
Abstract
Parkinson’s disease (PD) is characterized by α-synuclein (α-Syn)-positive intracytoplasmic inclusions, known as Lewy bodies. Although it is known that extracellular α-Syn is detected in the plasma and cerebrospinal fluid, its physiological significance remains unclear. Here, we show that extracellular α-Syn suppresses platelet-derived growth factor (PDGF)-induced chemotaxis in human neuroblastoma SH-SY5Y cells. The inhibitory effect was stronger in the mutant α-Syn(A53T), found in hereditary PD, and the degree of inhibition was time-dependent, presumably because of the oligomerization of α-Syn. PDGF-induced activation of Akt or Erk was not influenced by α-Syn(A53T). Further studies revealed that α-Syn(A53T) inhibited PDGF-induced Rac1 activation, whereas Cdc42 activation remained unaffected, resulting in unbalanced actin filament remodeling. These results shed light on the understanding of pathological as well as physiological functions of extracellular α-Syn in neuronal cells.
Similar content being viewed by others
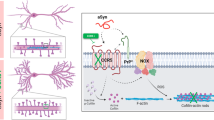


Introduction
Parkinson’s disease (PD) is the second most common progressive neurodegenerative disorder. The pathological hallmarks of PD are selective loss of the dopaminergic neurons in the substantia nigra pars compacta and the presence of α-synuclein (α-Syn)-positive intracytoplasmic inclusions, known as Lewy bodies, which develop in the cell body of affected neurons in both idiopathic1,2,3 and hereditary PD, i.e., missense mutations, α-Syn(A53T)4, α-Syn(A30P)5, and α-Syn(E46K)6 as well as multiplication in the α-Syn gene7,8.
α-Syn is a cytosolic protein that is highly expressed in neurons and enriched at synaptic terminals9. α-Syn is a natively unfolded molecule that can self-aggregate to form oligomers and fibrillar intermediates10,11. Subsequently, it has been suggested that oligomers rather than fibrillar structures might actually show toxicity to cells through binding to membrane lipids and causing membrane perturbations12,13,14. Further studies have revealed that α-Syn can be released from cultured cells by exocytosis15 or by exosomes16 and that α-Syn is detected in cerebrospinal fluid and plasma17,18. Furthermore, it has been reported that exogenous α-Syn causes abnormal cell migration in neuronal stem cells19 or in MDCK cells20. In light of this we reasoned that the extracellular effect of α-Syn would give us some potential hints for the understanding of pathogenesis of Parkinson’s disease. In the present studies we show evidence that extracellular α-Syn causes inhibition of PDGF-induced chemotaxis in SH-SY5Y cells through selective suppression of Rac1 activation required for cell migration.
Results
Impairment of PDGF-induced chemotaxis by α-Syn and α-Syn(A53T)
First, we tested the effect of extracellular α-Syn on cell motility. The ability of wild-type α-Syn and mutant protein, α-Syn(A53T) found in hereditary PD, to influence PDGF-induced chemotaxis of human neuroblastoma SH-SY5Y cells was tested using a two-chamber method. Intriguingly, PDGF-induced chemotaxis was strongly suppressed by the treatment of either purified recombinant α-Syn or α-Syn(A53T) (Fig. 1a,b) in a dose-dependent manner, with the inhibition by α-Syn(A53T) being three times stronger than that by the wild type (Fig. 1c). The effect of α-Syn(A53T) became more remarkable over time (Fig. 2a). This potentiation effect over time may partly due to the self-aggregation tendency of the protein (Fig. 3). Compared with the wild-type α-Syn, α-Syn(A53T) was more prone to form oligomeric structures in solution. On the other hand, an α-Syn mutant devoid of ganglioside-binding ability, α-Syn(K34A/Y39A/K45A)14-derived mutant α-Syn(A53T/K34A/Y39A/K45A), α-Syn(A53T)-AAA, lost its self-oligomerization activity. In contrast to the extracellular effect of α-Syn, expressed wild-type or mutant α-Syn within the cells had little or no effect on chemotaxis (Fig. 2b). The difference between exogenously added α-Syn and expressed cellular form of α-Syn may be ascribed to the conversion of monomeric to oligomeric forms including a dimer that occurs only in extracellular α-Syn (Fig. 4). This notion was further ascertained by the effect of α-Syn(A53T)-AAA. This oligomerization-deficient mutant was disabled to suppress PDGF-induced chemotaxis (Fig. 2c), indicating oligomerization/function relationship as suggested in the previous studies12,13. Next, to exclude the possibility that inhibition of chemotaxis can be seen only by these recombinant proteins expressed in E. coli but not by an endogenous protein from human sources, α-Syn was purified from human erythrocytes and tested for its ability to the chemotaxis. The erythrocyte α-Syn inhibited the PDGF-induced chemotaxis (Fig. 2c) to an extent similar to a recombinant wild-type α-Syn (Fig. 1b). These extracellular effects of α-Syn may not reflect its cellular toxicity because cells are viable even after 48 hr incubation with α-Syn(A53T) as judged by trypan blue exclusion assay (Table 1).

