only three environmental strains, we will focus on the comparison between clinical strains (n = 12) and foodborne strains (n = 21) in all of our analyses. Clinical strains had more prophages (mean: 4.15) than foodborne strains (mean: 2.81) with statistical significance (p < 0.05). The total genome sizes of prophages were 5.5–321.2 kb, contributing to 0.14–6.89% of C. sakazakii genome. As shown in Fig. 1b, the genome size of C. sakazakii increased with increase in total genome size of prophages, indicating a significant positive correlation. It implied that prophage is an important factor associated with the genome size of C. sakazakii. For the number of prophages in clinical strains was more than that of foodborne strains, the total genome size of prophages in clinical strains (mean: 125.79Kb) were also larger than that of foodborne strains (mean: 70.50Kb) with statistical significance (p < 0.05). Prophages can contribute important biological properties to their bacterial hosts, providing bacterial pathogens with virulence factors and accessory genes for the host fitness etc7,8. More prophages and prophages related genes may be benefit for the survival and pathogenicity of clinical strains.

(A) Frequencies of integrated prophages in the genomes of C. sakazakii strains. The strains isolated from clinical sample, food and environmental sample were colored with light red, light blue and black, respectively. (B) Correlation between genomic sizes of C. sakazakii and prophages. There was a significant positive correlation between the genome sizes of C. sakazakii strains and integrated prophages (Spearman’s ρ = 0.74, P < 10−4). The strains were colored the same as in Fig. 1A. (C) Contribution of prophage genes to genomic composition of C. sakazakii. The core genome corresponds to genes present in all strains, whereas the soft-core genome indicates genes present in more than 95% of the strains. The indispensable genome was split into two categories: prophages and other genes.
Pan genome analysis of C. sakazakii showed that there were 9245 gene families, including 2294 core genes, 3258 soft core genes (found in 95% of the strains, includes core genes), and 5987 indispensable genes. As shown in Fig. 1c, prophage genes contribute up to 16.54% (1529/9245) of pan genome and 25.54% (1529/5987) of indispensable genome. Moreover, 64.29% (983/1529) of the prophage genes were present in one or two C. sakazakii strains, suggesting their rapid loss upon acquisition, contributing to the open pan genome of this species. Some of the cargo genes carried by prophages, remaining in the chromosome of C. sakazakii, might be potential virulence factors. For example, a prophage in C. sakazakii strain cro360A2 contains genes encoding a protein homologous to the heat-labile enterotoxin A peptide and a prophage in strain BAA894 encodes a protein similar to the eae-like adhesion protein in Escherichia coli20. Both were recognized as virulence factors in enteropathogenic E. coli22,23. The results implied that prophages played an important role in genetic diversity and pathogenicity of C. sakazakii.
The identification of CRISPR-Cas systems in C. sakazakii
CRISPR arrays were found in 97.30% (36/37) of the strains. Moreover, 94.59% (35/37) of the strains had more than one CRISPR arrays. Ogrodzki et al. found 12 different CRISPR spacer arrays in four major sequence types (STs) according to the composition of spacers21. We also found a lot of different CRISPR spacer arrays in this study, moreover, all of them were located in conserved region of C. sakazakii genomes. As shown in Fig. 2a, five types of CRISPR arrays were detected in C. sakazakii according to the conserved location. Only CRISPR1, CRISPR2 and CRISPR3 had AT-rich leader sequences. CRISPR1 and CRISPR2 were simultaneously found in 86.49% (32/37) of the C. sakazakii strains. However, CRISPR3, CRISPR4, and CRISPR5 were only detected in a few strains (Fig. 2b). The spacers of CRISPR1, CRISPR2, and CRISPR3 had a high variability, implying their frequent gain and loss in C. sakazakii (Fig. 2c). Moreover, CRISPR2 had the largest average number of spacers. Only one cas gene locus, which was downstream of CRISPR1 and less than 20 kb upstream of CRISPR2, was detected in the C. sakazakii strains (Fig. 2a). The leader sequence and cas genes are two determinants strictly associated with active CRISPR loci9. CRISPR1 and CRISPR2 in C. sakazakii had leader sequences and cas genes, implying that they were active. As shown in Fig. 2a, the length of consensus repeat sequences in CRISPR1 and CRISPR2 was 29 bp, and their predicted RNA secondary structures were similar to repeat sequence cluster 2, associated with subtype I-E CRISPR-Cas system in E.coli24.

