
Abstract
Bactrocera correcta is one of the most destructive pests of horticultural crops in tropical and subtropical regions. Despite the economic risk, the population genetics of this pest have remained relatively unexplored. This study explores population genetic structure and contemporary gene flow in B. correcta in Chinese Yunnan Province and attempts to place observed patterns within the broader geographical context of the species’ total range. Based on combined data from mtDNA cox1 sequences and 12 microsatellite loci obtained from 793 individuals located in 7 countries, overall genetic structuring was low. The expansion history of this species, including likely human-mediated dispersal, may have played a role in shaping the observed weak structure. The study suggested a close relationship between Yunnan Province and adjacent countries, with evidence for Western and/or Southern Yunnan as the invasive origin of B. correcta within Yunnan Province. The information gleaned from this analysis of gene flow and population structure has broad implications for quarantine, trade and management of this pest, especially in China where it is expanding northward. Future studies should concentrate effort on sampling South Asian populations, which would enable better inferences of the ancestral location of B. correcta and its invasion history into and throughout Asia.
Similar content being viewed by others

Introduction
Bactrocera correcta (Bezzi) (Diptera: Tephritidae), the guava fruit fly, is one of the most destructive pests of many tropical and subtropical fruits and vegetables, such as guavas, mangoes, citruses, melons and chili peppers, causing production losses and quality degradation1,2. Considering that the species has a broad host range, is highly adaptable, and has a high reproductive ability and dispersal capacity3,4, it has been regulated as a quarantine pest by many countries, including China1,5. Bactrocera correcta was first reported in Bihar, India in 19166, where it is thought to have originally diverged from its common ancestor7,8. At present, it is distributed throughout South and East Asia, from Bhutan in the west to China and Vietnam in the east9,10,11,12. The species has been recorded in the United States, where it was first detected in California in 1986, but has not yet established due to timely prevention and control measures13. In China, B. correcta was first discovered in the Yunnan Province (Yuanjiang and Mosha) in 198214, and the infestation of this fly has become more entrenched in Yunnan in recent years15.
Yunnan is located in southwest China, adjacent to Myanmar, Laos and Vietnam, from which entry of exotic species into Yunnan has been facilitated by similar environmental conditions4. As such, Yunnan is considered to represent a transition zone for invasive fruit flies16, where the native ranges of some species blur into a northward-moving invasion front. One example of this is B. correcta (Bezzi), which first entered China through Yunnan17 and has since moved from Yuanjiang to Yuanmou in Yunnan province, and also to Panzhihua, Sichuan province4. Futhermore, ecological modelling data suggested that, at certain times of year, B. correcta is capable of establishment across the whole of China18. Given the notable rapid spread of B. correcta in China, its economic importance, and the risk of this species being introduced, establishing and invading other regions of China argue for greater understanding of population relationships and invasion routes.
Information regarding genetic diversity, genetic structure and gene flow are key issues when developing management strategies19,20. The mitochondrial (mtDNA) cytochrome oxidase subunit I (cox1) gene is easily amplified21, maternally inherited and relatively fast-evolving, which allows derivation of recent female-specific evolutionary histories. In contrast, microsatellites (SSRs) are nuclear, bi-parentally inherited, highly polymorphic and easy to isolate, making them particularly informative in the study of contemporary biological invasions16,22. Thus, these two sets of molecular markers are extensively used to study the population genetic structure of insect pests20,23,24, including Bactrocera fruit fly species16,25,26,27.
Here, we assess population genetic structure within Yunnan Province via dense geographical sampling to determine patterns of gene flow and dispersal within China. We then place these patterns in a broader geographical context by sampling other locations across the species’ distribution. In doing so, we build on previous localized evidence concerning the genetic structure and invasion history of B. correcta (e.g., within Thailand)28, and attempt to infer the route by which this species entered China. Specifically, we test whether B. correcta is expanding northward in China via gradual movement from adjacent populations or if new, adventive northern populations are derived from further away. Likewise, we seek to determine whether any locations within Yunnan Province show signs of having been the original invasive location in China and from which the invasion progressed. Additionally, we incorporate limited public data from South Asia (India, Sri Lanka) to address hypotheses concerning this region as the putative ancestral location of the species. Taken together, this study provides foundational data for understanding the population dynamics and genetic structure of B. correcta, which will contribute greatly toward development of control measures for this fly in Yunnan.