α-Syn-caused impairment of PDGF-induced chemotaxis.
(a) The effect of α-Syn and α-Syn(A53T) on chemotaxis of SH-SY5Y cells was measured using a two-chamber system. Cells were cultured for 18 hr without or with either 1 μM α-Syn or α-Syn(A53T) in the upper chamber. Cells migrated into the lower chamber in the absence (none) or presence of 20 ng/ml PDGF were observed under light microscopy after hematoxylin/eosin staining. Scale bars, 40 μm. (b) Cells were cultured for 18 hr with the buffer (control), 1 μM α-Syn or α-Syn(A53T) in the upper chamber and the cells migrated into the lower chamber in the absence (none) or presence of 20 ng/ml PDGF were counted. PDGF-induced chemotaxis was expressed as % chemotaxis. Values represent means ± s.e.m. of 3 independent experiments carried out in triplicate. Statistical significance was analysed by Student’s t-test (*P < 0.05; **P < 0.01). (c) Cells were cultured for 18 hr with various concentrations of either α-Syn (closed squares) or α-Syn(A53T) (closed circles) in the upper chamber and PDGF-induced chemotaxis was measured as in (b). Values represent means ± s.e.m. of 3 independent experiments carried out in triplicate.

Characterization of α-Syn-caused impairment of chemotaxis.
(a) Cells were preincubated for the indicated time under serum-starved conditions in the absence or presence of various concentrations of α-Syn(A53T) as indicated, trypsinised and cultured for 18 hr in the upper chamber. PDGF-induced chemotaxis was measured as in (Fig. 1b). Values represent means ± s.e.m. of 3 independent experiments carried out in triplicate. (b) Cells transiently transfected with vectors encoding mCherry (control), α-Syn or α-Syn(A53T) were cultured in the upper chamber and the cells expressing each protein migrated into the lower chamber in the absence (none) or presence of 20 ng/ml PDGF were counted. Values represent means ± s.e.m. of 3 independent experiments carried out in triplicate. (c) SH-SY5Y cells were cultured for 18 h in the upper chamber without (buffer control) or with 1 μM α-Syn purified from human erythrocytes or 1 μM α-Syn(A53T)-AAA. Cells, which migrated into the lower chamber in the absence (none) or presence of 20 ng/ml PDGF, were counted. Data are means ± s.e.m. of 3 independent experiments carried out in triplicate. Statistical significance was analysed by Student’s t-test (*P < 0.05).

Self-oligomerization tendency of wild-type α-Syn and its mutants.
One μM wild-type α-Syn, α-Syn(A53T), or α-Syn(A53T)-AAA was incubated in DMEM/F-12 medium for the indicated time periods at 37 °C. Samples were then subjected to SDS-PAGE followed by immunoblotting with anti-α-Syn antibody. Note that oligomeric forms of each protein increase in a time-dependent manner, especially in α-Syn(A53T) but not in ganglioside-binding mutant α-Syn(A53T)-AAA. The results are the representative of 3 independent experiments.

Propensity for oligomerization of wild-type α-Syn and α-Syn(A53T) especially incorporated from outside the cells.
SH-SY5Y cells were cultured with the vehicle (none), 1 μM wild-type α-Syn or 1 μM α-Syn(A53T) for various time periods as indicated. In some experiments cells were transfected with empty vector (pCMV5), vector encoding wild-type α-Syn, or α-Syn(A53T) and cultured for 48 hr. Cells were then lysed and subjected to immunoblotting with anti-α-Syn antibody. The results are the representative of 3 independent experiments.
