
Abstract
When produced biologically, especially by photosynthetic organisms, hydrogen gas (H2) is arguably the cleanest fuel available. An important limitation to the discovery or synthesis of better H2-producing enzymes is the absence of methods for the high-throughput screening of H2 production in biological systems. Here, we re-engineered the natural H2 sensing system of Rhodobacter capsulatus to direct the emission of LacZ-dependent fluorescence in response to nitrogenase-produced H2. A lacZ gene was placed under the control of the hupA H2-inducible promoter in a strain lacking the uptake hydrogenase and the nifH nitrogenase gene. This system was then used in combination with fluorescence-activated cell sorting flow cytometry to screen large libraries of nitrogenase Fe protein variants generated by random mutagenesis. Exact correlation between fluorescence emission and H2 production levels was found for all automatically selected strains. One of the selected H2-overproducing Fe protein variants lacked 40% of the wild-type amino acid sequence, a surprising finding for a protein that is highly conserved in nature. We propose that this method has great potential to improve microbial H2 production by allowing powerful approaches such as the directed evolution of nitrogenases and hydrogenases.
Similar content being viewed by others

Introduction
Biological H2 production is a promising source of renewable energy. Microorganisms produce H2 by the activity of nitrogenases and hydrogenases1. Nitrogenases catalyze the reduction of N2 with the limiting stoichiometry N2+8 H+ + 8e−+16MgATP+16H2O↔H2+2NH3+16MgADP+16Pi in a process known as biological nitrogen fixation, which produces H2 as a by-product2. On the other hand, hydrogenases catalyze the reversible 2 H+ + 2e−↔H2 reaction. H2 metabolism has been well studied in purple non-sulfur bacteria (PNS), a group of microorganisms notable for their metabolic versatility. PNS can grow photoautotrophically, photoheterotrophically, chemoorganotrophically, and chemolitotrophically with H2 as an electron donor and O2 as an electron acceptor3. H2 production by PNS generally occurs during photoheterotrophic anaerobic growth and is mainly due to nitrogenase4.
Rhodobacter capsulatus, the model PNS used in this study5, carries two genetically distinct nitrogenases (a Mo-nitrogenase and an Fe-only nitrogenase)6, which are differentially expressed depending on the Mo availability in the medium7, and two hydrogenases, a membrane-bound [Ni-Fe] hydrogenase for H2 uptake and a cytosolic [Ni-Fe] hydrogenase for H2 sensing8. Nitrogenases are two-component metalloproteins comprising an N2-reducing dinitrogenase (MoFe protein) and a dinitrogenase reductase (Fe protein) acting as an obligate electron donor9. These components are encoded by nifDK and nifH, respectively, in the case of the Mo-nitrogenases, and by anfDGK and anfH in the case of the Fe-only nitrogenases10. The uptake hydrogenase is a heterodimer of the hupA and hupB (formerly hupS and hupL) gene products11, in which activity is linked to the respiratory chain by the cytochrome b-containing protein HupC. Structural, maturation-related and regulatory genes for the uptake hydrogenase are clustered in the genome of R. capsulatus12 (Fig. S1).
In R. capsulatus, a two-component signal transduction system activates the transcription of the hup gene cluster in the presence of H2 13. The H2-sensing system comprises three elements: a cytosolic [Ni-Fe] hydrogenase, HupUV; a histidine kinase, HupT; and a response regulator, HupR. In the absence of H2, HupUV and HupT interact, causing HupT autophosphorylation and the transfer of a phosphate group to HupR, which in this state is unable to activate the transcription. In the presence of H2, HupUV binds H2 and HupT is released. In this state, phosphotransfer between HupT and HupR is not favored, and the unphosphorylated HupR binds to promoter DNA and activates the transcription of uptake hydrogenase genes. Similar regulatory systems are found in other bacterial species such as Bradyrhizobium japonicum14 and Ralstonia eutropha15.
Due to its great commercial significance, nitrogenase has been the subject of extensive biochemical, genetic and structural analyses. Nonetheless, it has proven difficult to find or engineer strains of microbes carrying nitrogenases with significantly increased H2 production efficiency. In this work, we present a method for the high-throughput selection of nitrogenase variants with enhanced H2 production. An R. capsulatus strain has been re-engineered to generate a fluorescent signal in response to nitrogenase-produced H2. A combination of nifH random mutagenesis and fluorescence-activated cell sorting (FACS) is then used to select the H2-overproducing nitrogenase variants in the R. capsulatus sensor strain (Fig. 1A).

Construction of a biosensor to select H2-overproducing nitrogenase variants.