Methods
Sample collection, DNA extraction, cox1 sequencing and microsatellite genotyping
Specimens of B. correcta were collected from 18 sites in China (Yunnan), Laos, Myanmar, Thailand, and Vietnam from 2008 to 201329 using methyl eugenol (ME) traps (Table 1 and Fig. 1)30. All flies were stored in 100% ethanol at −20 °C prior to DNA extraction. In addition to new data collected here, we added 12 cox1 sequences from GenBank for flies from India (accession numbers GU323781 and GU323782) and Sri Lanka (accession numbers JQ692856, JQ692753, JQ692641, JQ692784, JQ692787, JQ692756, JQ692631, JQ692832, JQ692711 and JQ692676)31.

Geographical location of 20 sampled sites across South and Southeast Asia.
Note: Insert figure: Yunnan Province, China. The map was created in ArcGIS 10.2 software (ESRI Inc., Redlands, CA, USA). URL http://www.esri.com/software/arcgis/arcgis-for-desktop.
Genomic DNA was extracted from each specimen using the Tissue/Cell DNA Mini Kit (Tiangen Biotech, Beijing, China). A 658 bp cox1 fragment was amplified and sequenced according to the method of Liu et al.32. Both directions of the cox1 sequence from each individual were assembled using DNAMAN 5.2 (Lynnon Corporation, Quebec, Canada). To delete low-quality sections, all sequences were aligned with the standard sequences of B. correcta from BOLD using Clustal X33 to generate 600-bp cox1 sequences. Unique sequences were deposited in GenBank with accession numbers KU669296–KU670076. Information about the twelve microsatellite loci and amplifying methods analyzed here34. Fluorescently labeled fragments were detected on an ABI PRISM 377 Genetic Analyzer, with ROX-500 size standard (Microread, Beijing, China). Allele size was analyzed by GeneScan V3.7 program (Applied Biosystems, Beijing, China).
Marker summary statistics and intra-population genetic diversity
For cox1 data, the nucleotide composition and variable positions were visualized using MEGA 635. The nucleotide diversity (π), haplotype diversity (Hd) and number of haplotypes for each geographical population of B. correcta were estimated using DNASP 5.136.
For microsatellite data, the number of alleles (NA), number of effective alleles (NE), observed heterozygosity (HO), expected heterozygosity (HE) were calculated each population using POPGENE 1.3237. Allelic richness (AR) and gene diversity (HS) were calculated using FSTAT 2.9.3.238. Frequency of null allele (AN) was estimated using GENEPOP 4.139. The same software was also used to check for genotypic linkage disequilibrium and for departure from Hardy-Weinberg Equilibrium (HWE) after sequential Bonferroni correction.
Population genetic structure
Pairwise FST was calculated for both types of markers using Arlequin 3.5 to measure the degree of genetic differentiation between pairs of populations and corrected for the effect of multiple tests by using a modification of the false discovery rate method40. Isolation-By-Distance (IBD) was examined by testing the correlation between FST/(1-FST)41 ln-scaled geographical distances using Mantel tests42 in Arlequin 3.5 with 1000 permutations. GoogleTM Earth 5.243 was used to determine the linear geographical distances between each pair of sampling sites of B. correcta, the correlation analysis results were plotted in SPSS v16.0 (SPSS Inc., Chicago, IL).
Evolutionary relationships among cox1 haplotypes were inferred using a haplotype network, constructed under the median-joining (MJ) method in NETWORK 4.644. Bayesian clustering of individuals based on microsatellite genotypes was performed in STRUCTURE 2.045 to infer genetic structure among the 18 studied populations of B. correcta. We set the number of clusters (K) from 1 to 10 and conducted 10 independent runs for each value of K. Each run consisted of a burn-in period of 50,000 steps, followed by 100,000 Markov chain Monte Carlo (MCMC) repetitions with a model allowing admixture. ΔK values46 were computed to select the most likely number of K using the online resource Structure Harvester47 that explained the structure in data. We then conducted model to summarize cluster membership coefficient matrices for each value of K with CLUMPP 1.1.248, and plotted using DISTRUCT 1.149. To identify the optimal number of groups (K) for sequences, spatial analysis of molecular variance was performed using SAMOVA 1.050 taking into account the longitude and latitude information. The supported groups K was selected with the highest Fct value associated with the subdivision scheme by repeating the analysis with K ranging from 2 to 9. Hierarchical analysis of molecular variance (AMOVA) was performed using Arlequin 3.5 to evaluate the distribution of molecular variance among groups, populations and individuals for both types of markers based on the grouping strategy from STRUCTURE and SAMOVA.