Dissection of PDGF signalling molecules sensitive to α-Syn(A53T)
To identify downstream signalling molecules, which are sensitive to α-Syn(A53T), PDGF signalling was dissected. Since a phosphoinositide product, PtdIns-3,4,5-trisphosphate, is known to accumulate during PDGF stimulation and this phosphorylated lipid in turn activates the protein kinase (Akt), which is implicated in triggering a cascade of responses including increased cell motility21, involvement of Akt was tested. Upon PDGF stimulation, cells underwent robust phosphorylation of Akt as demonstrated by immunoblot analysis using a specific anti-phosphoAkt antibody (Fig. 5a,b). Pretreatment of cells with α-Syn(A53T) had almost no effect on the phosphorylation of Akt. Next, another important signalling pathway after PDGF receptor activation includes mitogen-activated protein kinase (MAPK) that is also involved in the regulation of important cellular processes such as cell motility22. To assess the involvement of MAPK system, PDGF-induced activation of Erk was measured. PDGF caused a clear phosphorylation of Erk as judged by immunoblot analysis using an anti-phosphoErk antibody (Fig. 5c,d). In this system α-Syn(A53T) had little or no effect on the phosphorylation of Erk. These results indicate that α-Syn(A53T) does not interfere with PDGF-induced two major signalling pathways, i.e., the activation of Akt or Erk.

Both Akt and Erk phosphorylation after PDGF stimulation remains unchanged in α-Syn(A53T)-treated cells.
(a) SH-SY5Y cells grown to sub-confluence were serum-starved for 18 hr in the absence (vehicle, none) or presence of 1 μM α-Syn(A53T), and stimulated with 20 ng/ml PDGF for 5 min. Cells were lysed and subjected to immunoblot analysis with anti–phospho-Akt (Ser473) (p-Akt) or anti-Akt antibody. The results are the representative of 3 independent experiments. (b) PDGF-induced Akt phosphorylation as obtained in (a) was quantitated and expressed as relative to unstimulated controls. Values represent mean ± s.e.m. of 3 independent experiments. (c) Cells were treated in the absence (vehicle, none) or presence of 1 μM α-Syn(A53T) and stimulated with PDGF as in (a). Cells were lysed and subjected to immunoblot analysis with anti-phospho-Erk or anti-Erk antibody. The results are the representative of 3 independent experiments. (d) PDGF-induced Erk phosphorylation as obtained in (c) was quantitated and expressed as relative to unstimulated controls. Values represent mean ± s.e.m. of 3 independent experiments.
Impairment of Rac1 activation by α-Syn(A53T)
It is well known that the Rho family GTPases are regulated downstream of PDGF signalling and play critical roles in the agonist-induced chemotaxsis23,24,25. To identify the molecular basis for α-Syn(A53T)-mediated chemotaxis failure (Figs 1 and 2), we next studied the effect of α-Syn(A53T) on PDGF-induced activation of Rho family GTPases, including Cdc42, Rac1 and RhoA. We measured the activation of each GTPase using a FRET-based analysis. SH-SY5Y cells transiently expressing Raichu-Rac1, Raichu-Cdc42 or Raichu-RhoA, as reported previously26, were stimulated with PDGF, and the activity of each GTPase was monitored by ratiometric analysis in the living cells. PDGF caused both Rac1 and Cdc42 activation in a time-dependent manner (Fig. 6a,b). On the other hand, RhoA activation was not observed, which is consistent with a previous report27 (Fig. 6c). Importantly, pretreatment of the cells with α-Syn(A53T) almost completely blocked the PDGF-induced activation of Rac1 (Fig. 6a, closed circles). However, Cdc42 activation was insensitive to α-Syn(A53T) (Fig. 6b, closed circles). Impairment of PDGF-induced Rac1 activation by α-Syn(A53T) was further confirmed by a biochemical analysis. After stimulation of cells with PDGF, GTP-bound Rac1 was pulled-down by Cdc42/Rac-interactive binding domain of Pak1 fused to GST and detected by Rac1 antibody. α-Syn(A53T)-treatment resulted in almost complete block of Rac1 activation (Fig. 6d), strengthening the result obtained by a FRET-based analysis (Fig. 6a). To further support the idea that the ability of extracellular α-Syn(A53T) to attenuate PDGF-induced chemotaxis is mainly attributed to the impairment of Rac1 activation, the effect of Rac1 inhibitor NSC23766 on PDGF-induced chemotaxis was tested and compared with that of α-Syn(A53T). As expected, NSC23766 inhibited the chemotaxis to an extent similar to that caused by α-Syn(A53T) (Fig. 6e).

