
Abstract
A series of group-10 metal complexes 1–14 of oxoisoaporphine derivatives were designed and synthesized. 1–14 were more selectively cytotoxic to Hep-G2 cells comparing with normal liver cells. In vitro cytotoxicity results showed that complexes 1–6, 7, 8, 10 and 11, especially 3, were telomerase inhibitors targeting c-myc, telomeric, and bcl-2 G4s and triggered cell senescence and apoptosis; they also caused telomere/DNA damage and S phase arrest. In addition, 1–6 also caused mitochondrial dysfunction. Notably, 3 with 6-amino substituted ligand La exhibited less side effects than 6 with 8-amino substituted ligand Lb and cisplatin, but similar tumor growth inhibition efficacy in BEL-7402 xenograft model. Complex 3 has the potential to be developed as an effective anticancer agent.
Similar content being viewed by others
Introduction
G4s were non-canonical secondary structures formed by DNA sequences containing consecutive runs of guanosine. They differ from other nucleic acid secondary structures and are often associated with human diseases like cancer1, HIV2, and diabetes3, which make the G-quadruplex a potential therapeutic target. Recently, a variety of G4s, such as c-myc, c-kit-2, bcl-2, POT1, and c-kit-1, is considered as an appealing opportunity for drugs or compounds intervention in anticancer therapy4,5,6,7,8,9,10.
G-quadruplex structures have recently been found in telomeres and in promoter regions of certain genes. These G-quadruplexes are characterized by particular structures, and the formation or stabilization of G-quadruplexes in these regions may be specifically regulated. In particular, it is widely accepted that G4s in the c-myc (Pu27) gene play an important regulatory role in hTERT. Small molecule compounds were developed and synthesized against these targets to regulate telomerase activity, and thereby to selectively induce cancer cell apoptosis or/and senescence11,12. In addition, several G4 gene/oncogene promoters, such as those associated with the bcl-2 gene, have been associated with cell death/apoptosis and with diseases such as neurodegeneration, autoimmune deficiencies, and cancer13. Thus, designing and developing G4 ligands or G4s-based inhibitors are a novel potential anticancer strategy14,15,16,17,18.
Telomerase plays an key role in cancer biology and telomere maintenance19, so the design and synthesis of efficient telomerase inhibitors is a viable strategy towards developing anticancer drugs20,21. Some G4 ligands efficiently stabilize G-quadruplex DNA, which often leads to telomerase inhibition15,16,17,18.
Among the group-10 metals, Ni is unique in its structural versatility and redox activity. In contrast, Pd and Pt complexes have relatively rigid structures. For example, a square-planar geometry is common for PdII and PtII complexes. Group-10 metals share the same square-planar geometry, and they could all have the potential to be developed as antitumor agents22,23.
In the past decade, a large number of nickel(II)24,25,26,27,28, palladium(II)29,30, and platinum(II)31,32,33,34,35,36,37 complexes have been reported to inhibit telomerase activity and to stabilize G4s38,39,40. However, the antitumor activity and toxicology profiles of these metal complexes are still not satisfactory. It has been reported that alkaloids are an important source of G-quadruplex ligands, and they also exhibit significant anticancer bioactivities41. Nevertheless, there are only a few reported metal complexes with alkaloids as G-quadruplex ligands42,43. To combine the anticancer activity of group-10 metal complexes and those of alkaloids, we synthesized group-10 metal complexes with oxoisoaporphine ligands (6-amino-oxoisoaporphine, La; 8-amino-oxoisoaporphine, Lb; 8-chloro-oxoisoaporphine, Lc; and 10-chloro-11-amino-oxoisoaporphine, Ld)44,45,46. Our findings demonstrated that complexes 1–6, 7, 8, 10 and 11 selectively stabilized G4, inhibited telomerase activity, and exhibited remarkable in vitro cytotoxicity and tumor growth inhibiting activity, especially complex 3.