(A) Biosensor design. NifA-dependent expression libraries of nifH variants are randomly generated by error-prone PCR. In each R. capsulatus cell, HupUV detects H2 produced by a nitrogenase variant, and the signal is transduced to regulate the expression of lacZ, which serves as a reporter by catalyzing fluorescein isothiocyanate formation. FACS is then used to sort the cells and select those emitting fluorescence in a range at least an order of magnitude larger than the population average. The selected cells are finally subjected to further measurements of the fluorescence emission and H2 production. (B) Response of sensor strains to 10% H2 added to the culture gas phase. (C) Response of sensor strains to H2 produced by nitrogenase activity. Data represent the mean ± SD (n = 4). Strains: S1 (PhupA::lacZ), RC25 (ΔhupAB), S2 (PhupA::lacZ, ΔhupAB), RC54 (ΔhupR), RC24 (ΔhupT), RC54-S1 (PhupA::lacZ, ΔhupR), RC24-S1 (PhupA::lacZ, ΔhupT), and S3 (PhupA::lacZ, ΔhupAB, ΔnifH).
Results
Genetic modules for the selection of H2-overproducing nitrogenase variants
The construction of two genetic modules was required to perform high-throughput experiments to obtain nitrogenase variants with improved H2 production: a module expressing over a million random variants of dinitrogenase reductase (NifH) per experiment and a reporter module directing the emission of a visible signal in response to H2.
The reporter module was constructed in four steps. In a first step, an 874-bp DNA fragment comprising a promoter sequence upstream of hupA16 was translationally fused to lacZ in the replicative vector pMP220 to generate pRHB502. As illustrated in Fig. 1A, the expression from hupA promoter is activated by HupR in response to H2. The in vivo β-galactosidase activity of R. capsulatus cells harboring pRHB502 (RC4) was 600-fold higher than that of the control strain RC3 (carrying pMP220) and responded positively to the presence of 10% H2 in the culture gas phase, which confirmed the induction of the transcription from PhupA by H2 (Fig. S2A).
In a second step, the reporter dose was adjusted by integrating PhupA::lacZ between hypF and hupA in the R. capsulatus chromosome to generate the S1 strain (Fig. S1). The S1 strain exhibited a much lower β-galactosidase activity background level and a larger fold increase in the activity in response to the external H2 than RC4 (Fig. S2B).
In a third step, the reporter response to H2 was adjusted by mutating the genes involved in the H2 signal transduction pathway and metabolism. S1 derivative strains lacking hupAB structural genes for the uptake-hydrogenase (RC25-S1, also termed S2), the H2 response regulator encoding gene hupR (RC54-S1), or the histidine kinase gene hupT (RC24-S1) were generated, and their responses to H2 were analyzed (Fig. 1B and Fig. S3), obtaining the following results. First, the response to 10% exogenous H2 improved 5-fold in the ΔhupAB strain S2 compared to in S1; second, the constitutive activation of PhupA was observed in the ΔhupT strain RC24-S1; and third, no response to H2 was observed in the ΔhupR strain RC54-S1. These results were in agreement with previous reports13,16,17,18 indicating proper control by the R. capsulatus H2-sensing system and therefore permitting further H2 sensor development through the use of strain S2.
The deletion of hupAB genes in the RC25 strain completely eliminated its in vivo uptake hydrogenase activity (nil compared to 2504 ± 450 nmol H2 h−1 OD600−1 in the wild-type strain). Thus, in contrast to the wild-type strain, the S2 sensor strain (derived from RC25) evolved high levels of H2 under diazotrophic growth conditions (Fig. S2C) in spite of having similar levels of in vivo nitrogenase activity (Fig. S2D). In addition, the X-gal and H2-dependent signal-to-noise ratio was much better in S2 than in S1 cultures (Fig. S2E). It was therefore concluded that the elimination of the uptake hydrogenase activity would facilitate the detection of intracellular H2 produced during the nitrogen fixation process.
The S2 response to the H2 produced by the nitrogenase was evaluated in intact bacterial cells by using a fluorescent MUG-dependent β-galactosidase activity assay19, which permitted high-throughput growth and screening in a 96-well format as well as the recovery of viable selected clones. R. capsulatus strains were grown in 96-well microplates both under non-diazotrophic and diazotrophic conditions inside a glovebox, and their β-galactosidase activities were determined. Figure 1C shows that the β-galactosidase activity in S2 was 15-fold higher under diazotrophic conditions compared to non-diazotrophic conditions, validating its use as a biosensor. Importantly, all tested strains with mutations in H2 the metabolism or signal transduction pathway responded identically to nitrogenase-produced H2 and exogenous added H2 (compare Fig. 1C to Fig. S3).
