
Abstract
LncRNAs play pivotal roles in many important biological processes, but research on the functions of lncRNAs in human disease is still in its infancy. Therefore, it is urgent to prioritize lncRNAs that are potentially associated with diseases. In this work, we developed a novel algorithm, LncPriCNet, that uses a multi-level composite network to prioritize candidate lncRNAs associated with diseases. By integrating genes, lncRNAs, phenotypes and their associations, LncPriCNet achieves an overall performance superior to that of previous methods, with high AUC values of up to 0.93. Notably, LncPriCNet still performs well when information on known disease lncRNAs is lacking. When applied to breast cancer, LncPriCNet identified known breast cancer-related lncRNAs, revealed novel lncRNA candidates and inferred their functions via pathway analysis. We further constructed the human disease-lncRNA landscape, revealed the modularity of the disease-lncRNA network and identified several lncRNA hotspots. In summary, LncPriCNet is a useful tool for prioritizing disease-related lncRNAs and may facilitate understanding of the molecular mechanisms of human disease at the lncRNA level.
Similar content being viewed by others

Introduction
Recent studies have revealed that up to 70% of the human genome is transcribed into RNA, whereas protein-coding genes only make up less than 2% of the total genome. The majority of the transcriptional repertoire consists of non-coding RNAs (ncRNAs)1,2. Long non-coding RNAs (lncRNAs), which constitute the majority of ncRNAs, are a class of transcripts longer than 200 nt that lack protein-coding potential3,4. Accumulating evidence indicates that lncRNAs play pivotal roles in many important biological processes5,6. In particular, recent studies have suggested that lncRNAs are involved in the initiation and progression of a wide range of diseases7 and have been found to act as tumor suppressors or oncogenes8. For example, the lncRNA HOTAIR is dysregulated in several cancers, including colon, breast, pancreas, and liver cancers, and the overexpression of HOTAIR has been shown to drive breast cancer metastasis9. Therefore, identifying potential disease lncRNAs may facilitate understanding of the molecular mechanisms of human disease at the lncRNA level and may unveil new diagnostic and therapeutic opportunities. RNA-seq and microarray technologies have identified tens of thousands of human lncRNAs, but knowledge of disease-related lncRNAs is still limited. Therefore, it is a challenging task to prioritize lncRNAs associated with a high risk of disease for further functional investigation.
Recently, some computational methods have been proposed to predict disease-related lncRNAs. Some of these methods are based on the sequence or genomic locations of lncRNAs10,11,12. For example, a global disease-lncRNA associations have been predicted by LncRNADisease based on lncRNAs and their genomic loci related neighbor genes/miRNAs (within 2 kb) tend to be associated with the same disease11. A sequence based bioinformatics tool was proposed to predict lncRNA-disease associations using their interaction with disease-related miRNA12. LncRNA-mRNA co-expression-based methods have also been widely used to predict lncRNA function13,14 and to predict disease lncRNAs15. On the basis of the assumption that similar diseases tend to be associated with similar functional lncRNAs, several network-based methods have been developed to prioritize disease-related lncRNAs16,17,18,19. Sun et al. have presented the RLncD method using random walking on a lncRNA functional similarity network18. Zhou et al. have developed the RWRHLD method, involving random walking on a heterogeneous network integrating phenotype and disease information17,19. Most recently, IRWRLDA has been developed by integrating lncRNA expression similarity and disease semantic similarity, thereby effectively improving the prediction power20. On the basis of these previous results, we believe that associations between lncRNAs and genes, and disease-gene information are valuable information, and should be integrated in the study of lncRNA function. This integration is a feature unique to LncPriCNet.
It is reasonable to integrate multi-level data regarding genes, phenotypes, lncRNAs and their association information to prioritize disease-related lncRNA candidates. First, from a biological perspective, lncRNAs rarely function in isolation; instead, lncRNAs serve as regulators that may achieve regulatory specificity through modularity, assembling diverse combinations of proteins and possibly RNA and DNA interactions21. Some studies have suggested that lncRNAs may act synergistically22,23. Thus, a biological system can be intrinsically represented as an intricate network including multi-level information. The effects of one disease are not restricted to one or two lncRNAs but instead are spread among functionally related lncRNAs and genes. An integration strategy may provide more comprehensive and accurate information24. Second, although some experimentally determined disease-lncRNA associations have been collected11, completeness remains a distant goal. The integration strategy could use other information (such as disease-gene information and lncRNA-gene associations) to compensate for some missing information. Third, although it is important to investigate the functions of lncRNAs and uncover the mechanisms of biological processes, the functions of most lncRNAs remain unknown7. An integration strategy uses information about the genes associated with lncRNAs to facilitate interpretation. This integration strategy has been successfully used to disease metabolites prioritization and improve the prediction power25.
Assuming that functionally related lncRNAs and genes play roles in phenotypically similar diseases, we proposed a computational method called LncPriCNet (disease candidate LncRNAs Prioritization based on a Composite Network) to prioritize disease-related lncRNAs. First, we constructed a composite network integrating multi-level information including phenotypes, lncRNAs, genes and their associations. Fully considering the global functional interactions of the multi-level composite network, LncPriCNet prioritizes the candidate lncRNAs on the basis of their similarity to known disease information. LncPriCNet has better predictive power than previous methods. Importantly, by integrating multi-level information, LncPriCNet performs well even for diseases without known associated lncRNAs. When applied to a breast cancer RNA-Seq data set, LncPriCNet identified known disease-related lncRNAs as well as novel ones. Furthermore, a disease-lncRNA landscape was constructed and analyzed to provide a global view of disease lncRNAs.
Materials and Methods
Data sources and construction of a multi-level composite network
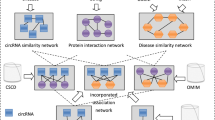
To construct a multi-level composite network, we collected experimentally validated or computationally predicted associations among phenotypes, genes and lncRNAs (Fig. 1a). The gene-gene associations were constructed on the basis of protein-protein interaction data downloaded from the HPRD database26. The disease-phenotype associations were obtained from the OMIM database27 by removing records with the prefixes “*” and “^”. The textual similarity between disease phenotypes was calculated by MimMiner28. The top five similar phenotypes were retained for analysis. RNA-seq data on 16 human tissues were obtained from the Human Body Map project (ERP000546). We used Tophat29 to perform read alignment and cufflinks30 to perform transcript assembly, and the expression levels of lncRNAs and coding genes were estimated as FPKM (fragments per kilobase of transcript per million fragments mapped). LncRNA-lncRNA and gene-lncRNA associations were measured using the Pearson correlation coefficient. The experimentally verified gene-lncRNA associations were also obtained if they were supported by more than two CLIP-seq experiments in the StarBase database31. Experimentally verified disease-lncRNA relationships were taken from the lncRNADisease database11. After removal of redundant records, 371 disease-lncRNA relations between 108 lncRNAs and 140 disease phenotypes were obtained. The phenotype-gene relations were extracted from the OMIM database by BioMart32.