Results and Discussion
Synthesis and Characterization
6-amino-oxoisoaporphine, La; 8-amino-oxoisoaporphine, Lb; 8-chloro-oxoisoaporphine, Lc; and 10-chloro-11-amino-oxoisoaporphine, Ld; were synthesized according to reported procedures44,45,46. Acylation of 4-chloro phenylethylamine or β-phenylethylamine with phthalic anhydride (or 3-chloro-phthalic anhydride) gave phenylethylphthalimide and its derivatives. The product was heated in sodium chloride and anhydrous aluminum chloride (1:5) at 180.0–220.0 °C for 3.0 h to yield o-isoquinolin-2-ylbenzoic acid or its derivatives. Cyclization of o-isoquinolin-2-ylbenzoic acid or its derivatives in concentrated sulfuric acid at 230.0 °C for 3.0 h afforded 6/8/10-chloro-oxoisoaporphine in 30.0–35.0% yield. 6/8-amino-oxoisoaporphine (La and Lb) was obtained by treating 6-chloro-oxoisoaporphine or 8-chloro-oxoisoaporphine (Lc) with concentrated ammonium hydroxide (50.0 mL) and stirring at 185.0 °C for 24.0 h in the reaction vessel. 10-chloro-11-nitro-oxoisoaporphine was obtained by nitration of 10-chloro-oxoisoaporphine with fuming nitric acid in the presence of concentrated sulfuric acid. Reaction of 10-chloro-11-nitro-oxoisoaporphine with Na2S yielded 10-chloro-11-amino-oxoisoaporphine (Ld) in 38.0% yield. Group-10 metal complexes [Ni(La)Cl2] (1), [Pd(La)Cl2] (2), [Pt(La)(DMSO)Cl]·H2O (3), [Ni(Lb)Cl2] (4), [Pd(Lb)Cl2] (5), [Pt(Lb)(DMSO)Cl]·H2O (6), [Pt(Lc)(DMSO)Cl] (7), [Pt(Lc)(en)]Cl (8) (en=1,2-ethylenediamine), [Pt(Lc)(pn)]Cl (9) (pn=1,3-propanediamine), [Pt(Lc′)(DMSO)Cl] (10), [Pt(Lc′)(en)]Cl (11) (en=1,2-ethylenediamine), [Ni(Lc)Cl2] (12), [Pd(Lc)Cl2] (13) and [Pt(Ld)Cl2] (14), were synthesized by the reactions of 6-amino-oxoisoaporphine (La), 8-amino-oxoisoaporphine (Lb), 8-chloro-oxoisoaporphine (Lc) and 10-chloro-11-amino-oxoisoaporphine (Ld) with NiCl2, PdCl2 and cis-[PtCl2(DMSO)2] under refluxing conditions, respectively, as depicted in Figs 1, 2 and 3. As shown in Figs 1, 2, 3 and S1–S65, the structures of ligands and their complexes 1–14 were characterized by IR, single crystal X-ray diffraction analysis, ESI-MS, NMR, and elemental analysis. Their metal centers are four-coordinate and approximately square planar.

Synthetic routes for group-10 metal(II) complexes 1–6 of 6-amino-oxoisoaporphine (La) and 8-amino-oxoisoaporphine (Lb).
Reagents and solvents are the following: (a) NiCl2 or PdCl2, ethanol/water (v/v = 20:1) (reflux); (b) cis-Pt(DMSO)2Cl2, ethanol/CH3CN (v/v = 20:1) (reflux).

Synthetic routes for group-10 metal(II) complexes 7–11 of 8-chloro-oxoisoaporphine (Lc).
Reagents and solvents are the following: (a) cis-Pt(DMSO)2Cl2, ethanol/water (v/v = 20:1) (80 °C); (b) 1,2-ethylenediamine or 1,3-propanediamine, anhydrous ethanol (reflux); (c) cis-Pt(DMSO)2Cl2, methanol/CH3CN (v/v = 20:1) (80 °C); (d) 1,2-ethylenediamine, anhydrous ethanol (reflux).

Synthetic routes for group-10 metal(II) complexes 12–14 of 10-chloro-11-amino-oxoisoaporphine (Ld).
Reagents and solvents are the following: NiCl2, PdCl2, or cis-Pt(DMSO)2Cl2, methanol/CH3CN/water (v/v/v = 15:5:1) (reflux).
Crystal Structures
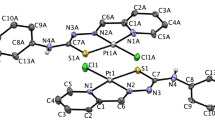
The details of the structural refinement parameters and the crystallographic data for ligand La, complexes 3, 7 and 10 are summarized in Tables S2, S4, S6 and S8 (Supporting Information), and the selected bond lengths are tabulated in Tables S1, S3, S5 and S7 (Supporting Information). The structure of ligand La is similar to that 6-hydroxyloxoisoaporphine, 1-azabenzanthrone and 10-chloro-1-azabenzanthrone43, due to the planar structure (Fig. 4). As shown in Fig. 4, the central Pt(II) in complex 3 was four-coordinate with one bidentate chelating planar ligand N^O-La, one chloride ligand, and one DMSO ligand, adopting an approximately square planar geometry. Furthermore, as shown in Fig. 4, complexes 7 and 10 are quite close to 1-azabenzanthrone and 6-hydroxyloxoisoaporphine platinum(II) complexes (Pt1 and Pt2) in structure with an organometallic C-Pt bond43. Such a feature may affect their cytotoxicities and metal DNA binding properties.