(A) Location of five CRISPR arrays in C. sakazakii. The order and orientation of genes and CRISPR arrays were drawn based on the genome of ATCCBAA894 strain. CRISPR1 was located between cas gene and a hypothetical gene. CRISPR2 was located between Y2-aiiA gene and fadM gene, less than 20 kb downstream of cas genes. In some cases, a hypothetical gene (gray dotted line) was found inserted between the Y2-aiiA and CRISPR2. The Weblogo and RNA secondary structure of consensus repeat sequences in CRISPR1 and CRISPR2 are indicated (below). CRISPR3, CRISPR4 and CRISPR5 were located between cueO gene and hypothetical gene, citB gene and speC gene, hxlR gene and cpdB gene, respectively. Hypothetical genes were indicated by a question mark. Genes with red, dark red, and gray edges represent the core, soft-core, and indispensable genes, respectively. L: AT-rich leader sequence region; Black diamond: repeat; Square: spacer. (B) Frequencies of five type of CRISPR arrays in Cronobacter sakazakii strains. (C) Number of spacers from five CRISPR arrays in Cronobacter sakazakii strains. CRISPR4 and CRISPR5 had a stable number of three and two spacers, respectively. The difference between the number of spacers in CRISPR1, CRISPR2, and CRISPR3 had statistically significance. **P < 0.001 (t-test, two tail).
As shown in Fig. 3, there were total 8 successive co-oriented cas genes: cas2, cas1, cas6, cas5, cas7, cse2, cas8e and cas3. In accordance with previous studies10,21, the CRISPR-Cas of C. sakazakii is subtype I-E. 89.19% (33/37) of the strains were found to have two common cas genes (cas1 and cas2), and the he signature cas gene of type I CRISPR-Cas system (cas3). Eight cas3 genes split into helicase cas3′ and HD nuclease cas3′′ genes. These split cas3 genes were also seen in subtype I-A CRISPR-Cas system10. One cas3′ gene further split into two small compartments (designated as cas3′a and cas3′b). The split genes were also seen in other cas genes (Fig. 3). Whether these genes can translate into active proteins is unknown. Three strains had single cas3 genes. Additionally, 91.89% (34/37) of the strains had both cas8e and cse2 genes, which encode for signature proteins of CRISPR-associated complex for antiviral defense (Cascade) effector complex of subtype I-E CRISPR-Cas system10. A ‘complete’ cas locus encompasses at least the full complement of genes for the main components of the interference module10. Whether these subtype I-E CRISPR-Cas system variants had activity to protect C. sakazakii strains away from phages attack are unknown.

Gene names follow the current nomenclature and classification. On the left is the arrangement of cas gene loci, whereas on the right is the number of strains containing the type of cas gene cluster. The cas genes are colored according to their different processes.
The correlation of phage infection and spacer presence in CRISPR-Cas systems of C. sakazakii
All predicted prophages (n = 131) sequences in C. sakazakii strains were extracted to build a local database. To evaluate the protection by CRISPR-Cas system against the foreigner nucleic sequences, we collected all spacers from the five CRISPR arrays to seek the similarity sequences from prophages and plasmids in ACLAME database and our local database in this study. All the spacers with matched protospacers in phages and plasmids belonged to CRISPR1 and CRISPR2, supporting our previous suggestion that CRISPR1 and CRISPR2 were active loci. In this study, we found the entire subtype I-E CRISPR-Cas system like E.coli and two active CRISPR arrays in C. sakazakii strains. Meantime, there were some spacers matched with phages in the CRISPR arrays, so we speculated that CRISPR-Cas system of C. sakazakii could provide defense from phage attacks through immunological memory saved in spacers.