(a) Construction of the multi-level composite network. This network is constructed by six sub-networks. White circle indicates lncRNA; white square indicates gene; white triangle indicates phenotype. The thickness of the edge indicates the weight score. (b) The flow chart by which LncPriCNet optimizes the candidate lncRNAs. First, the candidate lncRNAs of interest and seed nodes are mapped to the multi-level composite network. Then, a global extended RWR method is used to score the candidate lncRNAs according to their proximity to seed nodes. Finally, the candidate lncRNAs are ranked according to the scores. Purple circles represent the candidate lncRNAs of interest; red triangle indicates disease phenotype (phenotype seed) of interest from the OMIM data base; red squares represent known disease genes (gene seeds) from the OMIM database; and red circles indicate known disease lncRNAs (lncRNA seeds) from the lncRNADisease database.
Next, we integrated the above information to construct a weighted multi-level composite network. The edge weights were defined on the basis of the data sources: 1 for experimentally validated associations and correlation coefficients for computationally predicted associations. Let WG, WP, WL, WGL, WPL, WGP be the adjacency matrix of the gene network, phenotype network, lncRNA network, gene-lncRNA network, phenotype-lncRNA network and gene-phenotype network, respectively. Then, the adjacency matrix of the multi-level composite network can be defined as  , where the superscript T means the transpose of matrix.
, where the superscript T means the transpose of matrix.
LncPriCNet
We propose a novel global computational method to prioritize disease-related lncRNA (LncPriCNet), which extends the random walking with restart (RWR) algorithm to a multi-level network to capture global information (Fig. 1b). It simulates a random walker walking on the network, starting on a set of seed nodes, and at each step, it moves from the current nodes to their direct neighbor(s) randomly with a probability 1 − δ, then returns to the seed nodes with a restart probability δ. Let P0 be the initial probability vector, and let Pt represent a vector in which the i-th element is the probability of being at node i at step t. Then, the probability vector at step t + 1 is defined as follows:

where M is the transition matrix of the multi-level composite network, which can be calculated by the adjacency matrix W (the computational details will be described later). After several iterations, the probability will reach a steady state when the difference between P0 and Pt falls below 10−10 (measured by the L1 norm).
For one disease-phenotype of interest, the seed nodes defined in this study consist of this disease-phenotype (SP), and its corresponding known disease gene (SG) and lncRNA (SL). Suppose u0, v0, w0 are the initial probabilities of the gene network, phenotype network and lncRNA network, respectively. Here, u0 is calculated by assigning equal probability to all nodes in SG in the gene network with a sum equal to 1. Similarly, v0, w0 can be calculated. Then, the initial probability vector of the multi-level composite network is denoted as  . Here, α, β and 1 − α − β range from 0 to 1, and they represent the importance of the gene network, phenotype network and lncRNA network, respectively.
. Here, α, β and 1 − α − β range from 0 to 1, and they represent the importance of the gene network, phenotype network and lncRNA network, respectively.
Let the transition matrix  . MG, MP, ML denote the intra-subnetwork transition matrix, and other variables denote the inter-subnetwork transition matrix. Mij represents the transition probability from node i to node j. Suppose x, y, z are the jumping probability between the gene network and phenotype network, between the gene network and lncRNA network, and between the phenotype network and lncRNA network or vice versa, respectively. Then, the transition probability from gene i (gi) to gene j (gj) in the gene network can be computed as follows:
. MG, MP, ML denote the intra-subnetwork transition matrix, and other variables denote the inter-subnetwork transition matrix. Mij represents the transition probability from node i to node j. Suppose x, y, z are the jumping probability between the gene network and phenotype network, between the gene network and lncRNA network, and between the phenotype network and lncRNA network or vice versa, respectively. Then, the transition probability from gene i (gi) to gene j (gj) in the gene network can be computed as follows:

Similarly, the transition probability from phenotype i (pi) to phenotype j (pj) in the phenotype network can be calculated as follows:

The probability from lncRNA i (li) to lncRNA j (lj) in the lncRNA network can be computed as

The transition probability from gene i (gi) in the gene network to phenotype j (pj) in the phenotype network can be defined as

The transition probability from gene i (gi) in the gene network to lncRNA j (lj) in the lncRNA network can be described as

The transition probability from phenotype i (pi) in the phenotype network to gene j (gj) in the gene network can be defined as

The transition probability from phenotype i (pi) to lncRNA j (lj) can be described as

The transition probability from lncRNA i (li) to gene j (gj) can be defined as

The transition probability from lncRNA i (li) to phenotype j (pj) can be described as