The ORTEP drawings of La (A), complexes 3 (B), 7 (C) and 10 (D) showing atom labeling.
Stability of complexes 1–14 in Solution
The stability of complexes 1–14 in TBS (pH 7.35, 10 mM Tris-KCl-HCl buffer solution, 1% DMSO) was assessed by UV-Vis spectroscopy. Figures S66 and S67 showed no obvious spectral changes in the UV-Vis spectra of complexes 1–14 after 24 h, suggesting the compounds were stable in TBS. In addition, as shown in Fig. S68, after 24 h at room temperature, complexes 1–14 were also stable in DMSO for 24 h as shown by HPLC experiments.
Cytotoxicity
The cytotoxic activity of the La–Ld and complexes 1–14 were tested against a panel of cell lines such as Hep-G2, NCI-H460, SK-OV-3, HCT-8, BEL-7402 cells, and one normal cell line, HL-7702, and cell viability was determined using the MTT assay. Cisplatin was used as the positive control. The inhibitory rates of La–Ld and complexes 1–14 are listed in Table S9. The growth inhibitory rates of complexes 1–14 were higher than that of PdCl2, en, NiCl2, pn, La–Ld, and cis-Pt(DMSO)2Cl2 at 20.0 μM for 48 h, which were all relatively weak against normal HL-7702 cells. Moreover, the compounds affected the NCI-H460, Hep-G2, HCT-8, BEL-7402, and SK-OV-3 cells in a dose-dependent manner, compared with cisplatin, PdCl2, en, NiCl2, pn, and cis-Pt(DMSO)2Cl2. The IC50 values demonstrated that the ligands La–Ld and their complexes 1–14 were active against Hep-G2, SK-OV-3, BEL-7402, NCI-H460 and HCT-8 cell lines and that they possess great selectivity towards Hep-G2 cells as compared to other human tumor cell lines. Thus, complexes 1–14 were more specific against a particular cancer cell (Hep-G2 cells). The IC50 values for complexes 1–14 against Hep-G2 cells were 8.24, 15.22, 4.61, 18.16, 28.09, 14.25, 16.41, 10.15, 15.88, 12.18, 6.49, 28.76, 33.54 and 22.77 μM, respectively. Table 1 showed that 1–14 exhibited significantly enhanced cell cytotoxicity toward five tested human cancer cells as compared to La–Ld. In particular, the cytotoxicity of 3 was 2.1 times higher than that of cisplatin. Against Hep-G2, BEL-7402, SK-OV-3, HCT-8 and NCI-H460 cells, 3 showed higher cytotoxicity than La–Ld, 1, 2, 4–14, and cisplatin. As shown in Table S9 (Supporting Information) and Table 1, 1–14 displayed a synergistic effect as compared with the metal alone and the free corresponding 6-amino-oxoisoaporphine (La), 8-amino-oxoisoaporphine (Lb), 8-chloro-oxoisoaporphine (Lc) and 10-chloro-11-amino-oxoisoaporphine (Ld) ligands, respectively. In general, except for the Hep-G2 cells, the in vitro cytotoxicity of the group-10 metal complexes and La–Ld against the tested tumor cells followed the order of 1 > 4 > 12 > Ld > La > Lb, 2 > 5 > 13 > Ld > La > Lb and 3 > 11 > 8 > 10 > 6 > 9 > 7 > Lc > La > Lb (or 3 > 1 > 2 > La, 6 > 4 > 5 > Lb, 11 > 10 > 8 > 9 > 7 > Lc and 14 > 12 > 13 > Ld). Compared with the 6-hydroxyl-oxoisoaporphine organoplatinum(II) complex, the 6-amino-oxoisoaporphine platinum(II) complex 3 exhibited greater cytotoxicity against Hep-G2, SK-OV-3, NCI-H460 tumor cell lines43. In the case of SK-OV-3 and NCI-H460 cells, the 8-chloro substituent on complexes 7 and 10 led to greater cytotoxicity than that of 1-azabenzanthrone platinum(II) complex Pt143. This demonstrated the importance and the key role of halogen (e.g. Cl) substitution of 1-azabenzanthrone or oxoisoaporphine47. At the same time, Lc exhibited greater cytotoxicity than 1-azabenzanthrone did. Notably, complex 3 exhibited a broad spectrum of inhibition against five selected human cancer cells with IC50 values ranging from 4.61 to 14.17 μM. This indicated greater cytotoxic effects of complexes 7 and 10 on cancer cells. One of the features of complexes 3, 7, and 10 was the different substitution positions of the chloro or amino group on the phenyl ring of oxoisoaporphine or 1-azabenzanthrone, which may be contribute