Inhibition by α-Syn(A53T) of activation of Rac1 but not Cdc42 after PDGF stimulation.
(a–c) SH-SY5Y cells transiently expressing Raichu-Rac1 (a), Raichu-Cdc42 (b), or Raichu-RhoA (c), which had been cultured for 18 hr in the absence (open circles) or presence (closed circles) of 1 μM α-Syn(A53T), were stimulated with 20 ng/ml PDGF and analysed for ratiometric FRET in live cells. Arrows indicate the addition of PDGF. One of the representative quantification results of each GTPase activity (average of normalised FRET/CFP ratio) in 40 controls and 40 α-Syn(A53T)-treated cells from 3 independent experiments is shown. (d) SH-SY5Y cells were cultured in the absence of serum for 18 hr without or with 1 μM α-Syn(A53T) and then treated with 20 ng/ml PDGF for 4 min. Cells were lysed and a GTP-bound Rac1 was pulled down using GST-CRIB and detected by anti-Rac1 antibody (inset). The bands corresponding to GTP-bound Rac1 were quantitated and expressed as % of control. Data are means ± s.e.m. from 3 independent experiments. Statistical significance was analysed by Student’s t-test (*P < 0.05). (e) Cells were cultured for 18 hr with the buffer (control), 50 μM NSC23766 or 1 μM α-Syn(A53T) in the upper chamber and the cells migrated into the lower chamber in the absence (none) or presence of 20 ng/ml PDGF were counted. PDGF-induced chemotaxis was expressed as % chemotaxis. Values represent means ± s.e.m. of 3 independent experiments carried out in triplicate. Statistical significance was analysed by Student’s t-test (**P < 0.01).
It is well established that actin cytoskeleton remodeling is spatio/temporally regulated by Cdc42, Rac1 and RhoA, which are involved in filopodia, lamellipodia and stress fiber formation, respectively28. To verify the molecule/function relationship, PDGF-induced changes in actin cytoskeleton structures were characterized next. Fluorescent phalloidin staining showed that PDGF induced clear morphological changes including membrane-ruffled round cells (Fig. 7, see arrows). Intriguingly, PDGF-induced membrane-ruffled lamellipodia formation was strongly inhibited by α-Syn(A53T) treatment, instead resulting in prominent filopodia formation (Fig. 7, see arrowheads). These morphological changes matched well with the results that Rac1 but not Cdc42 activation was inhibited by extracellular α-Syn(A53T) (Fig. 6).

α-Syn(A53T)-caused abnormal actin cytoskeleton rearrangement.
SH-SY5Y cells, which had been cultured in the absence of serum for 18 hr without or with 1 μM α-Syn(A53T), were stimulated for 8 min without (none) or with 20 ng/ml PDGF. After fixation, actin filaments were stained by rhodamine-phalloidin. The results are the representative of 3 independent experiments. Arrows show PDGF-induced lamellipodia formation. Note that PDGF causes filopodia (see arrow heads) instead of lamellipodia in α-Syn(A53T)–treated cells. Bars, 10 μm.
Discussion
α-Syn-induced chemotaxis failure shown in the present studies may not be artefactual effects caused by a denatured protein expressed in E. coli but may represent a bona fide nature of this protein since α-Syn purified from human erythrocytes caused similar inhibitory effects on cell migration (Fig. 2c). In addition, the exracellular effects of α-Syn may not reflect its cellular toxicity because cells were viable even after 48 hr incubation with α-Syn(A53T) as judged by trypan blue exclusion assay (Table 1). The potency of α-Syn(A53T) to inhibit PDGF-induced chemotaxis was stronger than that of the wild-type (Fig. 1). Since oligomeric forms of α-Syn preferentially bind to membrane lipids and that α-Syn(A53T) has a higher propensity to form oligomers than a wild type13, it is reasonable to assume the difference in the potency to inhibit chemotaxis between α-Syn(A53T) and the wild type may be attributed to the tendency to their oligomerisation. To support this notion, α-Syn(A53T) in solution tends to aggregate more readily than a wild type as judged by SDS-PAGE followed by immuno-staining (Fig. 3). It has recently been suggested that α-Syn has an ability to interact with gangliosides in the cholesterol and sphingolipid-rich membrane microdomains known as lipid rafts and has a potency to alter the functions of several signaling molecules in the raft domains29. In this line, ganglioside binding mutant α-Syn(A53T)-AAA showed lack of self-oligomerization (Fig. 3) and of ability to inhibit chemotaxis (Fig. 2c). It is tempting to speculate that extracellular α-Syn may first interact with gangliosides in the lipid raft domains and its oligomerization causes perturbation of the raft microenvironment resulting in the alteration of the PDGF receptor signalling.