To further describe the equations for the transition probability, we take equation (10) as an example. Suppose the current node is a lncRNA i (li), and MLP (i, j) denotes the probability of moving from this lncRNA i (li) to a phenotype node j (pj). In this step,  means that no phenotypes directly link to this lncRNA, and thus the walker never moves to a phenotype node in this step, and the probability should be 0. Otherwise,
means that no phenotypes directly link to this lncRNA, and thus the walker never moves to a phenotype node in this step, and the probability should be 0. Otherwise,  means the walker could move to one of phenotype nodes. Then, the probability that the walker moves from this lncRNA i (li) to phenotype j (pj) should be calculated by multiplying the jumping probability z by a normalized adjacency matrix of the phenotype-lncRNA network
means the walker could move to one of phenotype nodes. Then, the probability that the walker moves from this lncRNA i (li) to phenotype j (pj) should be calculated by multiplying the jumping probability z by a normalized adjacency matrix of the phenotype-lncRNA network  .
.
LncPriCNet is performed until the probabilities tend to a steady state,  . Then, the candidate lncRNAs can be ranked according to w∞. In this study, we set the parameter δ to 0.7 and x, y, z, α, β to 1/3.
. Then, the candidate lncRNAs can be ranked according to w∞. In this study, we set the parameter δ to 0.7 and x, y, z, α, β to 1/3.
LncPriCNet is implemented in the R language. An R-based package of LncPriCNet is available at https://cran.r-project.org/.
Results
We first constructed a multi-level composite network integrating information on lncRNAs, genes, phenotypes and their associations (Fig. 1a). This network consisted of three types of node (gene, lncRNA and phenotype) and six types of association (gene-gene, lncRNA-lncRNA, phenotype-phenotype, phenotype-lncRNA, phenotype-gene and gene-lncRNA) (Table S1). Then, we developed a novel global computational method to prioritize disease-related lncRNAs on the basis of this multi-level composite network (LncPriCNet) (Fig. 1b). In this section, we first evaluated the performance of LncPriCNet and compared it with other methods. Then, we applied LncPriCNet to a breast cancer dataset to confirm its ability to find novel disease lncRNA candidates. Furthermore, a disease-lncRNA landscape was constructed and analyzed to provide a global view of disease-related lncRNAs.
Performance of LncPriCNet
To test the performance of LncPriCNet, we used leave-one-out cross validation (LOOCV) to assess whether LncPriCNet could identify known disease lncRNAs. First, we chose 53 phenotypes associated with at least two experimentally validated lncRNAs in the composite network. We obtained 284 known phenotype-lncRNA and 153 known phenotype-gene associations. For each round of cross-validation, one known disease lncRNA was selected as a test object, and links between this lncRNA and its corresponding target phenotype were removed. We defined the seed nodes as the target phenotype, other known lncRNAs associated with this phenotype and all known genes associated with this phenotype. Then, LncPriCNet was performed and returned a score for each non-seed lncRNA. The top-ranked lncRNAs were predicted to be related to the disease. Receiver operating characteristic analysis (ROC) and the area under the curve (AUC) were used to evaluate the overall performance of LncPriCNet. ROC was performed by plotting the true positive rate versus the false positive rate at various score threshold settings. The results showed that LncPriCNet achieved an AUC value up to 0.933 (Fig. 2a). Additionally, over 88.3% (251) of known disease-related lncRNAs were ranked in the top 10%, and even in the top 10, there were still 74.3% known disease-related lncRNAs. These results suggested that our strategy of using multi-level data from the composite network is effective in prioritizing disease lncRNAs.