to their various cytotoxicities and differing antitumor activities. The significantly improved biological behavior by complex 3 may be correlated with the substituted 6-amino in ligand La. This was similar to the behavior of the 5-amino-quinolone antimicrobial agents, which were reported to be more cytotoxic than the 5-substituted quinolone antimicrobial agents (e.g. substituent = CH3, CH2CH3, F, Cl and OH, etc.)48. On the other hand, compared with complex 3, the organometallic platinum(II) complexes 7 and 10 with C-Pt bond offered different possibilities for exploration as anticancer agents due to the large structural differences between different ligands (e.g. [(η6-arene)Ru(en)(Cl)]+, [(η6-Bip)Ru(en)Cl]PF6, Cp2TiCl2, oxoisoaporphine platinum(II) complexes Pt1 and Pt2, 6-amino-oxoisoaporphine and 8-chloro-oxoisoaporphine platinum(II) complexes, etc.) and different bonding modes (e.g. π-π-coordination, M-C multiple bonds)43,49. This might lead to different DNA binding modes (3 and 10 interacted with G4-DNA by covalent binding and by π-π stacking, respectively) and the weaker binding abilities of complexes 7 and 10 to G4 in comparison with complex 349. Therefore, complex 3 with the 6-amino substitution in ligand La exhibited higher selectivity for G4, stronger telomerase inhibition ability by directly down-regulating Pu27 G4 (the c-myc promoter/G4), and improved induction of cell apoptosis in Hep-G2 cells than complexes 7 and 10 did. Compounds 3, 6, 10 and 11 showed the highest in vitro cytotoxicity, therefore we selected 3 and 6 for investigating their cytotoxic mechanisms and in vivo tumor growth inhibition.
Cellular Uptake
In order to understand the uptake and distribution of cisplatin, complexes 1–6, 7, 8, 10 and 11 in Hep-G2 cells, an ICP-MS assay was performed to quantify the amount of metal (Pt, Ni or Pd) taken up by the cells. As shown in Figs 5A and S69A, treatment of Hep-G2 cells with complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), cisplatin (10 μM), 5 (28 μM), 6 (14 μM), 7 (16 μM), 8 (10 μM), 10 (12 μM), and 11 (6 μM) for 24 h led to a substantial increase in the cellular metal (Pt, Ni or Pd) concentrations as compared with untreated cells, demonstrating that each complex was readily internalized after 24 h. Treatment of cells with compound 3 ((9.57 ± 0.58 nmol platinum)/106 cells) led to notably higher cellular accumulation of metal (Pt) than treatment with cisplatin ((4.89 ± 0.43 nmol platinum)/106 cells), 1 ((4.41 ± 0.30 nmol nickel)/106 cells), 2 ((3.48 ± 0.26 nmol palladium)/106 cells), 4 ((4.05 ± 0.18 nmol nickel)/106 cells), 5 ((1.99 ± 0.47 nmol palladium)/106 cells), 6 ((4.86 ± 0.14 nmol platinum)/106 cells), 7 ((2.94 ± 0.35 nmol platinum)/106 cells), 8 ((3.59 ± 0.25 nmol platinum)/106 cells), 10 ((3.28 ± 0.14 nmol platinum)/106 cells) or 11 ((5.55 ± 0.11 nmol platinum)/106 cells).

Hep-G2 cells were treated with cisplatin (10 μM), 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM) and 6 (14 μM) at 37 °C for 24 h, respectively.
The distributions of metal (cisplatin, complexes 1–6, 7, 8, 10 and 11) in the nuclear fraction and mitochondrial fractions of Hep-G2 cells were studied after exposure of Hep-G2 cells to 1–6, 7, 8, 10, 11 and cisplatin for 24 h according to our previous reports43. As shown in Figs S69B and 5B, complexes 1, 3, 4, and 6 were accumulated to a large extent in the mitochondrial fraction and the nuclear fraction in Hep-G2 cells, whereas it only accumulated into the nuclear fraction when cells were treated with complex 7, 8, 10 or 11. In contrast, complexes 2, 5 and cisplatin were mainly accumulated in the mitochondrial fraction. Taken together, their difference of metal (Pt, Ni or Pd) distribution for complexes 1–6, 7, 8, 10, 11 and cisplatin could be attributed to their differences in cytotoxicity and the apoptotic pathways in Hep-G2 cells.