Demographic history
The demographic history analysis of all cox1 sequences from 20 populations (including India and Sri Lanka) were examined using mismatch distribution and neutrality tests in Arlequin 3.5 with 1000 bootstrap replicates. Six parameters were calculated: effective population size before expansion (θ0), effective population size after expansion (θ1), Time of populations expansion (T), Tajima’s D, Fu’s FS and sum of square deviation (SSD) between expected and observed mismatch distribution.
Inter-population migration rate estimates
The GENECLASS v2.0 program51 was used to assign/exclude populations as donors or receivers of individuals on the basis of multilocus genotypes. For each individual in a population of B. correcta, this program computes the probability that it belongs only to that population, the probability of being a migrant from each of the other populations and the probability of being a migrant to other populations16,25. We used the standard criterion, which applies Bayesian statistics to calculate probabilities52, and Monte Carlo resampling method53, which calculates the accurate inclusion/exclusion critical values. We simulated 10,000 genotypes for each population with a threshold probability value of 0.01.
Results
Marker summary statistics and intra-population diversity
In total, we sampled 781 B. correcta from 18 sites across China and Southeast Asia. Inclusion of 12 additional cox1 sequences from GenBank from India and Sri Lanka, produced a final alignment of 600 bp for 793 individuals. The A + T content was 63% (28.8% A and 34.2% T), higher than the G + C content (16.5% G and 20.5% C). This nucleotide composition is similar to B. dorsalis26 and B. cucurbitae27. Of the 600 nucleotide positions, 62 variable positions were observed (10.33%), including 19 singleton variable positions and 43 parsimony informative positions. Sixty-three haplotypes (designated H1-H63) were observed across the 20 populations of B. correcta. Of these, 30 haplotypes were shared by at least two populations (47.62%), with the most frequent haplotype H5 present in 17 populations. The number of haplotypes for per population ranged from 1 to 22 (Table 2). Three types of basic descriptive indices, namely haplotype diversity (Hd), nucleotide diversity ( ) and average number of nucleotide differences (k) were calculated to measure genetic diversity within populations (Table 2). Polymorphisms were found in 19 populations (Hd = 0.898, on average). Among them, Western Yunnan (YNRL, YNMS and YNBS) and SRIL showed a higher diversity, YNHH in the eastern displayed a lowest Hd and lowest nucleotide diversity was found in Vietnam neighboring to YNHH. However, there was no signal of decreased diversity in more northern populations of China compared with southern or western populations.
) and average number of nucleotide differences (k) were calculated to measure genetic diversity within populations (Table 2). Polymorphisms were found in 19 populations (Hd = 0.898, on average). Among them, Western Yunnan (YNRL, YNMS and YNBS) and SRIL showed a higher diversity, YNHH in the eastern displayed a lowest Hd and lowest nucleotide diversity was found in Vietnam neighboring to YNHH. However, there was no signal of decreased diversity in more northern populations of China compared with southern or western populations.
The same 781 individuals of B. correcta that were sequenced for cox1 were genotyped for 12 microsatellite loci. A total of 185 alleles were observed across 12 loci, ranging from 8 to 21 per locus (Supplementary Table S1). Consistent with the cox1 data, YNMS from the western Yunnan presented the highest genetic diversity (Hs = 0.714) while VIET was the lowest (Hs = 0.444) followed by YNHH (Hs = 0.617) (Table 3).
Population genetic structure
Genetic distance among populations estimated using pairwise FST values (Table 4) showed that, for cox1, YNHH and BURM were consistently significantly different to most other populations (ranging from 0.293 for YNRL/YNHH to 0.066 for YNJH/YNBS). Microsatellites by contrast suggested that most pairwise comparisons were significantly different; however, actual FST values were generally lower than 0.1, suggesting low population structure. An exception was the YNHH population, which was supported as more different to all other sites, with FST estimates ranging from 0.1 to 0.149, average 0.122. Mantel tests showed no significant relationship between genetic (FST/(1-FST)) and geographical distances. (cox1 data: r2 = 0.011, P = 0.146; microsatellite data: r2 = 0.00018, P = 0.869), indicating the absence of IBD (Supplementary Fig. S1).