It has previously been shown that suppression of neuronal stem cell migration along the rostral migratory stream was observed when the α-Syn-encoding retroviral vector was directly injected into the subventricular zone of mice in vivo19. On the contrary, it has recently been reported that neuron-derived α-Syn acts as a chemoattractant which directs microglial migration toward the injured neurons30. In this case α-Syn-induced cell migration required β1-integrin signalling and subsequent involvement of reactive oxygen species. Cell specific responses to α-Syn remains to be studied further.
In the present studies we have shown that PDGF-induced chemotaxis was inhibited by α-Syn and its hereditary mutant α-Syn(A53T) through the inhibition of Rac1 activation (Fig. 6a,d). This inhibition may not involve either Akt or MAPK since PDGF-induced phosphorylation of both Akt and Erk was insensitive to α-Syn(A53T) (Fig. 5). It has been previously shown that PDGF-induced migration and invasion were effectively inhibited by somatostatin treatment in SH-SY5Y cells27. In their studies somatostatin caused inhibition of Erk resulting in partial impairment of Rac activation in a pertussis toxin-sensitive manner. It is difficult to compare the results obtained from different conditions, however extracellular effect of α-Syn may need to be studied in terms of additional signaling events involving a pertussis toxin-sensitive phenomenon. More recently it has been shown that PDGF activates Src kinase, which in turn leads to activation of Rac31. In this context, an afadin-binding protein, afadin dilute domain-interacting protein (ADIP), interacted with Vav2, a GDP/GTP exchange factor for Rac, in a Src phosphorylation-dependent manner32. These results indicate that ADIP may be involved in PDGF-induced cell movement through Src/Rac signalling pathway and will be a potential target for α-Syn. Our present studies suggest that extracellular α-Syn first undergoes oligomerization at the lipid rafts in association with gangliosides and causes perturbation of the raft microenvironment resulting in altering the ability of signalling molecule(s), which may inhibit Rac1 full-activation. Further studies are necessary to clarify the molecular mechanism underlying extracellular α-Syn-induced pathophysiology including chemotaxis failure.
Methods
Antibodies and reagents
These antibodies and reagents were purchased from the following companies:
Anti-α-Syn mouse monoclonal antibody (clone 211) from Santa Cruz Biotechnology; anti–phospho-Akt (Ser473) (D9E) XP rabbit monoclonal antibody (Cell Signalling Technology); anti-Akt rabbit polyclonal antibody (BioLegend); anti–phospho-Erk (Biomol); rabbit polyclonal Erk1/2 antibody from Enzo Life Sciences.
Plasmids and mutations
Human α-Syn was amplified and subcloned into the bacterial expression vector pET3a or mammalian expression vector pCMV5. For α-Syn(A53T), alanine 53 was mutated to a threonine using a QuikChange site-directed mutagenesis protocol. pRaichu-Rac1, pRaichu-Cdc42 and pRaichu-RhoA were kindly provided by Dr. M. Matsuda (Kyoto University). α-Syn(A53T)-AAA was designed and expressed as reported previously14 with a slight modification that three point mutations were employed in α-Syn(A53T) instead of α-Syn. All the constructs were verified by sequencing.
Bacterial expression and purification of recombinant α-Syn and α-Syn(A53T)
Recombinant α-Syn, α-Syn(A53T) and α-Syn(A53T)-AAA were expressed in E. Coli and purified as described previously33. Briefly, α-Syn or α-Syn(A53T) cDNAs subcloned into pET3a was transformed in E. coli BL21 (DE3) and protein expression was induced by 0.1 mM IPTG for 3 hr. Bacterial pellets were resuspended in TE buffer (10 mM Tris-HCl, pH7.5 and 1 mM EDTA) containing 750 mM NaCl (TE-750 mM NaCl) with protease inhibitors, heated at 100 °C for 10 min, and centrifuged at 70,000×g for 30 min. The supernatant was dialysed against TE-20 mM NaCl, filtered by 0.22 μm filter and applied to a Mono S column (GE Healthcare). The unbound fractions were applied to a Mono Q column (GE Healthcare). α-Syn was eluted with a 0–0.5 M NaCl linear gradient. The fractions containing α-Syn were identified by Coomassie Brilliant Blue staining and immunoblot analysis following SDS-PAGE. Protein concentration was determined using Bradford protein assay kit (Bio-Rad).