As shown in Fig. 4A, only 11.47% (125/1090) of the spacer sequences in CRISPR1 and CRISPR2 were found to match sequences from phages and plasmids. It may not be exclude the reason for the lack of identified prophages and plasmids in public database. Of the matched sequences, 71.20% (89/125) were phages, suggesting that phages were an important type of HGT in C. sakazakii. Moreover, CRISPR2 had a higher proportion of spacers targeted phages (77.08%, 74/96) than plasmids (22.92%, 22/96). The spacers in CRISPR1 did not show the difference. Whether there is a priority to select CRISPR2 loci against phages need to be determined in the future.

(A) The pie chart shows the number of spacers from CRISPR1 and CRISPR2 with potential protospacer matches in phage and plasmid. (B) Matches of CRISPR spacers with the intact prophage in C. sakazakii ES15 strain (location: 1767028-1797368). The sequence regions of prophage matched with the spacers in CRISPR-Cas system are indicated by an arrow. The matched spacer was named based on the name of the isolated strain and the type of CRISPR locus. PLP: phage-like protein; Int: integrase; Tra: transposase; Ter: terminase; Coa: coat protein; Sha: tail shaft. All other genes on the black line encode hypothetical proteins. (C) Number of spacers from two active CRISPR arrays in C. sakazakii clinical strains and foodborne strains. The strains were colored the same as in Fig.1A. The average number of spacers in clinical strains was less than that of foodborne strains, and the difference had statistically significance. *P < 0.01 (t-test, two tail).
20.8% (26/125) of predicted prophages in C. sakazakii strains have found protospacers targeted by spacers from CRISPR1 and CRISPR2. As shown in Fig. 4B, one intact prophage from C. sakazakii ES15 strain (location: 1767028–1797368) was targeted by spacers, from its host strain and other strains, at five different sequence regions. From the targeted regions, two are in phage tail shaft protein, others in hypothetical non-structural protein. Four strains detected by the matched spacers were infected with the homologous phage. Especially, two different spacers targeting a sequence of prophage were integrated into the CRISPR2 of ES15 strain to enhance its immunological memory, to strengthen the defense in the future. Eight spacer sequences that matched this prophage were detected in strains without this prophage. This supported the idea that CRISPR-Cas system of C. sakazakii could provide defense from phage attacks through immunological memory saved in spacers. The number of spacers from two active CRISPRs in clinical strains was significantly less than that of foodborne strains (Fig. 4c). This result may explain the reason for the difference of the average number of prophages between clinical strains and foodborne strains. Touchon et al. reported a negative association between the number of spacers in CRISPR arrays and the number of prophages in lysogens25. However, there was no significant negative correlation between the number of spacers in CRISPR arrays and prophage frequencies in C. sakazakii (Supplemental Fig. 1). The reason for this inconsistency might be that the CRISPR-Cas system does not simply function as a defense system in C. sakazakii or that there are other efficient mechanisms against phages. All these speculations need to be determined in the future.
In conclusion, the universal infection of temperate phages is a major factor contributing to genetic diversity and pathogenicity of C. sakazakii. A solo class 1, subtype I-E CRISPR-Cas system and five types of CRISPR arrays were found in C. sakazakii. CRISPR1 and CRISPR2 loci with high variable spacers were active and showed potential protection against specific phage attacks through immunological memory saved in spacers. At the same time, we found that the number of spacers from active CRISPR loci in clinical strains is significant less than that of foodborne strains, it maybe a reason why clinical strains were found to have more prophages than foodborne strains. More prophages and their related genes may be benefit for the survival and pathogenicity of clinical strains. The rapid gain/loss of prophages and spacers in CRISPR loci is likely to drive the quick evolution of C. sakazakii. Our study is an important step towards better understanding the co-evolution of phages and C. sakazakii, and presents the importance of further research aimed at deciphering the mechanisms of prophages and CRISPR-Cas systems that affect the pathogenicity and environmental adaption of C. sakazakii.