(a) ROC curve for the predicted lncRNAs of 53 phenotypes. (b) ROC curve for the predicted lncRNAs of 20 phenotypes with two known lncRNAs. (c) ROC curve for the predicted lncRNAs of 42 phenotypes with only one known lncRNA. (d) The performance in hypothetical phenotypes without known disease lncRNAs.
To further investigate the performance of LncPriCNet in different disease classes, 53 phenotypes were grouped into 10 disease classes on the basis of previous work33 and manually annotated. LOOCV was performed for each disease class, and the AUC value was calculated. LncPriCNet achieved an AUC value over 0.7 in 8 disease classes, and in 4 classes, the AUC value was over 0.93 (Table S2). The metabolic disease class ranked in the top 1 (AUC = 1), and all six known metabolic lncRNAs were ranked in the top 3 by LOOCV. The cardiovascular disease class ranked in the top 2 (AUC = 0.999), and 68 (98.5%) known cardiovascular lncRNAs were ranked in the top 2 by LOOCV.
Method comparison
To highlight the superiority of integrating multi-level information, we compared LncPriCNet with RWRHLD and RlncD. All the three methods use random walking with restart on a network; LncPriCNet uses the phenotype-lncRNA-gene network, whereas RWRHLD uses the phenotype-lncRNA network19, and RlncD uses the lncRNA network18. The AUC values of RWRHLD and RlncD were 0.926 and 0.543, which was lower than that of LncPriCNet (Fig. 2a). When performed on different disease classes, LncPriCNet resulted in higher AUC values than the other two methods in 7 of 10 (70%) disease classes (Fig. S1 and Table S2).
Because the known lncRNA-disease associations remain limited, we further evaluated the performance of three methods for diseases lacking known lncRNAs. First, we extracted 20 phenotypes linked to only two known lncRNAs, after which 40 known disease lncRNAs and 40 known disease genes remained. The ROC curve obtained from LOOCV showed that LncPriCNet performed better than RWRHLD and RlncD (Fig. 2b). Second, we extracted 42 phenotypes with only one known disease lncRNA in the composite network, and we obtained 42 known lncRNAs and 71 known genes. As shown in Fig. 2c, LncPriCNet achieved an AUC value of 0.903, which was much higher than that of RWRHLD (AUC value of 0.765). Third, we assumed that all 53 phenotypes were linked to no known disease lncRNAs and performed LOOCV. For each run of LOOCV, all links between the phenotype and known lncRNAs were removed, and then the phenotype and known disease genes were used as seeds to recall the known lncRNAs. As shown in Fig. 2d, LncPriCNet achieved an AUC value of 0.889, whereas RWRHLD obtained an AUC value of only 0.758. RlncD completely lost efficiency under the last two conditions. These results showed that LncPriCNet performs better than the previous methods, especially for diseases with few known lncRNAs.
Parameters of LncPriCNet
LncPriCNet has five parameters: δ, x, y, z, α and β. Here, δ is the restart probability in the random walk method, and x, y, z are the jumping probability between gene network and phenotype network, between the gene network and lncRNA network, and between the phenotype network and lncRNA network or vice versa. The values of α, β and 1 − α − β range from 0 to 1, and they represent the importance of the gene network, phenotype network and lncRNA network, respectively. To investigate the possible effects of these parameters, different values were assigned to these parameters and LOOCV analysis was performed. The resulting AUC values varied from 0.879 to 0.943 (Tables S3 and S4), thus suggesting that LncPriCNet can achieve reliable and robust performance for different parameters.
Case study
Breast cancer is the leading cause of cancer mortality among women worldwide. We analyzed RNA-seq data consisting of 8 benign breast lesions, 8 ER-positive (ER+), 8 HER2-positive (HER2+), and 8 triple negative (TN) primary breast tumors (SRP019936). TopHat and cufflinks were used to align and assemble lncRNAs of each breast sample, and cuffdiff was used to identify differentially expressed lncRNAs for each type of breast cancer. We obtained 528 differentially expressed lncRNAs in ER+, HER+ and TN tumors. The breast cancer-related phenotype (MIM:114480), 11 known genes (BRCA2, PALB2, NBN, PIK3CA, RAD51, AKT1, CHEK2, XRCC3, BRCA1, BRIP1, FAM175A) and 11 known lncRNAs (BCYRN1, CDKN2B-AS1, GAS5, H19, HOTAIR, MIR31HG, MALAT1, MEG3, PVT1, UCA1, XIST) were used as seeds, and LncPriCNet, RWRHLD and RlncD were applied to score 528 differential lncRNAs. The top 10 lncRNAs for each method were predicted to be related to breast cancer, and their potential associations with breast cancer were investigated by manual literature mining (Table 1, Fig. 3a). The ranks of LncPriCNet and RWRHLD were similar, but they differed from the RlncD rank. Seven lncRNAs were ranked in the top 10 by both LncPriCNet and RWRHLD, and four of them had literature support (CBR3-AS1, ACTA2-AS1, TINCR, and TUSC8). Figure 3b illustrates the subnetwork of the top three lncRNAs and seed genes in LncPriCNet; the close connection indicated functional association in the same disease. CBR3-AS1 (PlncRNA-1), which was highly expressed in all three subtypes of breast cancer, was ranked first by LncPriCNet. It has been reported to be aberrantly expressed in both gastric cancer and prostate cancer, and silencing of CBR3-AS1 has been found to significantly reduce cell proliferation and induce apoptosis in prostate cancer cell lines34,35,36. High expression of ACTA2-AS1 (ZXF1) is related to a relatively poor prognosis and may promote invasion and metastasis in lung adenocarcinoma37. TINCR binds to staufen (STAU1) protein and mediates differentiated mRNA stabilization38. In addition, silencing TINCR expression inhibits cell proliferation, colony formation, tumorigenicity and apoptosis promotion39. TUSC8 ranked fourth and has been suggested to serve as an predictor for survival in cervical cancer40. Additionally, only LncPriCNet identified ZNRD1-AS1, which is involved in the occurrence and development of cancers by participating in the processes of DNA damage and repair and suppressing cell proliferation41,42,43.

(a) Venn diagram of the top 10 ranked lncRNAs identified by LncPriCNet and two other methods. The numbers in square brackets denote lncRNAs with literature support. (b) The subnetwork of the top three lncRNAs (CBR3-AS1, TINCR and ACTA2-AS1) and seed nodes. (c) The network of the top 10 ranked lncRNAs and enriched pathways of their co-expressed genes.
To investigate the functional mechanism of the predicted breast lncRNAs, pathway enrichment analysis was performed. For each lncRNA, first, genes linked with the lncRNA (co-expression score above 0.6 between the lncRNA and mRNA) in our multi-level network were obtained. Then, Subpathway-GM44 was applied to perform pathway enrichment analyses (p-value < 0.01). Next, a lncRNA-pathway network was constructed for better visualization (Fig. 3c). The first-ranked lncRNA CBR3-AS1 was linked to the TGF-beta signaling pathway. TGF-beta has a suppressive effect in the early stage of tumorigenesis and hence is regarded as a tumor suppressor; it promotes tumor progression and metastasis during later stages45. In breast cancer, the TGF-beta signaling pathway promotes the metastasis of cancer via regulating the epithelial-to-mesenchymal transition (EMT)46. TGF-beta also may serve as a predictive and prognostic marker of cancer stage47. The fifth-ranked lncRNA, BDNF-AS, had the highest degree in the lncRNA-pathway network. It is involved in many breast cancer-related pathways, including the ErbB signaling pathway48,49, MAPK signaling pathway50, and wnt signaling pathway51. The term “pathway in cancer” received the highest degree among all pathways (degree = 5). Interestingly, five lncRNAs, ranked from fifth to tenth by lncPriCNet (LINC00271, ITGA9-AS1, PSMD5-AS1, RP11-66B24.4, ZNRD1-AS1), were directly linked to this pathway. Four of them did not have literature support, thus suggesting that our method can capture novel disease lncRNAs.
A predicted landscape of disease-related lncRNAs
We further used LncPriCNet to infer relationships between all lncRNAs and 53 disease phenotypes to chart a predicted lncRNA-disease landscape. First, scores between all 10082 lncRNAs and 53 disease phenotypes were computed to construct a score matrix. Then, a two-way hierarchical clustering method was used to reveal the organization of the human phenotype-lncRNA relationships (Fig. 4a). The phenotypes clustered together tended to have a similar molecular or genetic basis. Phenotype clusters were annotated with enriched disease classes. LncRNA clusters were annotated with the most enriched KEGG pathways of their co-expressed genes (Spearman correlation coefficient between lncRNA and gene higher than 0.6). The predicted disease-lncRNA landscape revealed some highly scored modules, each consisting of a set of functionally related lncRNAs implicated in a set of disease phenotypes. For instance, as illustrated in the red circle in Fig. 4a, a cancer-related module consisted of lncRNAs in the cell cycle pathway. From the zoomed-in plot in Fig. 4b, we found that this module included three types of cancer and 146 highly scored lncRNAs. Further inspection of the biological functions of these 146 lncRNAs showed that they were significantly enriched in cancer-related pathways, such as the cell cycle pathway, oocyte meiosis pathway and p53 signaling pathway (Fig. 4c). These cancers also shared three seed genes (H19, MALAT1 and MEG3). The similar molecular basis and high connection in the network may contribute to the modularity.