Finally, the removal of endogenous nifH was necessary to use the sensor in combination with the genetic module consisting of an expression library of nifH variants (Fig. S4). The S3 sensor strain (ΔnifH ΔhupAB PhupA::lacZ) β-galactosidase activity levels in response to H2 were identical to those of S2 (Fig. 1B), demonstrating that the absence of nifH did not modify the capacity to detect H2. Importantly, the introduction of the pRHB576 expression vector carrying a wild-type copy of nifH under the control of its own promoter restored the in vivo nitrogenase activity of strain S3 (Fig. S5), validating its use to screen the activity of the nifH variants in S3. Interestingly, a general decrease in the nitrogenase activities was observed in the sensor S3-derived strains with respect to the wild-type R. capsulatus. This effect was associated with carrying the expression vector pBBR1MCS-3 (Fig. S6).
The second genetic module consisted of an expression library of randomly generated nitrogenase variants. The mutation analysis was initially constrained to nifH because the limiting steps in the nitrogenase catalysis are the dissociation of the NifH and NifDK components and the release of Pi from NifH2. Random nifH variants (nifHv) having an average of 5 amino acid changes (Table S1) were generated by error-prone PCR, ligated into pRHB602, and introduced into E. coli DH5α to obtain PnifH::nifH expression libraries (~ 4 × 106 clones per library). No obvious mutational hotspots were observed after sequencing 20 nifH variants per library.
Finally, the two genetic modules were combined by introducing the nifHv-expression libraries into R. capsulatus sensor strain S3. The mating process resulted in ~8 × 105 clones per library, a number that required high-throughput screening methods.
High-throughput selection of H2-overproducing nifH variants
R. capsulatus S3 libraries expressing nifH variants (nifHv-S3) were grown under nitrogenase-derepressing conditions inside a glovebox. After treatment with fluorescein di-β-D-galactopyranoside (FDG), culture samples were screened by FACS flow cytometry. Approximately 2 × 105 events were processed per sample. R. capsulatus cells emitting fluorescence at levels significantly higher than the main population (0.024% of total population, see P2 area in Fig. 2A) were sorted by the cytometer into separate wells of 96-well microplates containing growth medium.

High-throughput selection of cells carrying H2-overproducing NifH variants.
(A) Fluorescence-activated cell sorting. Dot-plot showing side-scattered light (SSC) versus fluorescence generated by fluorescein isothiocyanate (FITC) in pRHB602-S3 (no nifH), pRHB576-S3 (wild-type nifH), and nifHv-S3 (nifH variant pool) cell populations. P2 indicate the areas in which sorted cells exhibited fluorescence signals above the desired threshold. (B) β-Galactosidase activity of nifHv-S3 clones sorted by FACS as determined by MUG hydrolysis in a 96-well plate format. Blue and red bars represent pRHB602-S3 and pRHB576-S3 activities, respectively. Black bars represent activities of clones carrying nifH variants further selected to determine in vivo H2 production.
S3 derivatives transformed with empty expression vector (pRHB602-S3) or wild-type nifH-containing expression vector (pRHB576-S3) were used as negative and positive controls, respectively. pRHB602-S3 populations emitted the lowest average level of fluorescence, and only 0.008% of the population was found above the set threshold in the P2 area. On the other hand, the average fluorescence emission in the pRHB576-S3 populations was the highest, although only 0.0039% of the population was above the selective threshold.
The nifHv-S3 cells sorted by FACS were then cultured under nitrogenase-derepressing conditions using serine as a poor nitrogen source inside a glovebox for a secondary MUG-based fluorescence screen. Growth was observed in approximately 25% of the inoculated wells. Figure 2B shows the β-galactosidase activity levels of 67 nifHv-S3 cultures along with those of the pRHB602-S3 (blue) and pRHB576-S3 (red) cultures used as controls. The β-galactosidase activity was 2 to 12-fold higher in the nifHv-S3 cultures than in the pRHB576-S3 strain (carrying wild-type nifH). Importantly, 85% of the nifHv-S3 clones harbored an expression vector with a nifHv gene, as determined by PCR analysis. The strong correlation of the results from the FACS primary screening and the secondary MUG screening in the 96-well format validates the use of flow cytometry as a high-throughput method to screen nifHv-S3 libraries.
Nitrogenase-dependent H2 overproduction in selected strains with nifH variants
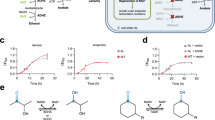
Based on the β-galactosidase activity levels, R. capsulatus strains V1, V7, V8, V10, V17, V18, V20 and V21 (Fig. 2B, black bars), carrying the corresponding nifH variants, were selected for in vivo H2 production assays. All these strains produced more H2 than the pRHB576-S3 strain carrying wild-type nifH (Fig. 3A), with the V1 and V7 strains consistently producing up to 10-fold more H2. Importantly, the acetylene reduction activity was almost abolished in the strains carrying H2-overproducing nifH variants (Fig. 3B), a result consistent with the random mutagenesis process designed to select only for improved H2 production. The ratio of H2 to ethylene production in V7 strain was 6000-fold higher than in pRHB576-S3. Neither H2 nor ethylene was produced in the absence of nifH (see strain pRHB602-S3 in Fig. 3), indicating that the H2 production was dependent on the nifH expression.