Selectivity for Binding with G4 by Complexes
The formation and stabilization of duplex DNA and G4 after treatment with complexes 1–6, 7 and 10 were first examined by various spectroscopic methods. As shown in Figs S70 and S71, FID curves (Supporting Information) were obtained by plotting the percent displacement of TO (thiazole orange) against the concentration of complexes 1–6, 7 and 10. The concentrations of complexes 1–6, 7 and 10 required to decrease the fluorescence by 50%, reflecting binding to G-quadruplex (G4DC50) or duplex (ds26DC50 or/and ctDNADC50) structures, are reported in Table 2. Complexes 1–6, 7 and 10, and especially 3, appear to be strong binders of HTG21 and Pu27 G-quadruplex structures with a G4DC50 of 0.75–70.83 μM, whereas 1–6, 7 and 10 exhibit the poor affinity for the ctDNA structures. Overall, the FID assay indicated that the selectivity of complexes 1–6, 7 and 10 for Pu27 and HTG21 G4s over duplex DNA (ctDNA) was generally moderate, and they were also the most efficient TO-displacers, with a ctDNADC50/G4DC50 ratio (or Est. Sel.)43,50 within the range of 1.67 and 130.49 fold, and 1.41 and 64.81 fold, respectively. As expected, complex 3 showed higher selectivity for quadruplex-DNA than 1, 2, 4–6, 7 and 10 did. Because of the weak binding to G4-DNA by 4–6, 7 and 10 in the FID assay, only 1–3 were selected for G-quadruplex and duplex DNA binding studies using FRET and CD spectroscopy. Similar trends were observed in the FRET-melting assay. As shown in Fig. 6 and Figs S72–S74, the ΔTm values were 2.06, 0.80 and 5.39 °C for F21T, 7.48, 4.98 and 11.50 °C for FPu18T, and 2.68, 1.89 and 6.32 °C for FMidG4T for 1.0 μM of complexes 1, 2 and 3, respectively. In contrast, the ΔTm values were 1.33, 1.45 and 0.19 °C (F32T + H20M) DNA (or duplex DNA) for complexes 1, 2 and 3 treated were under the same conditions, respectively, suggesting that complexes 1, 2 and 3 exerted a stronger stabilizing effect on HTG21 (telomeric), FPu18T (Pu39, bcl-2) and Pu27 (c-myc) G4s over (F32T + H20M) DNA/duplex DNA than complexes 1 and 2 did (Figs S72–S76). In addition, the ESI-MS spectra of Pu27 G-quadruplex treated with complexes 3 and 6 indicated that complexes 3 and 6 were able to form adducts with Pu27 G4 {3 + Pu27 G4: ESI-MS: m/z 9290.47 [Pu27 G4 + Pt(La)(DMSO) + CH3CN + H2O-2H]− (Calcd. m/z 9290.41); 6 + Pu27 G4: ESI-MS: m/z 9267.58 [Pu27 G4 + Pt(Lb)(DMSO)Cl-H]− (Calcd. m/z 9267.62); Pu27 G4: ESI-MS: m/z 8713.53 [Pu27-H]− (Calcd. m/z 8713.49)}, as shown in Fig S77. Moreover, the ESI-MS spectra of Pu27 G-quadruplex treated with complex 10 indicated that it was able to form adducts with Pu27 G4 {10 + Pu27 G4: ESI-MS: m/z 9051.91 [Pu27 G4 + Pt(Lc′)(DMSO)Cl + CH3CN-H]− (Calcd. m/z 9051.87); Pu27 G4: ESI-MS: m/z 8423.38 [Pu27-H]− (Calcd. m/z 8423.30)}. This finding demonstrated that complex 10 interacted with Pu27 G4 DNA most likely by π-π stacking, which was different from 6-amino-oxoisoaporphine platinum(II) complex 3 and the oxoisoaporphine platinum(II) complexes Pt1 and Pt243. Comparison of the ESI-MS spectra before and after complex treated showed that 3 was coordinated to the guanine of G4-DNA (Fig. S77), whereas 6 and 10 might have been interacting with the π-π stacking of the G-quadruplex10,33. FID and ESI-MS spectra experiments showed that the organometallic platinum(II) complexes 7 and 10 with a direct metal-carbon bond offers different possibilities for exploration as anticancer agents due to the large structural differentiation between the different ligands (e.g. [(η6-arene)Ru(en)(Cl)]+, [(η6-Bip)Ru(en)Cl]PF6, Cp2TiCl2, oxoisoaporphine platinum(II) complexes Pt1 and Pt2, 6-amino-oxoisoaporphine and 8-chloro-oxoisoaporphine