(a) Hierarchical clustering of the LncPriCNet scores between 53 phenotypes and 10082 lncRNAs. The color of each cell represents the LncPriCNet score of a lncRNA (row) for a phenotype (column). Phenotype clusters were annotated with enriched disease categories (bottom), and lncRNA clusters were annotated with the most enriched pathways of their co-expressed genes (right). The red circled region indicates a module composed of lncRNAs involved in the cell cycle process. (b) Zoom-in plot of the red circled region, involving 3 type of cancers and 156 high-risk lncRNAs. (c) Enriched pathways for the co-expressed genes of 156 high-risk lncRNAs.
Furthermore, we investigated the number of diseases in which each lncRNA was involved. Figure 5a shows that the majority of lncRNAs were not related to any disease, whereas some lncRNAs were related to multiple diseases. We extracted 20 lncRNA hotspots that were associated with most diseases and investigated their ranks in all 53 diseases (Fig. 5b). The highest-risk lncRNAs (MEG3 and CDKN2B-AS1) were involved in 32, 42, or 42 diseases when the top 10, 50, or 100 lncRNAs were regarded as disease-related. MEG3 is a maternally expressed imprinted gene that interacts with cAMP, p53, and GDF15 and modulates the activity of TGF-b genes by binding to distal regulatory elements, thereby playing a role in cell proliferation control52,53. Studies have reported that CDKN2B-AS1 is involved in various neoplasms52,54. In addition, we tested whether high-risk lncRNAs identified by LncPriCNet were more likely to be dysregulated in diseases. For this purpose, we downloaded the differentially expressed lncRNAs in six types of tumors55 and compared them with the top 50 lncRNAs ranked by LncPriCNet. As illustrated in Fig. 5c, high-risk lncRNAs significantly overlapped with dysregulated lncRNAs in all six tumors (Fisher’s Exact Test p-value < 0.01).