Characterization of selected nifHv-S3 strains.
H2 production (A) and acetylene reduction activity (B) of highly fluorescent R. capsulatus strains carrying nifH variants. Strains pRHB602-S3 (no nifH) and pRHB576-S3 (wild-type nifH) were used as controls. Data represent the mean ± SD (n = 4). (C) Stereoview overlap of 3D-structural models for R. capsulatus NifH (purple) and the NifH-V7 variant (green). Both proteins are expected to form homodimers with a [4Fe-4S] cluster at the subunit interface. (D) Correlation of H2 production (nmol ml−1 hour−1) and presence of NifH-V7. Data represent the mean ± SD (n = 4). (E) Immunodetection of nitrogenase components NifH and NifDK in strains carrying or lacking the NifH-V7 variant. pRHB602-S3 and pRHB576-S3 were used as controls. Lower panel shows protein loading in each sample as determined by the Coomassie staining of the corresponding SDS gels.
The nature of the mutations in the nifH-V1 and nifH-V7 variants was determined by DNA sequencing (Table S2 and Fig. S7). A frame shift mutation was found in codon 172 of nifH-V7 that would result in a NifH protein lacking 124 amino acids at its C-terminus. In addition, NifH-V7 would carry G32C and A107T amino acid substitutions. This was surprising because NifH is a highly conserved protein, and such a drastic truncation would be expected to yield an inactive protein. An overlap of the three-dimensional models of R. capsulatus NifH and NifH-V7 is shown in Fig. 3C. The overall protein overlap (4–171 amino acid residues) and an RMSD value of 0.900 Angstroms for the protein backbone atoms indicate very similar model architectures. However, subtle differences were observed in the loops near the [4Fe-4S] cluster, where residues K43, A44, A100, G101, R102 and G115 did not overlap.
To confirm that the H2 overproduction in V7 was dependent on the activity of both Mo-nitrogenase component proteins, the following two experiments were performed. First, the nifH-V7 expression plasmid was cured from the V7 strain by passing five times under a nonselective media. Plasmid elimination was confirmed by PCR analysis and by growth inhibition on media supplemented with tetracycline (Tc). The resulting strain, V7C, was unable to produce H2 above the levels of the pRHB602-S3 control strain (0.42 nmol H2 h−1 OD600−1 ml−1) (Fig. 3D). The reintroduction of the nifH-V7 expression plasmid by mating completely restored the H2-producing activity (compare H2 productions of V7 and V7′ in Fig. 3D). The presence of a truncated form of NifH in V7 and its elimination in V7C were confirmed by an immunoblot analysis (Fig. 3E). This analysis also showed that all strains accumulated similar levels of nitrogenase component proteins, ruling out the possibility of increased accumulation underlying the H2 overproduction by V7.
Second, R. capsulatus sensor strains lacking either nifH (strain S3) or nifHDK (strain S5) were complemented with an expression plasmid harboring nifH-V7. While the S3-V7 cultures produced large amounts of H2, background levels were detected in the S5-V7 cultures (Fig. S8). This result demonstrates that the NifH-V7 dependent H2 production also requires the presence of NifDK and is not due to the activity of other cellular proteins, such as the Fe-only nitrogenase.
Discussion
There are many reports of metabolic engineering aiming to increase microbial H2 production. Genetic approaches include deleting hydrogenase and/or nitrogenase structural and regulatory genes20,21,22,23,24,25, eliminating Rubisco26, lowering intracellular O2 levels27, re-engineering hydrogenase to increase its tolerance to O228, and modifying nitrogenase substrate selectivity by site-directed mutagenesis29,30. Nonetheless, it has been difficult to find or engineer vastly improved H2-overproducing enzymes or microbial strains.
Directed evolution mimics biological evolution in the laboratory and is an effective approach to change enzyme properties such as catalytic turnover31. Directed evolution requires a strategy to generate libraries with a large number of variants and a method capable of screening or selecting for the best variants in a large pool. In this work, we have combined the random mutagenesis of nifH and high-throughput screening to improve nitrogenase H2-producing activity. We hypothesized that the nitrogenase H2 production could be greatly improved if no selection was applied to maintain the N2-reducing activity. The nifH gene was selected to obtain a proof of concept because the limiting steps in nitrogenase catalysis are the NifH/NifDK complex dissociation and the release of Pi from NifH2. FACS flow cytometry was used as a high-throughput method to select H2-overproducing NifH variants from the pool. A genetic module endowing a NifH-expressing sensor strain with the capacity to emit light in proportion to the amount of H2 detected was required for this selection. The screening of bacterial libraries by FACS flow cytometry had been performed before in directed evolution procedures32.