Methods
Bacterial strains, genome sequencing, and pan genome analysis
17 C. sakazakii strains, isolated from several types of food in China, were selected (Supplemental Table S1). 13 of them belonged to three major sequence types (ST4, ST1 and ST8) of C. sakazakii strains in China, three were our newly discovered sequence types (ST266, ST283, ST287), one were ST641. The genomes of C. sakazakii strains were fragmented using NEBNext dsDNA Fragmentase (NEB, USA) and Bsp143I (Sau3AI; Fermentas, Lithuania). DNA fragments (500 bp) were purified for preparation of a sequencing library, using QIAGEN GeneRead Library Prep (I) kit (Qiagen, Germany). Samples were subjected to 2 × 250 bp paired-end sequencing, using the Illumina Hiseq 2500 instrument, to generate 1 million reads with 100-fold coverage. The raw data for each bacterium were error-corrected and assembled using SPAdes 3.6.226. The final assemblies were filtered to contain ≥200 bp contigs. Genome annotation was performed using Prokka 1.1127. Pan genome analysis was performed by GET_HOMOLOGUES software28, using the standard described by Tettelin and collaborators29.
Identification of prophages and CRISPR-Cas system
The whole genome sequences of 17 strains achieved from our study and 42 strains available in NCBI genome database (the strains had more than one submitted genome sequences, only the sequence with fewer scaffolds was selected) were extracted as our sequence set. For many public clinical sequences were from a neonatal intensive care unit outbreak in France 1994, they were multiple isolates from the same strains, then we selected the reference strains as represent according to the primary paper for our analyses (detail in Supplemental Table S2)30,31. The prophages were identified using PHASTER32. We removed prophages with a large number of insertion sequences (IS; >25% of the predicted ORFs). Microsoft Excel was used for all the statistical analysis.
CRISPR array can be detected by CRISPRFinder33. CRISPR arrays with two or more identical spacer lengths were identified. The graphical representations (logos) of the patterns in the alignments of all the consensus repeats of specific CRISPR array in C. sakazakii were visualized with WebLogo 3.034. Secondary structure prediction of the most frequently occurring repeat sequence was performed using Mfold35. We extracted 500 bp, downstream and upstream of the CRISPR array, to find the AT-rich leader region. For cas genes, 394 profiles were used for PSI-BLAST analysis of the potential cas genes in genomes of C. sakazakii strains with e-value 10–6, as described previously10. The potential cas genes related to CRISPR array were identified, and their conserved domain was analyzed using the Conserved Domain Database36.
Determining spacer matches
We extracted all sequences of predicted prophages (n = 131) from C. sakazakii strains to build a local database. The similarity search of identified spacer sequences was performed by using BLAST with ACLAME database37 and local database in this study. We considered matches to be not less than 84% (minimum of 27/32 matching nucleotides).
Nucleotide sequence accession numbers
The GenBank accession numbers of the 17 whole genome sequences of C. sakazakii strains reported in this article are provided in Supplemental Table S1.
Additional Information
How to cite this article: Zeng, H. et al. The driving force of prophages and CRISPR-Cas system in the evolution of Cronobacter sakazakii. Sci. Rep. 7, 40206; doi: 10.1038/srep40206 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
26 April 2017
A correction has been published and is appended to both the HTML and PDF versions of this paper. The error has not been fixed in the paper.
26 April 2017
Scientific Reports 7: Article number: 40206; published online: 06 January 2017; updated: 26 April 2017. This Article contains an error in Figure 2, where the translation orientation of the hypothetical gene adjacent to CRISPR3 is reversed. In addition, the location of citB and speC beside CRISPR4, along with the sequence logo of CRISPR4 repeats should be reversed.
References
Xu, X. et al. Prevalence, molecular characterization, and antibiotic susceptibility of Cronobacter spp. in Chinese ready-to-eat foods. International journal of food microbiology 204, 17–23, doi: 10.1016/j.ijfoodmicro.2015.03.003 (2015).
Jason, J. Prevention of invasive Cronobacter infections in young infants fed powdered infant formulas. Pediatrics 130, e1076–1084, doi: 10.1542/peds.2011–3855 (2012).
Bowen, A. B. & Braden, C. R. Invasive Enterobacter sakazakii disease in infants. Emerging infectious diseases 12, 1185–1189, doi: 10.3201/eid1208.051509 (2006).