Cell cultures and transfections
SH-SY5Y cells obtained from American Type Culture Collection (ATCC, CRL-2266) were maintained in DMEM/F-12 medium (Wako Pure Chemical Industries) containing 10% fetal bovine serum and 1% penicillin/streptomycin at 37 °C in 5% CO2. Cells were plated onto 35 mm glass-bottom culture dishes (MatTek) before transfection. Transient transfection was carried out using FuGENE HD (Promega). All experiments were performed 2 to 3 days after transfection.
Cell migration
Cell migration was assessed using 24-well Transwell plates (Coring). The insert membranes (8.0 μm pore size) were coated with collagen type I (50 μg/mL in 5% acetic acid) for 2 hr. SH-SY5Y cells were trypsinised and resuspended in serum-free DMEM/F-12 containing 0.1% fatty acid-free bovine serum albumin (BSA) (Sigma). The cells were then added to the upper wells of Transwell plate (Corning) at 5 × 105 cells per insert. In some experiments 1 μM α-Syn, 1 μMα-Syn(A53T) or 50 μM NSC23766 (Calbiochem) were included in the upper wells as specified in the figure legends. PDGF (20 ng/ml, Wako Pure Chemical Industries) were added to the lower wells. After 18 hr at 37 °C the migrated cells were detached from the membranes by trypsinisation and the number of the cells was counted by a hemocytometer. In some experiments the trypsinised cells were seeded on 35 mm glass-bottom culture dishes (MatTek) and cultured for 24 hr, followed by fixation and staining using anti-α-Syn antibody.
Immunocytochemistry
For cell morphological studies SH-SY5Y cells were serum-starved in the absence or presence of purified α-Syn for 18 hr and stimulated with 20 ng/ml PDGF for 8 min, followed by fixation with 4% paraformaldehyde in PBS (pH 7.4) for 15 min. After washing twice with PBS, the cells were permeabilised with PBS containing 0.1% Triton X-100 for 10 min, then blocked with 1% BSA for 10 min. The cells were then incubated with rhodamine-phalloidin (Sigma) for 1 hr and washed three times with PBS. In some of the experiments SH-SY5Y cells transiently expressing α-Syn were fixed, permeabilised and treated with 1% BSA. The cells were then incubated with the anti-α-Syn antibody (diluted 1:200) in PBS for 1 hr. After washing in PBS, the cells were incubated with Alexa-Fluor 488 goat anti-mouse IgG (diluted 1:1,000, Cell Signalling Technology) for 30 min. After washing three times with PBS, the fluorescence was observed under a confocal laser scanning microscope (LSM 510 META, Carl Zeiss) using a 63× oil plan-apochromat objective.
Ratiometric analysis of Rac1, Cdc42 and RhoA activity in living cells
SH-SY5Y cells were seeded on glass-bottom dishes and transfected with expression vectors for Raichu-Rac1, Raichu-Cdc42 or Raichu-RhoA. After 24 hr, the cells were serum-starved for 18 hr in the presence or absence of 1 μM α-Syn(A53T). Medium was replaced by HEPES-buffered medium consisting of 10 mM HEPES/NaOH (pH 7.4), 130 mM NaCl, 4.7 mM KCl, 1 mM CaCl2, 1.2 mM KH2PO4, 1.2 mM MgSO4, 10 mM glucose with or without 1 μM α-Syn(A53T), and the cells were stimulated with 20 ng/ml PDGF. The cells were imaged on an Olympus IX microscope (Olympus Optical Co.) equipped with a cooled CCD camera, Cascade II 512 (Photometrics), and the imaging system was controlled by MetaFluor fluorescence ratio imaging software (Molecular Devices). Each cell imaging session was repeated at least six times.