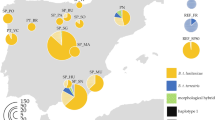
The median-joining network constructed from 63 haplotypes demonstrated that several haplotypes were highly common and shared by many locations. In particular, H4 and H5, which were separated by a single mutation, were common across the sampled range and were connected to several low frequency tip haplotypes, implying that they may represent putative ancestral haplotypes (Fig. 2, Supplementary Fig. S2). Myanmar was the only location did not share either of these common haplotypes, but does share haplotypes with Yunnan and Thailand (H14, H24, H28, H46). Western Yunnan (Sites YNBS, YNMS and YNRL) displayed a high level of genetic variability in possessing 15 unique haplotypes, one of which is shared exclusively with Sri Lanka (H38).

Median-Joining haplotype network of B. correcta based on mtDNA cox1 data.
Note: Size of nodes and pie segments were proportional to haplotype frequency; small black circles represent median vectors (roughly equivalent to hypothetical unsampled haplotypes); length of the branched is proportional to number of mutational changes between haplotypes.
Bayesian clustering analysis of microsatellite genotypes implemented in STRUCTURE showed that the maximum value for the estimated likelihood of K was found at K = 2 (Supplementary Fig. S3). Visualisation of cluster membership coefficients suggests that flies from the Site YNHH formed a single cluster separate to all other locations (Fig. 3). SAMOVA analysis also suggested the same grouping strategy, with the greatest proportion of variation among groups under a K = 2 hypothesis (FCT = 0.12284) (Supplementary Table S2). AMOVA analyses performed according to these two groups (YNHH separated from all other populations) showed genetic differentiation among groups accounted for 12.34% and 7.46% for cox1 and microsatellite data, respectively. All fixation indices, including FCT, FSC, FST, FIS and FIT were highly significant (P < 0.01) (Supplementary Table S3).

Bayesian results based on STRUCTURE among 18 populations of B.correcta at K = 2, individuals were grouped by collection site according to Table 1, each individual was represented by a vertical bar displaying membership coefficients.
Demographic history
Neutrality tests performed on the total cox1 dataset produced significant negative Tajima’s D and Fu’s FS values (Table 2) and the mismatch distribution was unimodal (Supplementary Fig. S4), supporting a model of population expansion (PSSD > 0.05). Ratios between estimated effective population size after expansion (θ1) and effective population size before expansion (θ0), which can serve as an estimate of the extent of population growth, indicated that B. correcta exhibited a certain degree of population growth in all the populations (Table 2).
Inter-population migration rate estimates
Bi-directional migration rates among populations of B. correcta, estimated based on microsatellite data, show marked variation across the sampled geographical distribution, ranging from 0 (YNHH into YNMS) to 0.727 (TCHM into VIET). In these analyses, migration rate estimates below 0.100 imply restricted gene flow, whereas moderate to high gene flow is suggested by values greater than 0.100. Interestingly, the migration rates from the YNHH and VIET populations to other populations were all below 0.100, except for YNHH into VIET (m = 0.122). Meanwhile, estimated migration rates were also very low between BURM and other populations. The probability with which individuals were assigned to their own populations varied from 0.344 (YNWS) to 0.501 (BURM) (Table 5).
Discussion
Understanding population structure and gene flow among regionsare very important aspects in the management of quarantine fruit flies. In this study, we obtained data from both mitochondrial and nuclear DNA markers of an extensive sampling of B. correcta in Yunnan Province, China, and neighboring countries. Our aim was to examine population structure and gene flow within Yunnan and to place Chinese diversity and structure in a regional context.
The clearest signal of population structure across the sampled locations supported the population YNHH, from eastern Yunnan, as being significantly structured from all other populations. This pattern was exemplified by low levels of estimated gene flow between this site and all others, and manifests in this site being supported as a separate genetic entity in Bayesian clustering analysis of microsatellite genotypes. Yunnan Province is characterized geologically as a longitudinal range-gorge region, where the mountain chains tend to run south to north, potentially blocking the spread of mobile insects eastward4,54. Genetic differentiation may thus be the result of natural barriers that limited gene flow from Eastern Yunnan westward and drove divergence of this population. Alternatively, this pattern may be representative of a separate origin for the YNHH population from a currently unsampled population, but presumably not from nearby.