Friedemann, M. Epidemiology of invasive neonatal Cronobacter (Enterobacter sakazakii) infections. European journal of clinical microbiology & infectious diseases: official publication of the European Society of Clinical Microbiology 28, 1297–1304, doi: 10.1007/s10096-009-0779-4 (2009).
Singh, N., Goel, G. & Raghav, M. Insights into virulence factors determining the pathogenicity of Cronobacter sakazakii. Virulence 6, 433–440, doi: 10.1080/21505594.2015.1036217 (2015).
Brum, J. R., Hurwitz, B. L., Schofield, O., Ducklow, H. W. & Sullivan, M. B. Seasonal time bombs: dominant temperate viruses affect Southern Ocean microbial dynamics. The ISME journal 10, 437–449, doi: 10.1038/ismej.2015.125 (2016).
Fortier, L. C. & Sekulovic, O. Importance of prophages to evolution and virulence of bacterial pathogens. Virulence 4, 354–365, doi: 10.4161/viru.24498 (2013).
Bobay, L. M., Touchon, M. & Rocha, E. P. Pervasive domestication of defective prophages by bacteria. Proceedings of the National Academy of Sciences of the United States of America 111, 12127–12132, doi: 10.1073/pnas.1405336111 (2014).
Szczepankowska, A. Role of CRISPR/cas system in the development of bacteriophage resistance. Advances in virus research 82, 289–338, doi: 10.1016/B978-0-12-394621-8.00011-X (2012).
Makarova, K. S. et al. An updated evolutionary classification of CRISPR-Cas systems. Nature reviews. Microbiology 13, 722–736, doi: 10.1038/nrmicro3569 (2015).
Westra, E. R., Buckling, A. & Fineran, P. C. CRISPR-Cas systems: beyond adaptive immunity. Nature reviews. Microbiology 12, 317–326, doi: 10.1038/nrmicro3241 (2014).
Barrangou, R. CRISPR-Cas systems and RNA-guided interference. Wiley interdisciplinary reviews. RNA 4, 267–278, doi: 10.1002/wrna.1159 (2013).
Semenova, E. et al. Highly efficient primed spacer acquisition from targets destroyed by the Escherichia coli type I-E CRISPR-Cas interfering complex. Proceedings of the National Academy of Sciences of the United States of America 113, 7626–7631, doi: 10.1073/pnas.1602639113 (2016).
Xue, C. et al. CRISPR interference and priming varies with individual spacer sequences. Nucleic acids research 43, 10831–10847, doi: 10.1093/nar/gkv1259 (2015).
van der Oost, J., Jore, M. M., Westra, E. R., Lundgren, M. & Brouns, S. J. CRISPR-based adaptive and heritable immunity in prokaryotes. Trends in biochemical sciences 34, 401–407, doi: 10.1016/j.tibs.2009.05.002 (2009).
Wiedenheft, B., Sternberg, S. H. & Doudna, J. A. RNA-guided genetic silencing systems in bacteria and archaea. Nature 482, 331–338, doi: 10.1038/nature10886 (2012).
Garneau, J. E. et al. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468, 67–71, doi: 10.1038/nature09523 (2010).
Grim, C. J. et al. Pan-genome analysis of the emerging foodborne pathogen Cronobacter spp. suggests a species-level bidirectional divergence driven by niche adaptation. BMC Genomics 14, 366, doi: 10.1186/1471-2164-14-366 (2013).
Joseph, S. et al. Comparative analysis of genome sequences covering the seven cronobacter species. PLoS One 7, e49455, doi: 10.1371/journal.pone.0049455 (2012).
Kucerova, E. et al. Genome sequence of Cronobacter sakazakii BAA-894 and comparative genomic hybridization analysis with other Cronobacter species. PLoS One 5, e9556, doi: 10.1371/journal.pone.0009556 (2010).
Ogrodzki, P. & Forsythe, S. J. CRISPR-cas loci profiling of Cronobacter sakazakii pathovars. Future microbiology, doi: 10.2217/fmb-2016-0070 (2016).
Liu, D. et al. Discovery of the cell-penetrating function of A2 domain derived from LTA subunit of Escherichia coli heat-labile enterotoxin. Applied microbiology and biotechnology 100, 5079–5088, doi: 10.1007/s00253-016-7423-x (2016).