Rac1 pull-down assay
SH-SY5Y cells (1 × 107 cells) were serum-starved for 18 hr in the presence or absence of 1 μM α-Syn(A53T) and treated with 20 ng/ml PDGF for 4 min. Cells were lysed for 5 min with the ice-cold cell lysis buffer (25 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM MgCl2, 1% NP-40, 5% glycerol and protease inhibitor cocktail (Nacalai Tesque)) containing 10 μg of GST-fused Cdc42/Rac-interactive binding domain of PAK1 (GST-CRIB). Cell lysates were then centrifuged for 5 min at 10,000×g at 4 °C, and the supernatants were mixed with glutathione-Sepharose beads (GE Healthcare UK Ltd.) with mild rotation for 30 min at 4 °C. After washing the beads with the cell lysis buffer, the bound proteins were resolved on 12.5% SDS-polyacrylamide gel electrophoresis and immunoblotted using a mouse monoclonal anti-Rac1 antibody (1:1000, BD Biosciences).
Purification of human erythrocyte α-Syn
Native α-Syn was purified from freshly collected human erythrocytes from healthy volunteers by a non-denaturing method described previously34.
Detection of protein phosphorylation by immunoblotting
SH-SY5Y cells were incubated with 1 μM α-Syn for 18 hr under serum-starved condition and stimulated with 20 ng/ml PDGF for 5 min. The cells were then solubilised and separated on 12.5% SDS-PAGE gels, blotted onto polyvinylidene difluoride membrane (Millipore), probed with the anti-phospho-Akt, anti-Akt, anti-phospho-Erk, or anti-Erk antibody, followed by HRP-conjugated secondary antibodies treatment and visualized using SuperSignal West Dura Extended Duration Substrate (Thermo Fisher Scientific). Images were acquired using LAS1000 (Fujifilm).
Statistical analysis
Results are expressed as means ± s.e.m. Data were analysed by Student’s t-test. P-values <0.05 were considered significant. Error bars indicate s.e.m.
Additional Information
How to cite this article: Okada, T. et al. Impairment of PDGF-induced chemotaxis by extracellular α-synuclein through selective inhibition of Rac1 activation. Sci. Rep. 6, 37810; doi: 10.1038/srep37810 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
06 April 2018
Scientific Reports 6: Article number: 37810; published online: 25 November 2016; updated: 06 April 2018. The original version of this Article contained an error in the indexing of the author Shaymaa Mohamed Mohamed Badawy. This error has now been corrected.
References
Spillantini, M. G. et al. Alpha-synuclein in Lewy bodies. Nature 6645, 839–840 (1997).
Baba, M. et al. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am. J. Pathol. 152, 879–884 (1998).
Takeda, A. et al. Abnormal accumulation of NACP/alpha-synuclein in neurodegenerative disorders. Am. J. Pathol. 152, 367–372 (1998).
Polymeropoulos, M. H. et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047 (1997).
Krüger, R. et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 18, 106–108 (1998).
Zarranz, J. J. et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 55, 164–173 (2004).
Fuchs, J. et al. Phenotypic variation in a large Swedish pedigree due to SNCA duplication and triplication. Neurology 68, 916–922 (2007).
Singleton, A. B. et al. Alpha-synuclein locus triplication causes Parkinson’s disease. Science 302, 841–841 (2003).
Totterdell, S., Hanger, D. & Meredith, G. E. The ultrastructural distribution of alpha-synuclein-like protein in normal mouse brain. Brain Res. 1004, 61–72 (2004).
Weinreb, P. H. et al. NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 35, 13709–13715 (1996).
Hashimoto, M. et al. Human recombinant NACP/alpha-synuclein is aggregated and fibrillated in vitro: relevance for Lewy body disease. Brain Res. 799, 301–306 (1998).
Conway, K. A. et al. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. USA 97, 571–576 (2000).
Giannakis, E. et al. Dimeric structures of alpha-synuclein bind preferentially to lipid membranes. Biochim. Biophys. Acta 1778, 1112–1119 (2008).
Fantini, J. & Yahi, N. Molecular basis for the glycosphingolipid-binding specificity of α-synuclein: key role of tyrosine 39 in membrane insertion. J. Mol. Biol. 408, 654–669 (2011).
Lee, H. J., Patel, S. & Lee, S. J. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J. Neurosci. 25, 6016–6024 (2005).
Emmanouilidou, E. et al. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 30, 6838–6851 (2010).
Borghi, R. et al. Full length alpha-synuclein is present in cerebrospinal fluid from Parkinson’s disease and normal subjects. Neurosci. Lett. 287, 65–67 (2000).