A number of H2-overproducing NifH variants were obtained. Perhaps the most interesting was the V7 variant, which lacks 124 amino acid residues at the C-terminus of the protein (Fig. 3C). In wild-type NifH, ATP binding and hydrolysis is required for electron transfer to NifDK during catalysis2. Interestingly, NifH-V7 lacks all residues shown to enable hydrogen bonding to the adenine base (Asp185, Gln218, and Gln236 in the Azotobacter vinelandii NifH), suggesting that NifH-V7 might have lost its specificity for Mg·ATP over other nucleotides33. The mechanism by which NifH-V7 is capable of sustaining H+ reduction merits further investigation.
Other biological and chemical methods have also been developed to detect H2-producing microorganisms. Chemochromic transparent sensor films that turn blue in the presence of H2 and indicators consisting of a coloring agent and a water-soluble derivative of Wilkinson’s catalyst have been used for phenotypic screenings of Chlamydomonas reinhardtii34,35 and R. capsulatus36, respectively. Moreover, a biosensor to screen algal H2 production was developed starting from the R. capsulatus H2-sensing system37,38. However, all these methods suffer from limitations in the number of clones that could be screened per experiment, making directed evolution techniques impossible to apply.
Conclusions
In this work, we have re-engineered the natural H2 sensing system of R. capsulatus and combined it with FACS for the high-throughput selection of nitrogenase variants with enhanced H2 production that independently retain their N2 fixation activity. This method allows screening 105–106 variants per experiment, thus permitting enzyme improvement by directed evolution. This technology possesses great potential to identify nitrogenase amino acid substitutions leading to H2-overproducing variants that could be mimicked in nitrogenases from other microorganisms, expanding the impact of the findings. In addition, it might be used for the genome-wide screening of mutations leading to enhanced H2 production in R. capsulatus.
Materials and Methods
β-galactosidase activity assays
Transcription from PhupA was estimated by measuring β-galactosidase activity of R. capsulatus strains carrying PhupA::lacZ transcriptional fusions. Cells were cultured at 30 °C for 7 h, transferred to assay tubes (0.7 ml of culture), permeabilized by addition of 20 μl of chloroform and 10 μl of 0.1% SDS, and β-galactosidase activity estimated at 28 °C as described39.
When the 96-well microplate format was used, β-galactosidase activity assays were carried out as described in19 with modifications. R. capsulatus cultures were incubated overnight under diazotrophic conditions inside a glove box in a 96-well plate (black/clear Optilux™ flat bottom; BD Biosciences) covered with a transparent adhesive sealer. One hundred and twenty μl of each culture were transferred to a 96-well microplate containing 100 μl of Z-Buffer in each well39, then supplemented with 25 μl of 4-methylumbelliferone β-D-galacto-pyranoside (MUG; 1 mg/ml stock solution in dimethyl sulfoxide), and incubated at room temperature for 2 h in darkness. MUG hydrolysis by β-galactosidase was quantified by fluorescence emission at 445 nm (372 nm excitation wavelength) in a Genios Pro (Tecan) microplate fluorometer.
Construction of random mutagenesis libraries
Random mutagenesis of nifH was carried out by Error-Prone PCR using PCR GeneMorph® II Random Mutagenesis Kit (Stratagene) according to the manufacturer’s instructions. Reaction mixtures contained 1 μl DNA template (0.4 ng of pRHB529 including 0.1 ng of nifH), 5 μl of 10X Mutazyme II reaction buffer, 1 μl of 40 mM dNTP mix (200 μM each final), 0.5 μl of primer mix (250 ng/μl of each primer), 1 μl of Mutazyme II DNA polymerase (2.5 U/μl), and 41.5 μl of H2O. Primers P19 and P20 were used to amplify nifH gene by Mutazyme II DNA polymerase. PCR conditions used were: 95 °C for 2 min, followed by 30 cycles of 95 °C for 30 sec, 55 °C for 30 sec, and 72 °C for 1 min, and finished with an incubation at 72 °C for 10 min. Amplified DNA was digested with NdeI and XbaI, ligated into pRHB602, and introduced into E. coli DH5α competent cells (NEB, C2987I) by heat shock to generate a expression library of nifH variants. On average, ~ 4 × 106 transformants were recovered per 100 ng DNA.
In vivo hydrogenase activity assays
Hydrogenase activity was measured by using a Clark-type hydrogen microelectrode (Unisense) with O2 as electron acceptor40. When necessary, hydrogenase expression was induced by injecting 9 ml of H2 into 100 ml-capped vials containing 10 ml of R. capsulatus cultures and incubating under culture conditions for 6 h.
In vivo nitrogenase activity assays
To determine acetylene reduction activity in R. capsulatus cultures grown under diazotrophic conditions, 1-ml samples were transferred to 9-ml sealed vials with a 94% N2/6% acetylene gas phase and incubated at 30 °C in the light for 1 h. Ethylene formation was detected in 50 μl samples withdrawn from the gas phase by using a Shimadzu GC-2014 gas chromatographer equipped with a 9-ft long, 1/8-in diameter Porapak R column. In vivo nitrogenase activity units are defined as nmol ethylene formed per min per ml of culture at an OD600 equal to 1.