Kaper, J. B. The locus of enterocyte effacement pathogenicity island of Shiga toxin-producing Escherichia coli O157:H7 and other attaching and effacing E. coli. Japanese journal of medical science & biology 51 Suppl, S101–107 (1998).
Kunin, V., Sorek, R. & Hugenholtz, P. Evolutionary conservation of sequence and secondary structures in CRISPR repeats. Genome biology 8, R61, doi: 10.1186/gb-2007-8-4-r61 (2007).
Touchon, M., Bernheim, A. & Rocha, E. P. Genetic and life-history traits associated with the distribution of prophages in bacteria. The ISME journal, doi: 10.1038/ismej.2016.47 (2016).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of computational biology: a journal of computational molecular cell biology 19, 455–477, doi: 10.1089/cmb.2012.0021 (2012).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069, doi: 10.1093/bioinformatics/btu153 (2014).
Contreras-Moreira, B. & Vinuesa, P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Applied and environmental microbiology 79, 7696–7701, doi: 10.1128/AEM.02411-13 (2013).
Tettelin, H. et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proceedings of the National Academy of Sciences of the United States of America 102, 13950–13955, doi: 10.1073/pnas.0506758102 (2005).
Caubilla-Barron, J. et al. Genotypic and phenotypic analysis of Enterobacter sakazakii strains from an outbreak resulting in fatalities in a neonatal intensive care unit in France. J Clin Microbiol 45, 3979–3985, doi: 10.1128/JCM.01075-07 (2007).
Masood, N. et al. Genomic dissection of the 1994 Cronobacter sakazakii outbreak in a French neonatal intensive care unit. BMC Genomics 16, 750, doi: 10.1186/s12864-015-1961-y (2015).
Arndt, D. et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic acids research, doi: 10.1093/nar/gkw387 (2016).
Grissa, I., Vergnaud, G. & Pourcel, C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic acids research 35, W52–57, doi: 10.1093/nar/gkm360 (2007).
Crooks, G. E., Hon, G., Chandonia, J. M. & Brenner, S. E. WebLogo: a sequence logo generator. Genome research 14, 1188–1190, doi: 10.1101/gr.849004 (2004).
Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic acids research 31, 3406–3415 (2003).
Marchler-Bauer, A. et al. CDD: NCBI’s conserved domain database. Nucleic acids research 43, D222–226, doi: 10.1093/nar/gku1221 (2015).
Leplae, R., Lima-Mendez, G. & Toussaint, A. ACLAME: a CLAssification of Mobile genetic Elements, update 2010. Nucleic acids research 38, D57–61, doi: 10.1093/nar/gkp938 (2010).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31601571, 31371780), the Natural Science Foundation of Guangdong Province (2016A030310315), the Science and Technology Planning Project of Guangdong Province (2014A040401057) and the Natural Science Foundation of Guangdong Academy of Science (qnjj201603).
Author information
Authors and Affiliations
Contributions
H.Z. and Q.W. designed the study. H.Z. and J.Z. performed experiment, analyzed data and wrote the paper. C.L., T.X., N.L. and Y.Y. performed some experiments. All authors approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zeng, H., Zhang, J., Li, C. et al. The driving force of prophages and CRISPR-Cas system in the evolution of Cronobacter sakazakii. Sci Rep 7, 40206 (2017). https://doi.org/10.1038/srep40206
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep40206
This article is cited by
-
Genome-Wide Identification and Analysis of Chromosomally Integrated Putative Prophages Associated with Clinical Klebsiella pneumoniae Strains
Current Microbiology (2021)
-
Characterization of integrated prophages within diverse species of clinical nontuberculous mycobacteria
Virology Journal (2020)
-
Screening and characterization of prophages in Desulfovibrio genomes
Scientific Reports (2018)
-
Genomic characterization of malonate positive Cronobacter sakazakii serotype O:2, sequence type 64 strains, isolated from clinical, food, and environment samples
Gut Pathogens (2018)
-
Comparative genomics of Australian and international isolates of Salmonella Typhimurium: correlation of core genome evolution with CRISPR and prophage profiles
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