Elsewhere across the sampled distribution, there were a small number of locations that were supported as significantly structured. Firstly, Myanmar, situated southwest of Yunnan and west of Southeast Asia also exhibited some evidence for significant genetic differentiation from other populations. This pattern may be associated with significant mountain ranges and/or reduced trade with other Asian countries that act to limit direct and indirect gene flow between Myanmar and surrounding regions. Myanmar displayed a close relationship with adjacent western Yunnan and Thailand, and high gene flow was inferred from these locations into Myanmar, in contrast to minimal migration in the opposite direction. This pattern has also been described in the highly invasive species B. dorsalis16,25, which is otherwise largely panmictic across much of Asia. Secondly, Vietnam is supported by microsatellite data as significantly different from most other sampled populations. Like Myanmar, there was reduced migration out of Vietnam to other locations, along with reduced diversity. It is unclear what might drive this pattern; however, a combination of trade practices and geographical barriers to dispersal may each play a role, or alternatively it could be driven by the small sample size for this population.
More broadly, resolved patterns are consistent with recent demographic expansion in this species. Significant negative Tajima’s D and Fu’s Fs indices, unimodle mismatch distributions and non-significant SSD values all supported the hypothesis of a sudden population expansion model. As B. correcta is a phytophagous and highly polyphagous fruit fly, presence of abundant hosts may aid its regional dispersal. Combined with highly frequent fruit trade between Yunnan and other Asian countries, this likely presents advantageous conditions for the long-distance dispersal of this fly3,55,56, which is considered to be on-going.
Within China, B. correcta was first recorded in southern Yunnan Province in 198214, a region that represents one of the gateways into Southeast Asia with potential invasion via trade routes along the Makhong River or the Kunming-Bangkok international highway57,58. Our data provides no insight on the origins of the Chinese populations other than that they do not differ greatly in genetic profile from other locations across southeast Asia. Nevertheless, western Yunnan exhibited higher diversity than other Chinese populations. It is generally accepted that fruit flies invading new areas are most likely to first establish in regions that have abundant host plants and suitable climate59, characteristics that are exemplified in western Yunnan. Further, given that older populations often demonstrate higher levels of genetic diversity than more recently established populations60,61, we argue that our genetic data supports a scenario of B. correcta having established first in western Yunnan before expanding into other regions in China.
Patterns across the rest of Yunnan Province shed some light on movement among populations and the potential invasion history of B. correcta in the region. In particular, populations in southern and eastern Yunnan displayed lower genetic diversity than western populations. This suggests that these populations may be the result of gradual eastward migration from an initial invasion into western Yunnan. This is contrary to previous hypotheses that the region was an original entry point for B. correcta into China, which were based on its geographical proximity and horticultural trade relationships with neighbouring countries, along with the first detection of B. correcta in China occurring in southern Yunnan. Instead, this scenario finds little support in our data, although it cannot be conclusively excluded.
Our inclusion of Indian and Sri Lankan cox1 sequence data allows some comment on the hypothesis that Bactrocera species may have originated in India7,8. We can propose the type of evidence that would be required to reject this hypothesis specifically, low genetic diversity and external/tip haplotypes in the cox1 network for South Asian locations. In marked contrast: and despite low sample size, there was high haplotype diversity (7 haplotypes from 12 sequences across both sites), with these being mostly internal to the network and often shared between many other locations. Thus, these data provide some evidence for these populations being more ‘ancestral’ than Southeast Asian populations, indicatingthat we cannot reject the idea that Bactrocera originated in India. The limited data for India and Sri Lanka restricts our deductive capacity. Further sampling will be required to determine the geographical origin of B. correcta and resolve patterns of gene flow and dispersal between India and other Asian countries.
We applied two sets of molecular markers to avoid any bias due to the use of only a single marker. Mitochondrial DNA corresponds to the maternal lineage, making it sensitive to selective neutrality, loss of mutation-drift equilibrium and male-to-female sex ratio balance62, and is particularly informative for inferring phylogeographical patterns. Nuclear microsatellites, on the other hand, are biparentally inherited and evolve faster than mtDNA, making them more suitable for analysis of contemporary gene flow. Hence, each marker type provides resolution of patterns at slightly different evolutionary scales, and the limited observed discrepancies are likely to be due to the different evolutionary history of each marker. For example, many more pairwise comparisons of population structure (using FST indices) were significant for microsatellites than for cox1. We argue that this is most likely driven by the faster mutation rate of the former, although it may possibly represent some degree of female-biased dispersal.