platinum(II) complexes, etc.) and their different bonding modes (e.g. π-π-coordination, M-C multiple bonds)43,49. This might lead to their different DNA binding modes (3 and 10 interacted with G4-DNA by the covalent binding and π-π stacking, respectively) and weaker binding abilities of complexes 7 and 10 to G4 in comparison with complex 349. Furthermore, the CD spectra revealed positive peaks near 265 and 300 nm, as shown in Fig. S78, which indicated that the Pu27 sequence in TBS contained both parallel and antiparallel G4s before treated with the complex43,51,52. After treatment with compounds 1–3, the CD intensity of the positive peak near 265 nm decreased, which revealed a structural transformation from a mixture of parallel and antiparallel G4s to an antiparallel G-quadruplex. However, the CD spectrum Pu27 G4 in the presence of complexes 1–3 suggested a mixture G-quadruplexes DNA of parallel and antiparallel G-quadruplex conformations, likely because 1–3 preferentially folded the Pu27 G4 DNA into a mixed-type or hybrid G4 structure in these experimental conditions51,52. In all, it is clear that the parallel and antiparallel (mixed-type or hybrid G4 structure) conformation of the G4 DNA after treatment with 1–3 was favorable for the binding of Pu27 G4 in TBS, which also suggested that complexes 1–3 might interact with the loops and grooves of G-quadruplex DNA10,24,33,38,51,52. In addition, in the presence of K+, the CD data indicated that HTG21, and Pu39 DNAs (sequence) exhibited a mixture G-quadruplex DNA in parallel and antiparallel G-quadruplex conformations24,38,51,52, similar to 1–3 treated Pu27 G4 (Figs S78–S80 and Table S10). In general, complex 3 was considered a highly selective ligand for HTG21 (telomeric), FPu18T (Pu39, bcl-2) and Pu27 (c-myc) G4s.

ΔTm data (°C) of 1.0 μM duplex DNA (F32T + H20M DNA), F21T (HTG21 G4), FMidG4T (Pu39 G4) and FPu18T (c-myc/Pu27 G4) G4s after treated with complexes 1–3 (1.0 μM) evaluated by RT-PCR.
Telomere Damage
Previous studies have demonstrated that TRF1 and TRF2 regulate the two major functions of telomeres53: TRF1 controls telomere length by inhibiting telomerase at chromosome ends and perhaps by inhibiting C-strand synthesis54, and TRF2 controls chromosome end protection55, which could induce telomere damage and DNA damage by its effect on ATM, 53BP1, γ-H2AX, Rif1, and Mre11, etc56,57,58. Herein, to assess the effects of complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM) and 6 (14 μM) on telomere dysfunction in Hep-G2 cells, TRF1, 53BP1, and TRF2 telomere damage factors was analyzed by Western blot (Fig. 7). Figure 7 illustrated that complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM) and 6 (14 μM) led to a 33.27 ± 8.32%, 154.42 ± 18.77% and 143.74 ± 12.32%, 127.90 ± 15.65%, 104.34 ± 9.65% and 111.94 ± 10.06%, 339.14 ± 15.06%, 243.69 ± 7.23% and 281.57 ± 11.06%, 185.36 ± 12.14%, 50.97 ± 7.04% and 9.78 ± 17.02%, 118.87 ± 16.07%, 18.12 ± 10.13% and 10.54 ± 15.38%, and 499.63 ± 9.11%, 153.05 ± 13.35% and 60.01 ± 6.35% increase of 53BP1, TRF1 and TRF2 protein expression levels, respectively, demonstrating that their abilities to induce telomere damage53 were 43.73%, 28.69%, 65.33%, 30.77%, 23.73% and 42.65%, respectively. The results suggested that the selectivity of complex 3 for interacting with telomeric regions was higher than that of complexes 1, 2 and 4–6, and the different selectivities for telomeric regions were in the following order: 3 > 1 > 6 > 4 > 2 > 5 (or 1 > 4, 2 > 5 and 3 > 6).

The expression of TRF2, 53BP1, and TRF1 in Hep-G2 cells after treated with complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM) and 6 (14 μM) for 24 h was analyzed by Western blot, respectively.