(a) The number of disease phenotypes involving each lncRNA, with three different rank cutoffs. (b) The stacked plot of 20 lncRNAs, which are predicted to be associated with most phenotypes. (c) High-risk lncRNAs (top 50) are significantly overlapped with dysregulated lncRNAs in all six tumors (BRCA: Breast carcinoma; COAD: colon adenocarcinoma; LUSC: lung squamous cell carcinoma; LUAD: lung adenocarcinoma; PRAD: prostate adenocarcinoma; KIRC: kidney renal clear cell carcinoma).
Discussion
In this study, we present a novel computational method, LncPriCNet, to prioritize and predict disease candidate lncRNAs by integrating multi-level information regarding genes, lncRNAs, phenotypes and their associations. LncPriCNet showed clearly higher predictive power than previously published methods. More importantly, a breast cancer case study showed that LncPriCNet was able to capture well-documented lncRNAs as well as identify and infer the functional roles of novel lncRNAs. Furthermore, a human disease-lncRNA landscape revealed some phenotype modules with a similar molecular basis. The top 50 lncRNAs related to all 53 diseases were listed in (Supplementary Dataset 1). To further evaluate the predictions of the novel disease lncRNA, we also performed some literature research to the predicted top five diseae-related lncRNAs. For example, the top-ranked lncRNA PVT1 in hepatocellular carcinoma has been found to promote the proliferation and stem cell-like properties of hepatocellular carcinoma cells by stabilizing NOP256 and might serve as a recurrence and supplementary diagnosis biomarker57,58. The top-ranked lncRNA HOTAIR in bladder cancer has been reported to be correlated with disease progression and may serve as a prognostic biomarker59,60. The serum levels of SNHG5, ranked fifth by LncPriCNet in melanoma, have been found to be significantly higher in patients with melanoma than in normal subjects and may serve as a new tumor marker of malignant melanoma61. Apart from cancer, the top-ranked lncRNA H19 associated with myocardial infarction has been reported to bind directly to miR-103/107 and regulate FADD expression and necrosis. The modulation of its levels may provide a new approach for preventing myocardial infarction62. LncRNAs GAS5, ranked fourth in atherosclerosis, is significantly increased in the plaques of atherosclerosis patients compared with normal subjects and may lead to new clinical applications63. LINC00929, which is significantly highly expressed in hereditary hemorrhagic disease64, was predicted as the top lncRNA by LncPriCNet. Furthermore, MALAT1, ranked second in Parkinson disease (PD), has been reported to show decreased expression in PD and to inhibit α-synuclein protein expression, thereby providing a neuroprotective effect in PD65. The fourth-ranked lncRNA in diabetes is MIAT, whose knockdown ameliorates diabetes mellitus-induced retinal microvascular dysfunction in vivo and inhibits endothelial cell proliferation, migration, and tube formation in vitro66. More literature citations supporting novel predictions are listed in Table S5. It is possible to select high-ranked lncRNAs from the prioritized list and test their causality through appropriate experiments. We also found disease lncRNA hotspots, such as MEG3 and CDKN2B-AS1, which have been reported to be involved in many diseases. Further analysis of six tumors showed that high-risk lncRNAs identified by LncPriCNet tend to be dysregulated in tumors, thus further supporting that LncPriCNet can identify disease-related LncRNAs.
The outstanding performance of LncPriCNet can be attributed to two aspects. First, LncPriCNet takes advantage of the multi-level information of the composite network. Abnormal phenotypes are usually a consequence of perturbed transcriptional levels, including not only coding genes but also lncRNAs. In addition, lncRNAs rarely perform biological functions alone but instead act as key regulators, such as scaffolds or sponges, that regulate genes21. The close connections of various levels may compensate for some missing information. For example, LncPriCNet performs well by using other information when information regarding known disease-associated lncRNAs is lacking. Second, LncPriCNet extends the RWR method and enables it to be used on a more complicated network model. It uses a global distance measure to prioritize candidate lncRNAs on the basis of their global similarity to known disease lncRNAs and genes, thereby capturing multi-level information of the composite network. This approach ensures that candidate lncRNAs are ranked according to the interaction information in the entire composite network rather than merely the local environment. However, there are still some limitations to this methodology. LncPriCNet relies on the topology of the composite network, and therefore the incompleteness and bias of the data may limit its performance. It will be improved when more accurate and complete resources are available.
Currently, the functions of most lncRNAs remain unknown, and the knowledge of disease-related lncRNAs are very limited. Candidate lncRNAs may be obtained by differential expression analysis or genome-wide association study analysis, but there are still too many candidates to experimentally validate. LncPriCNet prioritizes these candidate lncRNAs, allowing biologists to select high-ranking disease lncRNAs and test their functions. Furthermore, the lncRNA-disease and lncRNA-pathway network might provide clues as to the functional mechanisms of lncRNAs. Overall, LncPriCNet is a useful tool for disease lncRNA prioritization and provides better understanding of the molecular mechanisms of human disease at the lncRNA level, which may uncover new diagnostic and therapeutic opportunities. The strategy of the multi-level composite network could be used in other fields of biomedicine, such as disease, drug and target discovery.
Additional Information
How to cite this article: Yao, Q. et al. Global Prioritizing Disease Candidate lncRNAs via a Multi-level Composite Network. Sci. Rep. 7, 39516; doi: 10.1038/srep39516 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Djebali, S. et al. Landscape of transcription in human cells. Nature 489, 101–108, doi: 10.1038/nature11233 (2012).
Derrien, T. et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome research 22, 1775–1789, doi: 10.1101/gr.132159.111 (2012).
Kapranov, P. et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 316, 1484–1488, doi: 10.1126/science.1138341 (2007).
Mattick, J. S. & Rinn, J. L. Discovery and annotation of long noncoding RNAs. Nature structural & molecular biology 22, 5–7, doi: 10.1038/nsmb.2942 (2015).