In conclusion, we provide evidence based on combined independent molecular markers that there is high gene flow among most Southeast Asian populations of B. correcta, with exceptions in eastern Yunnan, Myanmar and Vietnam. We provide support for hypotheses of first entry of this species into China occurring in western Yunnan, with gradual dispersal eastward. Given current global warming trends, which will render more of northern China ecologically suitable for B. correcta18, and the notable rapid spread of this invasive species, we argue that this species warrants urgent attention to understand and manage this invasion front. Moreover, early detection and control measures need to be enhanced to avoid or slow the rate of new invasions in China.
Additional Information
How to cite this article: Qin, Y.-J. et al. Genetic diversity and population structure in Bactrocera correcta (Diptera: Tephritidae) inferred from mtDNA cox1 and microsatellite markers. Sci. Rep. 6, 38476; doi: 10.1038/srep38476 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
White, I. M. & Elson-Harris, M. M. Fruit Flies of Economic Significance: their Identification and Bionomics. Wallingford UK: CAB International (1992).
Allwood, A. J. et al. Host plant records for fruit flies (Diptera: Tephritidae) in Southeast Asia. Raffles. B. Zool. 47(Supplement 7), 1–92 (1999).
Liu, X. F. & Ye, H. Research progress of Bactrocera correcta (Bezzi) in Yunnan. Chinese J. Biol. Control. 25, 86–90 (2009).
Liu, X. F., Jin, Y. & Ye, H. Recent spread and climate ecological niche of the invasive guava fruit fly, Bactrocera correcta, in mainland China. J. Pest Sci. 86, 449–458 (2013).
Liu, J. Q., Deng, Y. L., Li, Z. H., Bai, Y. H. & Huang, L. G. Advances in molecular biology of Bactrocera (Bactrocera) correcta. Plant Protection. 35, 11–14 (2009).
Bezzi M. On the fruit-flies of the genus Dacus occurring in India, Burma and Ceylon. B. Entomol. Res. 7, 99–121 (1916).
Clarke, A. R. et al. Invasive phytophagous pests arising through a recent tropical evolutionary radiation: the Bactrocera dorsalis complex of fruit flies. Annu. Rev. Entomol. 50, 293–319 (2005).
Bezzi, M. Indian tephritids (fruit flies) in the collection of the Indian Museum, Calcutta. Mem. Indian Mus. 3, 153–175 (1913).
CPC, CABI. Crop Protection Compendium. http://www.cabi.org/cpc/datasheet/8703 (2015).
Drew, R. A. I. & Raghu, S. The fruit fly fauna (Diptera: Tephritidae: Dacinae) of the rainforest habitat of the western Ghats, India. Raffles. B. Zool. 50, 327–352 (2002).
Liang, G. Q., Yang, G. H., Liang, F., Situ, B. L. & Liang, X. D. Insects of Bactrocera (Dipetra: Tephritidae) in Asia-Pacific area Guangdong Science and Technology Press, Guangdong (1996).
Wang, X. J. & Zhao, M. Z. Species of genus Dacus Fabricius (Diptera: Tephritidae) from China. Acta Zootaxonomica Sinica 14, 209–219 (1989).
Foote, R. H., Blanc, F. L. & Norrbom, A. L. Handbook of the fruit flies (Diptera: Tephritidae) of America north of Mexico Comstock Publishing Associates, New York (1993).
Wang, X. J. & Zhao, M. Z. Notes on the genus Dacus Fabricius in China with descriptions of five new species. Acta Zoologica Sinica. 14, 209–219 (1989).
Liu, J. H., Shi, W. & Ye, H. Population genetics analysis of the origin of the Oriental fruit fly, Bactrocera dorsalis Hendel (Diptera: Tephritidae), in northern Yunnan province, China. Entomol. Sci. 10, 11–19 (2007).
Shi, W., Kerdelhue, C. & Ye, H. Genetic structure and inferences of potential source areas for Bactrocera dorsalis (Hendel) based on mitochondrial and microsatellite markers. Plos One 7, e37083 (2012).
Liu, X. F., Wang, D. M. & Ye H. Overview on research of Bactrocera correcta (Bezzi). Trop. Agric. Sci. Technol. 28, 30–33 (2005).