To determine H2 production in R. capsulatus cultures grown under diazotrophic conditions, 16-ml samples were transferred to 23-ml sealed vials with a 100% N2 atmosphere and incubated at 30 °C in the light for 48 h. H2 formation was detected in 250 μl samples withdrawn from the gas phase by using a Shimadzu GC-8A gas chromatographer equipped with a 6-ft long, 1/8-in diameter Molecular Sieve column. Activity units are defined as nmol H2 formed per h in a culture at an OD600 equal to 1.
Protein methods
Protein concentration was determined by the bicinchoninic acid method (Pierce) with bovine serum albumin as the standard41. For SDS-PAGE, cells from 1-ml culture samples were collected by centrifugation, resuspended in 2 × Laemmli sample buffer supplemented with 0.1 M dithiothreitol (to a concentration equivalent to an OD600 of 4), and electrophoresed in 12% acrylamide/bisacrylamide (29:1) gels. For immunoblot analysis proteins were transferred to nitrocellulose membranes for 40 min at 20 V using a Transfer-Blot® Semi Dry system (Bio-Rad). Immunoblot analyses were carried out with antibodies raised against a 1:1 mixture of A. vinelandii and Rhodospirillum rubrum NifH proteins (1:2,500 dilution) or with antibodies raised against R. capsulatus NifDK (1:2,000 dilution; antibody kindly donated by Yves Jouanneau, CNRS, Grenoble). Secondary alkaline phosphatase-conjugated anti-rabbit antibodies (Sigma, A3687) were used at 1:5,000 dilution.
Flow cytometry
Cells from 50-ml R. capsulatus cultures under diazotrophic conditions inside a glove box were collected by centrifugation in Falcon tubes for 15 min at 4 °C, 4500 × g, resuspended in 5 ml PBS supplemented with 10% glycerol, and incubated for 30 min at 4 °C. Cells were then collected, resuspended in 1 ml of an 8:1:1 mixture of PBS, fluorescein di-β-D-galactopyranoside (FDG) and propidium iodide (PI), and incubated at 37 °C for 30 min to facilitate FDG entrance into the cells. FDG releases fluorescein when cleavage by β-galactosidase42. Cells were collected by centrifugation, resuspended in RCV medium supplemented with Tc, and analyzed in a FACSVantage (sorter) flow cytometer using an argon ion laser to excite the fluorochrome (488 nm). Cells exhibiting high fluorescence levels were sorted and recovered in 96-well microplates containing YPS medium supplemented with Tc.
NifH 3-D models
3-D model of R. capsulatus NifH and the V7 variant were generated by homology modeling (http://swissmodel.expasy.org/) using the A. vinelandii NifH structures as template (1NIP for wild-type NifH and 1G5P for NifH-V7, respectively). Both models yielded NifH homodimer with capability to ligate [4Fe-4S] clusters.
Additional Information
How to cite this article: Barahona, E. et al. Hydrogen overproducing nitrogenases obtained by random mutagenesis and high-throughput screening. Sci. Rep. 6, 38291; doi: 10.1038/srep38291 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Hallenbeck, P. C. & Ghosh, D. Advances in fermentative biohydrogen production: the way forward? Trends Biotechnol. 27, 287–297 (2009).
Hoffman, B. M., Lukoyanov, D., Yang, Z. Y., Dean, D. R. & Seefeldt, L. C. Mechanism of nitrogen fixation by nitrogenase: the next stage. Chem. Rev. 114, 4041–4062 (2014).
Hunter, C. N., Daldal, F. C. T. M. & Beatty, J. T. In Advances in Photosynthesis and Respiration Vol. 28 (Springer, Dordrecht, The Netherlands, 2008).
Kranz, R. G. & Haselkorn, R. Anaerobic regulation of nitrogen-fixation genes in Rhodopseudomonas capsulata. Proc. Natl. Acad. Sci. USA. 83, 6805–6809 (1986).
Strnad, H. et al. Complete genome sequence of the photosynthetic purple nonsulfur bacterium Rhodobacter capsulatus SB 1003. J. Bacteriol. 192, 3545–3546 (2010).
Scolnik, P. A. & Haselkorn, R. Activation of extra copies of genes coding for nitrogenase in Rhodopseudomonas capsulata. Nature 307, 289–292 (1984).
Masepohl, B. & Hallenbeck, P. C. Nitrogen and molybdenum control of nitrogen fixation in the phototrophic bacterium Rhodobacter capsulatus. Adv. Exp. Med. Biol. 675, 49–70 (2010).