El-Agnaf, O. M. et al. Alpha-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J. 17, 1945–1947 (2003).
Tani, M. et al. Ectopic expression of α-synuclein affects the migration of neural stem cells in mouse subventricular zone. J. Neurochem. 115, 854–863 (2010).
Sousa, V. L. et al. α-synuclein and its A30P mutant affect actin cytoskeletal structure and dynamics. Mol. Biol. Cell 20, 3725–3739 (2009).
Cantley, L. C. The phosphoinositide 3-kinase pathway. Science 296, 1655–1657 (2002).
Chang, L. & Karin, M. Mammalian MAP kinase signalling cascades. Nature 410, 37–40 (2001).
Ridley, A. J. & Hall, A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 70, 389–399 (1992).
Ridley, A. J. et al. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell 70, 401–410 (1992).
Nobes, C. D. & Hall, A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81, 53–62 (1995).
Nakamura, T., Aoki, K. & Matsuda, M. Monitoring spatio-temporal regulation of Ras and Rho GTPase with GFP-based FRET probes. Methods 37, 146–153 (2005).
Pola, S., Cattaneo, M. G. & Vicentini, L. M. Anti-migratory and anti-invasive effect of somatostatin in human neuroblastoma cells: involvement of Rac and MAP kinase activity. J. Biol. Chem. 278, 40601–40606 (2003).
Etienne-Manneville, S. & Hall, A. Rho GTPases in cell biology. Nature 420, 629–635 (2002).
Martinez, Z., Zhu, M., Han, S. & Fink, A. L. GM1 specifically interacts with alpha-synuclein and inhibits fibrillation. Biochemistry 46, 1868–1877 (2007).
Wang, S. et al. Alpha-synuclein, a chemoattractant, directs microglial migration via H2O2-dependent Lyn phosphorylation. Proc. Natl. Acad. Sci. USA 112, E1926–E1935 (2015).
Ouyang, M., Sun, J., Chien, S. & Wang, Y. Determination of hierarchical relationship of Src and Rac at subcellular locations with FRET biosensors. Proc. Natl. Acad. Sci. USA 105, 14353–14358 (2008).
Fukumoto, Y., Kurita, S., Takai, Y. & Ogita, H. Role of scaffold protein afadin dilute domain-interacting protein (ADIP) in platelet-derived growth factor-induced cell movement by activating Rac protein through Vav2 protein. J. Biol. Chem. 286, 43537–43548 (2011).
Maguire-Zeiss, K. A. et al. Identification of human alpha-synuclein specific single chain antibodies. Biochem. Biophys. Res. Commun. 349, 1198–1205 (2006).
Bartels, T., Choi, J. G. & Selkoe, D. J. α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477, 107–110 (2011).
Acknowledgements
We thank R. Kharbas for careful reading of the manuscript. This work was supported in part by a Grant-in-Aid for Scientific Research (C) to T.O., a Grant-in-Aid for challenging Exploratory Research to S.N., a Grant-in-Aid for Scientific Research (C) to T.K. from the Ministry of Education, Culture, Sports, Science and Technology of Japan; the Osaka Medical Research Foundation for Intractable Diseases to T.K.; the GSK Japan Research Grant 2015 to T.K., and also supported in part by Subsidy for research and development from Hyogo Foundation for Science and Technology to T.O.
Author information
Authors and Affiliations
Contributions
T.O. and S.N. designed the study. T.O. performed FRET analysis. C.H. performed chemotaxis experiments. L.Z., S.M.M.B., and T.K. performed biochemical and immunocytochemical analysis. S.N. wrote the manuscript together with contributions from T.O. and T.K.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Okada, T., Hirai, C., Badawy, S. et al. Impairment of PDGF-induced chemotaxis by extracellular α-synuclein through selective inhibition of Rac1 activation. Sci Rep 6, 37810 (2016). https://doi.org/10.1038/srep37810
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep37810
This article is cited by
-
The Small GTPase RAC1/CED-10 Is Essential in Maintaining Dopaminergic Neuron Function and Survival Against α-Synuclein-Induced Toxicity
Molecular Neurobiology (2018)
-
Extracellular α-synuclein induces sphingosine 1-phosphate receptor subtype 1 uncoupled from inhibitory G-protein leaving β-arrestin signal intact
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