(A,C) TRF2, 53BP1, and TRF1 protein levels in Hep-G2 cells were analyzed by western blot. (B,D) The whole-cell extracts were prepared and analyzed by Western blot analysis using antibodies against TRF2, 53BP1, and TRF1. The same blots were stripped and re-probed with a β-actin antibody to show equal protein loading. Western blot bands from three independent measurements were quantified with Image J. in (B,D).
Senescence Induction
Since complexes 1–6 were found to be one most promising complexes as c-myc, telomeric, and bcl-2 G4 ligands/G4s-based inhibitors and a telomerase inhibitor in all studies, the following cellular senescence were carried out. These giant/flat cells59 also stained positively for SA-β-Gal activity after continuous treatment of Hep-G2 cells with 0.5 μM complexes 1–6 for 7 days (Fig. 8), respectively, suggesting complexes 1–6 can induce accelerated senescence in Hep-G2 cells. In addition, these results also indicated that 3, featuring a 6-amino substitution in the ligand La, induced cell senescence more readily than complexes 1, 2 and 4–6.

Senescence induced by complexes 1–6 (0.5 μM) or 0.1% DMSO (control) on Hep-G2 cells for 7 days, and examined by Fluorescence microscope (Nikon Te2000 microscope, 100×) with stained β-galactosidase.
Effects of Tested Complexes on hTERT and c-myc in Hep-G2 Cells
Previous studies suggested that the hTERT gene core promoter contains two E-box binding sites of c-myc promoter region, which was also related to cell apoptosis and/or senescence37,52,60. Herein, we confirmed the expression levels of two genes (hTERT and c-myc) mRNA in Hep-G2 ells treated with complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM), 6 (14 μM), 7 (16 μM), 8 (10 μM), 10 (12 μM), and 11 (6 μM) for 24 h (Figs 9 and S81), respectively. As shown in Figs 9 and S81, the down-regulation of complex 3 on two mRNA in Hep-G2 cells was more than that of complexes 1, 2, 4–6, 7, 8, 10 and 11, and the results showed the different effects on hTERT and c-myc promoter in the following order: 3 > 1 > 6 > 4 > 2 > 5 (or 1 > 4, 2 > 5, 3 > 6) and 3 > 11 > 6 > 8 > 10 > 7 (or 11 > 8 > 10 > 7). As shown in Figs 10 and S82, similar trends were observed in Hep-G2 cells by the Western blot assay.

Complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM) and 6 (14 μM) effect on hTERT, bcl-2 and c-myc mRNA expression levels in Hep-G2 cells.

Western blot analysis of hTERT, bcl-2, and c-myc in Hep-G2 cells after 24 h incubation with complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM) and 6 (14 μM) for 24 h, respectively.
(A,C) hTERT, bcl-2, and c-myc protein levels in Hep-G2 cells were analyzed by western blot. (B,D) The whole-cell extracts were prepared and analyzed by Western blot analysis using antibodies against hTERT, bcl-2, and c-myc. The same blots were stripped and reprobed with a β-actin antibody to show equal protein loading. Western blotting bands from three independent measurements were quantified with Image J. in (B,D).
Transcription and Translation of bcl-2 Gene
Recent studies demonstrate that inhibition of bcl-2 expression can decrease cellular proliferation and enhance the efficacy of chemotherapy13. Herein, we were also interested in bcl-2 gene transcription and translation levels in Hep-G2 cells after incubation with complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM), 6 (14 μM), 8 (10 μM) and 11 (6 μM). As shown in Figs 9 and 10, S81 and S82, complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM), 6 (14 μM), 8 (10 μM) and 11 (6 μM) could down-regulate the bcl-2 gene in Hep-G2 cells, and complex 3 decreased the transcription and protein level of bcl-2 more than that of complexes 1, 2, 4–6, 8 and 11, which inhibited the expression of bcl-2 via its specific interaction with Pu39 G413.