Vignais, P. M. & Billoud, B. Occurrence, classification, and biological function of hydrogenases: an overview. Chem. Rev. 107, 4206–4272 (2007).
Bulen, W. A. & LeComte, J. R. The nitrogenase system from Azotobacter: two-enzyme requirement for N2 reduction, ATP-dependent H2 evolution, and ATP hydrolysis. Proc. Natl. Acad. Sci. USA. 56, 979–986 (1966).
Loveless, T. M. & Bishop, P. E. Identification of genes unique to Mo-independent nitrogenase systems in diverse diazotrophs. Can. J. Microbiol. 45, 312–317 (1999).
Vignais, P. M. & Colbeau, A. Molecular biology of microbial hydrogenases. Curr. Issues Mol. Biol. 6, 159–188 (2004).
Colbeau, A. et al. Organization of the genes necessary for hydrogenase expression in Rhodobacter capsulatus. Sequence analysis and identification of two hyp regulatory mutants. Mol. Microbiol. 8, 15–29 (1993).
Vignais, P. M., Elsen, S. & Colbeau, A. Transcriptional regulation of the uptake [NiFe]hydrogenase genes in Rhodobacter capsulatus. Biochem. Soc. Trans. 33, 28–32 (2005).
Durmowicz, M. C. & Maier, R. J. Roles of HoxX and HoxA in biosynthesis of hydrogenase in Bradyrhizobium japonicum. J. Bacteriol. 179, 3676–3682 (1997).
Buhrke, T., Lenz, O., Krauss, N. & Friedrich, B. Oxygen tolerance of the H2-sensing [NiFe] hydrogenase from Ralstonia eutropha H16 is based on limited access of oxygen to the active site. J. Biol. Chem. 280, 23791–23796 (2005).
Colbeau, A. & Vignais, P. M. Use of hupS::lacZ gene fusion to study regulation of hydrogenase expression in Rhodobacter capsulatus: stimulation by H2 . J. Bacteriol. 174, 4258–4264 (1992).
Colbeau, A. et al. Rhodobacter capsulatus HypF is involved in regulation of hydrogenase synthesis through the HupUV proteins. Eur. J. Biochem. 251, 65–71 (1998).
Dischert, W., Vignais, P. M. & Colbeau, A. The synthesis of Rhodobacter capsulatus HupSL hydrogenase is regulated by the two-component HupT/HupR system. Mol. Microbiol. 34, 995–1006 (1999).
Vidal-Aroca, F. et al. One-step high-throughput assay for quantitative detection of β-galactosidase activity in intact Gram-negative bacteria, yeast, and mammalian cells. BioTechniques 40, 433–440 (2006).
Jahn, A., Keuntje, B., Dörffler, M., Klipp, W. & Oelze, J. Optimizing photoheterotrophic H2 production by Rhodobacter capsulatus upon interposon mutagenesis in the hupL gene. Appl. Microbiol. Biotechnol. 40, 687–690 (1994).
Liu, T., Li, X. & Zhou, Z. Improvement of hydrogen yield by hupR gene knock-out and nifA gene overexpression In Rhodobacter sphaeroides 6016. Int. J. Hydrogen Energy 35, 9603–9610 (2010).
Masukawa, H., Mochimaru, M. & Sakurai, H. Disruption of the uptake hydrogenase gene, but not of the bidirectional hydrogenase gene, leads to enhanced photobiological hydrogen production by the nitrogen-fixing cyanobacterium Anabaena sp. PCC 7120. Appl. Microbiol. Biotechnol. 58, 618–624 (2002).
Zhu, R., Wang, D., Zhang, Y. & Li, J. Hydrogen production by draTGB hupL double mutant of Rhodospirillum rubrum under different light conditions. Chinese Science Bulletin 51, 2611–2618 (2006).
Kim, E., Lee, M., Kim, M. & Lee, J. Molecular hydrogen production by nitrogenase of Rhodobacter sphaeroides and by Fe-only hydrogenase of Rhodospirillum rubrum. Int. J. Hydrogen Energy 33, 1516–1521 (2008).
Rey, F. E., Heiniger, E. K. & Harwood, C. S. Redirection of metabolism for biological hydrogen production. Applied and environmental microbiology 73, 1665–1671 (2007).
Wang, D., Zhang, Y., Welch, E., Li, J. & Roberts, G. P. Elimination of Rubisco alters the regulation of nitrogenase activity and increases hydrogen production in Rhodospirillum rubrum. Int. J. Hydrogen Energy 35, 7377–7385 (2010).
Melis, A., Zhang, L., Forestier, M., Ghirardi, M. L. & Seibert, M. Sustained photobiological hydrogen gas production upon reversible inactivation of oxygen evolution in the green alga Chlamydomonas reinhardtii. Plant Physiol. 122, 127–136 (2000).