Fatica, A. & Bozzoni, I. Long non-coding RNAs: new players in cell differentiation and development. Nature reviews. Genetics 15, 7–21, doi: 10.1038/nrg3606 (2014).
Batista, P. J. & Chang, H. Y. Long noncoding RNAs: cellular address codes in development and disease. Cell 152, 1298–1307, doi: 10.1016/j.cell.2013.02.012 (2013).
Wapinski, O. & Chang, H. Y. Long noncoding RNAs and human disease. Trends in cell biology 21, 354–361, doi: 10.1016/j.tcb.2011.04.001 (2011).
Rinn, J. L. & Chang, H. Y. Genome regulation by long noncoding RNAs. Annual review of biochemistry 81, 145–166, doi: 10.1146/annurev-biochem-051410-092902 (2012).
Gupta, R. A. et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464, 1071–1076, doi: 10.1038/nature08975 (2010).
Li, J. et al. A bioinformatics method for predicting long noncoding RNAs associated with vascular disease. Science China. Life sciences 57, 852–857, doi: 10.1007/s11427-014-4692-4 (2014).
Chen, G. et al. LncRNADisease: a database for long-non-coding RNA-associated diseases. Nucleic acids research 41, D983–986, doi: 10.1093/nar/gks1099 (2013).
Wang, J. et al. LncDisease: a sequence based bioinformatics tool for predicting lncRNA-disease associations. Nucleic acids research, doi: 10.1093/nar/gkw093 (2016).
Liao, Q. et al. Large-scale prediction of long non-coding RNA functions in a coding-non-coding gene co-expression network. Nucleic acids research 39, 3864–3878, doi: 10.1093/nar/gkq1348 (2011).
Necsulea, A. et al. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature 505, 635–640, doi: 10.1038/nature12943 (2014).
Wang, L. et al. Genome-wide screening and identification of long noncoding RNAs and their interaction with protein coding RNAs in bladder urothelial cell carcinoma. Cancer letters 349, 77–86, doi: 10.1016/j.canlet.2014.03.033 (2014).
Chen, X. & Yan, G. Y. Novel human lncRNA-disease association inference based on lncRNA expression profiles. Bioinformatics 29, 2617–2624, doi: 10.1093/bioinformatics/btt426 (2013).
Yang, X. et al. A network based method for analysis of lncRNA-disease associations and prediction of lncRNAs implicated in diseases. PloS one 9, e87797, doi: 10.1371/journal.pone.0087797 (2014).
Sun, J. et al. Inferring novel lncRNA-disease associations based on a random walk model of a lncRNA functional similarity network. Molecular bioSystems 10, 2074–2081, doi: 10.1039/c3mb70608g (2014).
zhou, M. Prioritizing candidate disease-related long non-coding RNAs by walking on the heterogeneous lncRNA and disease network, doi: 10.1039/C4MB00511B 10.1039/c4mb00511b (2015).
Chen, X., You, Z. H., Yan, G. Y. & Gong, D. W. IRWRLDA: improved random walk with restart for lncRNA-disease association prediction. Oncotarget, doi: 10.18632/oncotarget.11141 (2016).
Guttman, M. & Rinn, J. L. Modular regulatory principles of large non-coding RNAs. Nature 482, 339–346, doi: 10.1038/nature10887 (2012).
Li, Y. et al. Construction and analysis of lncRNA-lncRNA synergistic networks to reveal clinically relevant lncRNAs in cancer. Oncotarget 6, 25003–25016, doi: 10.18632/oncotarget.4660 (2015).
Ma, W. et al. Fine-scale chromatin interaction maps reveal the cis-regulatory landscape of human lincRNA genes. Nature methods 12, 71–78, doi: 10.1038/nmeth.3205 (2015).
Ritchie, M. D., Holzinger, E. R., Li, R., Pendergrass, S. A. & Kim, D. Methods of integrating data to uncover genotype-phenotype interactions. Nature reviews. Genetics 16, 85–97, doi: 10.1038/nrg3868 (2015).
Yao, Q. et al. Global Prioritization of Disease Candidate Metabolites Based on a Multi-omics Composite Network. Scientific reports 5, 17201, doi: 10.1038/srep17201 (2015).
Keshava Prasad, T. S. et al. Human Protein Reference Database–2009 update. Nucleic acids research 37, D767–772, doi: 10.1093/nar/gkn892 (2009).
Amberger, J. S., Bocchini, C. A., Schiettecatte, F., Scott, A. F. & Hamosh, A. OMIM.org: Online Mendelian Inheritance in Man (OMIM(R)), an online catalog of human genes and genetic disorders. Nucleic acids research 43, D789–798, doi: 10.1093/nar/gku1205 (2015).
van Driel, M. A., Bruggeman, J., Vriend, G., Brunner, H. G. & Leunissen, J. A. A text-mining analysis of the human phenome. European journal of human genetics: EJHG 14, 535–542, doi: 10.1038/sj.ejhg.5201585 (2006).
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111, doi: 10.1093/bioinformatics/btp120 (2009).
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28, 511–515, doi: 10.1038/nbt.1621 (2010).
Li, J. H., Liu, S., Zhou, H., Qu, L. H. & Yang, J. H. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic acids research 42, D92–97, doi: 10.1093/nar/gkt1248 (2014).
Smedley, D. et al. BioMart–biological queries made easy. BMC genomics 10, 22, doi: 10.1186/1471-2164-10-22 (2009).
Goh, K. I. et al. The human disease network. Proceedings of the National Academy of Sciences of the United States of America 104, 8685–8690, doi: 10.1073/pnas.0701361104 (2007).
Gao, J., Cao, R. & Mu, H. Long non-coding RNA UCA1 may be a novel diagnostic and predictive biomarker in plasma for early gastric cancer. International journal of clinical and experimental pathology 8, 12936–12942 (2015).
Martens-Uzunova, E. S. et al. Long noncoding RNA in prostate, bladder, and kidney cancer. European urology 65, 1140–1151, doi: 10.1016/j.eururo.2013.12.003 (2014).
Cui, Z. et al. The prostate cancer-up-regulated long noncoding RNA PlncRNA-1 modulates apoptosis and proliferation through reciprocal regulation of androgen receptor. Urologic oncology 31, 1117–1123, doi: 10.1016/j.urolonc.2011.11.030 (2013).
Zhang, L., Zhou, X. F., Pan, G. F. & Zhao, J. P. Enhanced expression of long non-coding RNA ZXF1 promoted the invasion and metastasis in lung adenocarcinoma. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie 68, 401–407, doi: 10.1016/j.biopha.2014.03.001 (2014).
Kretz, M. et al. Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature 493, 231–235, doi: 10.1038/nature11661 (2013).
Xu, T. P. et al. SP1-induced upregulation of the long noncoding RNA TINCR regulates cell proliferation and apoptosis by affecting KLF2 mRNA stability in gastric cancer. Oncogene 34, 5648–5661, doi: 10.1038/onc.2015.18 (2015).
Liao, L. M. et al. Low expression of long noncoding XLOC_010588 indicates a poor prognosis and promotes proliferation through upregulation of c-Myc in cervical cancer. Gynecologic oncology 133, 616–623, doi: 10.1016/j.ygyno.2014.03.555 (2014).
Hong, L. et al. ZNRD1 gene suppresses cell proliferation through cell cycle arrest in G1 phase. Cancer biology & therapy 4, 60–64 (2005).
Guo, L. et al. Expression quantitative trait loci in long non-coding RNA ZNRD1-AS1 influence cervical cancer development. American journal of cancer research 5, 2301–2307 (2015).
Wen, J. et al. Expression quantitative trait loci in long non-coding RNA ZNRD1-AS1 influence both HBV infection and hepatocellular carcinoma development. Molecular carcinogenesis 54, 1275–1282, doi: 10.1002/mc.22200 (2015).