Qin, Y. J. et al. The potential geographic distribution of Bactrocera correcta (Diptera: Tephrididae) in China based on eclosion rate model. Appl. Entomol. Zool. 1–11 (2015).
Estoup, A. & Guillemaud, T. Reconstructing routes of invasion using genetic data: why, how and so what? Mol. Ecol. 19, 4113–4130 (2010).
Roderick, G. K. Geographic structure of insect populations: gene flow, phylogeography, and their uses. Annu. Rev. Entomol. 41, 263–290 (1996).
Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome C oxidase subunit I from diverse metazoan invertebrate. Molecular Marine Biology and Biotechnology. 3, 294–299 (1994).
Nardi, F., Carapelli, A., Dallai, R., Roderick, G. K. & Frati, F. Population structure and colonization history of the olive fly, Bactrocera oleae. Mol. Ecol. 14, 2729–2738 (2005).
Roderick, G. K. Tracing the origin of pests and natural enemies: genetic and statistical approaches. In Genetics, Evolution, and Biological Control CAB International, Wallingford, UK, 97–112 (2004).
Smith, P. T. & Mitochondrial D. N. A. variation among populations of the glassy-winged sharpshooter, Homalodisca coagulate. J. Insect Sci. 5, 41 (2005).
Aketarawong, N. et al. Inferences on the population structure and colonization process of the invasive oriental fruit fly, Bactrocera dorsalis (Hendel). Mol. Ecol. 16, 3522–3532 (2007).
Li, Y. L., Wu, Y., Chen, H., Wu, J. J. & Li, Z. H. Population structure and colonization of Bactrocera dorsalis (Diptera: Tephritidae) in China inferred from mtDNA COI sequences. J. Appl. Entomol. 136, 241–251 (2012).
Wu, Y., McPheron, B. A., Wu, J. J. & Li, Z. H. Genetic relationship of the melon fly, Bactrocera cucurbitae (Diptera: Tephritidae) inferred from mitochondrial DNA. Insect Sci. 19, 195–204 (2012).
Kunprom, C., Sopaladawan, P. N. & Pramual, P. Population genetics and demographic history of guava fruit fly Bactrocera correcta (Diptera: Tephritidae) in northeastern Thailand. Eur. J. Entomol. 112, 227 (2015).
Buahom, N. Population genetic structure of Bactrocera correcta (Diptera: Tephritidae) in main distributions areas. China Agricultural University, Ph. D thesis (In Chinese) (2011).
ArcGIS (version 10.2). ESRI Inc., Redlands, CA, USA. http://www.esri.com/software/arcgis/arcgis-for-desktop.
Khamis, F. M. et al. Taxonomic Identity of the Invasive Fruit Fly Pest, Bactrocera invadens: Concordance in Morphometry and DNA Barcoding. Plos One. 7, E44862 (2012).
Liu, L. J. et al. Identification of Bactrocera invadens (Diptera: Tephritidae) from Burundi, based on morphological characteristics and DNA barcode. Afr. J. Biotechnol. 10, 13623–13630 (2011).
Thomson, J. D., Higgins, D. G. & Gibson, T. J. Clustal W: improving the sensitive of progressive multiple sequence alignments through sequence weighting, position specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680 (1994).
Buahom, N. et al. Polymorphic microsatellite markers in the guava fruit fly, Bactrocera correcta (Diptera: Tephritidae). Appl. Entomol. Zool. 48, 409–412 (2013).
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 (2011).
Librado, P. & Rozas, J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 25, 1451–1452 (2009).
Yeh, F. C., Yang, R. C. & Boyle, T. Popgene version 1.31, free software. University of Alberta, Canada (1999).
Goudet J. FSTAT (Version 2.9.3). Institute of Ecology, Biology Building, University of Lausanne. http://www2.unil.ch/popgen/softwares/fstat.htm (2002).
Raymond, M. & Rousset, F. GENEPOP (Version 1.2): Population genetics software for exact tests and ecumenicism. J. Hered. 86, 248–249 (1995).
Rice, W. R. Analyzing tables of statistical tests. Evolution. 43, 223–225 (1989).
Rouset, F. Genetic differentiation and estimation of gene flow from F-statistics under isolation by distances. Genetics. 145, 1219–1228 (1997).
Legendre, P. & Legendre, L. Numerical Ecology: Second English Edition Elsevier, Amsterdam (1998).