Flanagan, L. A., Wright, J. J., Roessler, M. M., Moir, J. W. & Parkin, A. Re-engineering a NiFe hydrogenase to increase the H2 production bias while maintaining native levels of O2 tolerance. Chem. Commun. 52, 9133–9136 (2016).
Christiansen, J., Cash, V. L., Seefeldt, L. C. & Dean, D. R. Isolation and characterization of an acetylene-resistant nitrogenase. J. Biol. Chem. 275, 11459–11464 (2000).
Masukawa, H., Inoue, K., Sakurai, H., Wolk, C. P. & Hausinger, R. P. Site-directed mutagenesis of the Anabaena sp. strain PCC 7120 nitrogenase active site to increase photobiological hydrogen production. Appl. Environ. Microbiol. 76, 6741–6750 (2010).
Packer, M. S. & Liu, D. R. Methods for the directed evolution of proteins. Nat. Rev. Genet. 16, 379–394 (2015).
Griswold, K. E. et al. Evolution of highly active enzymes by homology-independent recombination. Proc. Natl. Acad. Sci. USA. 102, 10082–10087 (2005).
Peters, J. W., Boyd, E. S., Hamilton, T. & Rubio, L. M. In Nitrogen cycling In bacteria: molecular analysis (ed Moir, J. W. B. ) 59–99 (Caister Academic Press, 2011).
Flynn, T. M. Accumulation of O2-tolerant phenotypes in H2-producing strains of Chlamydomonas reinhardtii by sequential applications of chemical mutagenesis and selection. Int. J. Hydrogen Energy 27, 1421–1430 (2002).
Seibert, M., Benson, D. K. & Flynn, T. M. Method and apparatus for rapid biohydrogen phenotypic screening of microorganisms using a chemochromic sensor. US patent 6, 277,589 B1 (2001).
Katsuda, T., Ooshima, H., Azuma, M. & Kato, J. New detection method for hydrogen gas for screening hydrogen-producing microorganisms using water-soluble wilkinson’s catalyst derivative. J. Biosci. Bioeng. 102, 220–226 (2006).
Wecker, M. S. A. & Ghirardi, M. L. High-throughput biosensor discriminates between different algal H2-photoproducing strains. Biotechnol. Bioeng. 111, 1332–1340 (2014).
Wecker, M. S. A., Meuser, J. E., Posewitz, M. C. & Ghirardi, M. L. Design of a new biosensor for algal H2 production based on the H2-sensing system of Rhodobacter capsulatus. Int. J. Hydrogen Energy 36, 11229–11237 (2011).
Miller, J. H. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press. 466 (1972).
Albareda, M. et al. Dual role of HupF in the biosynthesis of [NiFe] hydrogenase in Rhizobium leguminosarum. BMC Microbiol. 12, 256 (2012).
Smith, P. K. et al. Measurement of protein using bicinchoninic acid. Anal. Biochem. 150, 76–85 (1985).
Plovins, A., Alvarez, A. M., Ibañez, M., Molina, M. & Nombela, C. Use of fluorescein-di-beta-D-galactopyranoside (FDG) and C12-FDG as substrates for beta-galactosidase detection by flow cytometry in animal, bacterial, and yeast cells. Appl. Microbiol. Biotechnol. 60, 4638–4641 (1994).
Acknowledgements
Flow cytometry was performed at the Flow Cytometry Unit of the Interdepartmental Investigation Service (SIdI)- Faculty of Medicine (Universidad Autónoma de Madrid). We thank Laura Molero Martín for help in the design of the flow cytometry protocol. We thank Luis Fernández Pacios for developing the model of the V7 NifH variant. This work was supported by the European Research Council Starting Grant 205442 and by MINECO Grant BIO2014-59131-R.
Author information
Authors and Affiliations
Contributions
E.B. performed molecular biology, cellular biology, flow cytometry and biochemical assays. E.J.V. contributed biochemical assays. E.B. and L.M.R performed the experimental design and data analysis and wrote the paper. L.M.R initiated and directed this research.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Barahona, E., Jiménez-Vicente, E. & Rubio, L. Hydrogen overproducing nitrogenases obtained by random mutagenesis and high-throughput screening. Sci Rep 6, 38291 (2016). https://doi.org/10.1038/srep38291
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep38291
This article is cited by
-
Structural consequences of turnover-induced homocitrate loss in nitrogenase
Nature Communications (2023)
-
Debottlenecking the biological hydrogen production pathway of dark fermentation: insight into the impact of strain improvement
Microbial Cell Factories (2022)
-
Understanding potential-dependent competition between electrocatalytic dinitrogen and proton reduction reactions
Nature Communications (2021)
-
Efforts toward optimization of aerobic biohydrogen reveal details of secondary regulation of biological nitrogen fixation by nitrogenous compounds in Azotobacter vinelandii
Applied Microbiology and Biotechnology (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