Li, C. et al. Subpathway-GM: identification of metabolic subpathways via joint power of interesting genes and metabolites and their topologies within pathways. Nucleic acids research 41, e101, doi: 10.1093/nar/gkt161 (2013).
Drabsch, Y. & ten Dijke, P. TGF-beta signalling and its role in cancer progression and metastasis. Cancer metastasis reviews 31, 553–568, doi: 10.1007/s10555-012-9375-7 (2012).
Shipitsin, M. et al. Molecular definition of breast tumor heterogeneity. Cancer cell 11, 259–273, doi: 10.1016/j.ccr.2007.01.013 (2007).
de Kruijf, E. M. et al. The prognostic role of TGF-beta signaling pathway in breast cancer patients. Annals of oncology: official journal of the European Society for Medical Oncology/ESMO 24, 384–390, doi: 10.1093/annonc/mds333 (2013).
Moasser, M. M. The oncogene HER2: its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene 26, 6469–6487, doi: 10.1038/sj.onc.1210477 (2007).
Tebbutt, N., Pedersen, M. W. & Johns, T. G. Targeting the ERBB family in cancer: couples therapy. Nature reviews. Cancer 13, 663–673, doi: 10.1038/nrc3559 (2013).
Balko, J. M. et al. Activation of MAPK pathways due to DUSP4 loss promotes cancer stem cell-like phenotypes in basal-like breast cancer. Cancer research 73, 6346–6358, doi: 10.1158/0008-5472.CAN-13-1385 (2013).
Moon, R. T., Kohn, A. D., De Ferrari, G. V. & Kaykas, A. WNT and beta-catenin signalling: diseases and therapies. Nature reviews. Genetics 5, 691–701, doi: 10.1038/nrg1427 (2004).
Benetatos, L., Vartholomatos, G. & Hatzimichael, E. MEG3 imprinted gene contribution in tumorigenesis. International journal of cancer. Journal international du cancer 129, 773–779, doi: 10.1002/ijc.26052 (2011).
Mondal, T. et al. MEG3 long noncoding RNA regulates the TGF-beta pathway genes through formation of RNA-DNA triplex structures. Nature communications 6, 7743, doi: 10.1038/ncomms8743 (2015).
Shi, X., Sun, M., Liu, H., Yao, Y. & Song, Y. Long non-coding RNAs: a new frontier in the study of human diseases. Cancer letters 339, 159–166, doi: 10.1016/j.canlet.2013.06.013 (2013).
Yan, X. et al. Comprehensive Genomic Characterization of Long Non-coding RNAs across Human Cancers. Cancer cell 28, 529–540, doi: 10.1016/j.ccell.2015.09.006 (2015).
Wang, F. et al. Oncofetal long noncoding RNA PVT1 promotes proliferation and stem cell-like property of hepatocellular carcinoma cells by stabilizing NOP2. Hepatology 60, 1278–1290, doi: 10.1002/hep.27239 (2014).
Ding, C. et al. Long non-coding RNA PVT1 is associated with tumor progression and predicts recurrence in hepatocellular carcinoma patients. Oncology letters 9, 955–963, doi: 10.3892/ol.2014.2730 (2015).
Yu, J. et al. The long noncoding RNAs PVT1 and uc002mbe.2 in sera provide a new supplementary method for hepatocellular carcinoma diagnosis. Medicine 95, e4436, doi: 10.1097/MD.0000000000004436 (2016).
Berrondo, C. et al. Expression of the Long Non-Coding RNA HOTAIR Correlates with Disease Progression in Bladder Cancer and Is Contained in Bladder Cancer Patient Urinary Exosomes. PloS one 11, e0147236, doi: 10.1371/journal.pone.0147236 (2016).
Sun, X. et al. Long non-coding RNA HOTAIR regulates cyclin J via inhibition of microRNA-205 expression in bladder cancer. Cell death & disease 6, e1907, doi: 10.1038/cddis.2015.269 (2015).
Ichigozaki, Y. et al. Serum long non-coding RNA, snoRNA host gene 5 level as a new tumor marker of malignant melanoma. Experimental dermatology 25, 67–69, doi: 10.1111/exd.12868 (2016).
Wang, J. X. et al. MicroRNA-103/107 Regulate Programmed Necrosis and Myocardial Ischemia/Reperfusion Injury Through Targeting FADD. Circulation research 117, 352–363, doi: 10.1161/CIRCRESAHA.117.305781 (2015).
Chen, L. et al. Global transcriptomic study of atherosclerosis development in rats. Gene 592, 43–48, doi: 10.1016/j.gene.2016.07.023 (2016).
Torring, P. M. et al. Long non-coding RNA expression profiles in hereditary haemorrhagic telangiectasia. PloS one 9, e90272, doi: 10.1371/journal.pone.0090272 (2014).
Zhang, Q. S. et al. Beta-asarone protects against MPTP-induced Parkinson’s disease via regulating long non-coding RNA MALAT1 and inhibiting alpha-synuclein protein expression. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie 83, 153–159, doi: 10.1016/j.biopha.2016.06.017 (2016).
Yan, B. et al. lncRNA-MIAT regulates microvascular dysfunction by functioning as a competing endogenous RNA. Circulation research 116, 1143–1156, doi: 10.1161/CIRCRESAHA.116.305510 (2015).
Vita, M. & Henriksson, M. The Myc oncoprotein as a therapeutic target for human cancer. Seminars in cancer biology 16, 318–330, doi: 10.1016/j.semcancer.2006.07.015 (2006).
Chen, C. R., Kang, Y. & Massague, J. Defective repression of c-myc in breast cancer cells: A loss at the core of the transforming growth factor beta growth arrest program. Proceedings of the National Academy of Sciences of the United States of America 98, 992–999, doi: 10.1073/pnas.98.3.992 (2001).
Acknowledgements
This work was supported by the National Institutes of Health National Natural Science Foundation of China [31501077], National Key Scientific Instrument and Equipment Develeopment Project of China [2012YQ03026108] and National Grand Program on Key Infectious Diseases [2015ZX10004801-005].
Author information
Authors and Affiliations
Contributions
Q.L.Y. designed the study; Q.L.Y. and L.L.W. performed experiments, analyzed data. Q.L.Y. wrote the manuscript; Q.L.Y. developed the R package; All (J.L., L.G.Y., Y.D.S., Z.L., S.H., F.Y.F.) authors contributed to biological interpretation of the results. Y.X.L. & H.L. supervised research and revised the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Yao, Q., Wu, L., Li, J. et al. Global Prioritizing Disease Candidate lncRNAs via a Multi-level Composite Network. Sci Rep 7, 39516 (2017). https://doi.org/10.1038/srep39516
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep39516
This article is cited by
-
Association filtering and generative adversarial networks for predicting lncRNA-associated disease
BMC Bioinformatics (2023)
-
BiGAN: LncRNA-disease association prediction based on bidirectional generative adversarial network
BMC Bioinformatics (2021)
-
A machine learning framework that integrates multi-omics data predicts cancer-related LncRNAs
BMC Bioinformatics (2021)
-
A novel mechanism by which ACTA2-AS1 promotes cervical cancer progression: acting as a ceRNA of miR-143-3p to regulate SMAD3 expression
Cancer Cell International (2020)
-
Prediction of lncRNA-disease associations by integrating diverse heterogeneous information sources with RWR algorithm and positive pointwise mutual information
BMC Bioinformatics (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