Google Earth (version 5.2). Google Inc, Mountain View, CA, USA (2011).
Bandelt, H. J., Forster, P. & Rohl, A. Median-Joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 191, 37–48 (1999).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics. 155, 945–959 (2000).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Earl, D. A. Structure Harvester: a website and program for visualizing structure output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361 (2011).
Jakobsson, M. & Rosenberg, N. A. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 23, 1801–1806 (2007).
Rosenberg, N. A. Distruct: a program for the graphical display of population structure. Mol. Ecol. Notes. 4, 137–138 (2004).
Dupanloup, I., Schneider, S. & Excoffier, L. A simulated annealing approach to define the genetic structure of populations. Mol. Ecol. 11, 2571–81 (2002).
Piry, S. et al. GENECLASS 2: a software for genetic assignment and first-generation migrant detection. J. Hered. 95, 536–539 (2004).
Rannala, B. & Moutain, J. L. Detecting immigration by using multilocus genotypes. P. Natl. Acad. Sci. USA 94, 9197–9201 (1997).
Paetkau, D., Slade, R., Burden, M. & Estoup, A. Genetic assignment for the direct, real-time estimation of migration rate: a simulation-based exploration of accuracy and power. Mol. Ecol. 13, 55–65 (2004).
He, D. M. et al. A study of ecosystem changes in longitudinal range-gorge region and transboundary eco-security in southwest China. Advances in Earth Science. 20, 932–943 (2005).
Jiang, X. L. Risk analysis of quarantine fruit flies along Yunnan border. Journal of Southwest Agricultural University. 24, 402–405 (2002).
Li, J. Y. The study of relationship between border trade and economic growth in Yunnan. Journal of Management. 26, 28–37 (2013).
Duan, X. M. & Liu, W. Conservation planning of plant diversity in Jinghong City of Xishuangbanna. Journal of Anhui Agricultural Science. 38, 9333–9335 (2010).
Zhou, W. L. & Zhuang, L. J. The study on situation and countermeasures of agricultural product trade between Yunnan and ASEAN. Commercial Research. 3, 185–188 (2009).
Duyck, P. F., David, P. & Quilici, S. A review of relationship between interspecific competition and invasions in fruit flies (Diptera: Tephritidae). Ecology Entomology. 29, 511–520 (2004).
Gasparich, G. E. et al. Population genetic structure of the Mediteranean fruit fly (Diptera: Tephritidae) and implications for worldwide colonization pattern. Ann. Entomol. Soc. Am. 90, 790–797 (1997).
Lanzavecchia, S. B. et al. Origin and distribution of Ceratitis capitata mitochondrial DNA haplotypes in Argentina. Ann. Entomol. Soc. Am. 101, 627–638 (2008).
Rollins, L. A., Woolnough, A. P., Sinclair, R., Mooney, N. J. & Sherwin, W. B. Mitochondrial DNA offers unique insights into invasion history of the common starling. Mol Ecol. 20, 2307–2317 (2011).
Acknowledgements
We would like to thank all other members from the Plant Quarantine and Invasion Biology Laboratory (CAUPQL) of China Agricultural University. This research was supported by the National Natural Science Foundation of China (No. 30971916) and the National Public Welfare Program (No. 201010042). Thanks the comments from the anonymous reviewers on the earlier version.
Author information
Authors and Affiliations
Contributions
Z.H.L. and N.B. conceived the ideas for the study; N.B., Z.H.L., J.Q.L., Y.D., Y.L.D. and X.L.J. collected the samples; N.B. performed the experiments; N.B., Y.J.Q., Y.W., J.Q.L. and M.N.K. analyzed the data; Y.J.Q., N.B., A.R.M., M.N.K. and Z.H.L. composed the manuscript. All authors have read and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Qin, YJ., Buahom, N., Krosch, M. et al. Genetic diversity and population structure in Bactrocera correcta (Diptera: Tephritidae) inferred from mtDNA cox1 and microsatellite markers. Sci Rep 6, 38476 (2016). https://doi.org/10.1038/srep38476
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep38476
This article is cited by
-
Genetic Analysis of Populations of the Peach Fruit Fly, Bactrocera zonata (Diptera: Tephritidae), in Iran
Neotropical Entomology (2019)
-
Microsatellite evidence of dispersal mechanism of red swamp crayfish (Procambarus clarkii) in the Pearl River basin and implications for its management
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