Transfection Assay
To further demonstrate the role of the c-myc (Pu27 or c-myc G4) promoter on the telomerase and/or hTRET gene in Hep-G2 cells, transfection assays of the c-myc promoter gene vector and EGFP gene vector were performed as Chalfie, Chen, and He have reported43,61,62. As shown in Figs 11(A) and S83(A and C), the transfection of a EGFP gene vector was successful when Hep-G2 cells showed green fluorescence after EGFP plasmid transfection. Moreover, the Hep-G2 cells were then transfected by c-myc plasmid and treated with complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM), 6 (14 μM), 7 (16 μM), 8 (10 μM), 10 (12 μM), and 11 (6 μM). Consequently, they were examined under the Luciferase reporter gene assay kit and demonstrated a remarkable decrease in emission of bright green fluorescence, as shown in Figs 11(B) and S83(B and D). As a result, treatment with complex 3 (5 μM) reduced the fluorescence emission by 58.5%, whereas complexes 1 (8 μM), 2 (15 μM), 4 (18 μM), 5 (28 μM), 6 (14 μM), 7 (16 μM), 8 (10 μM), 10 (12 μM), and 11 (6 μM) only reduced the fluorescence emission by 30.6%, 3.9%, 58.5%, 25.3%, 2.2%, 29.0%, 7.0%, 19.8%, 15.6% and 44.1% under the same conditions, respectively. This further confirmed the superior efficacy of complex 3 (5 μM) in inhibiting telomerase activity via directly down-regulating c-myc/Pu27 G4 in Hep-G2 cells, even at lower concentration. Taken together, these results demonstrated that the inhibitory effects of complex 3 in the Hep-G2 cells were more than that of 1, 2, 4–6, 7, 8, 10 and 11, which were in agreement with all the Western blot and RT-PCR results.

The successful transfection of 2.0 μg EGFP plasmid vector (A) and c-myc promoter plasmid vector (B) in Hep-G2 cells treatment of complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM) and 6 (14 μM) for 24 h was examined by fluorescence microcopy or/and Multimodel Plate Reader with luciferase reporter gene assay kit, respectively.
TRAP-Silver Staining Assay
A large number of studies have demonstrated that G4 ligands, such as c-myc, telomeric, and bcl-2 G4s have been previously reported to inhibit telomerase activity and induce cell apoptosis and/or senescence43,63,64. Herein, it was of interest to compare the ability of complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM), 6 (14 μM), 7 (16 μM), 8 (10 μM), 10 (12 μM), and 11 (6 μM) on the telomerase activity as examined by the TRAP-silver staining assay. As shown in Figs 12 and S84, all the results demonstrated that the inhibitory ratio for telomerase activity induced by 5 μM of complex 3 reached 58.72%, whereas that induced by complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM), 6 (14 μM), 7 (16 μM), 8 (10 μM), 10 (12 μM), and 11 (6 μM) only reached 45.44%, 15.33%, 58.72%, 16.21%, 14.88%, 54.76%, 15.57%, 22.94%, 16.84% and 44.99%, respectively. This was consistent with the levels of c-myc, hTERT, and bcl-2 determined using RT-PCR, the transfection assay, and Western blotting.

The influence of complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM) and 6 (14 μM) on telomerase activity in Hep-G2 cells for 24 h, respectively.
Cell Cycle
Many studies have demonstrated that telomerase activity is related to cell cycle arrest. Various compounds can cause S phase and/or G2-M phase arrest in human tumor cells65,66. Cell cycle analysis (Figs 13 and S85–S87) clearly showed that complexes 1–6, 8 and 11 caused S phase arrest in Hep-G2 cells for 24 h, in comparison with the control cells. As shown in Figs 13 and S84–S86, S populations of 28.73%, 39.73%, 39.99%, 27.45%, 33.00%, 35.13%, 29.08% and 37.31% for complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM), 6 (14 μM), 8 (10 μM) and 11 (6 μM), respectively, were observed, as compared with an S population of 22.15% (or 17.20%) for untreated cells.

The different phase percentages of Hep-G2 cell cycle treated with complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM), 6 (14 μM), 8 (10 μM) and 11 (6 μM) for 24 h, respectively.
Effect on Cell Cycle Protein Regulators
It is known that cdc25 A, CDK2, cyclin D, cyclin B, and cyclin A play an important role in G1 arrest and/or S phase progression. Therefore, whether complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM), 6 (14 μM), 8 (10 μM) and 11 (6 μM) treatment effected on these five proteins in Hep-G2 cells was determined by Western blotting. As shown in Fig. 14 and Fig. S88, treatment with complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM), 6 (14 μM), 8 (10 μM) and 11 (6 μM) led to a decrease in these proteins in the Hep-G2 cells, likely due to complexes 1–6, 8 and 11, which caused Hep-G2 cell cycle arrest at S phase67. Because the CDK inhibitors p53, p21 or/and p27 are known to inhibit CDK activity68, we subsequently examined the effect of complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM), 6 (14 μM), 8 (10 μM) and 11 (6 μM) on these protein in Hep-G2 cells. Figures 14 and S88 shows that the levels of p53, p27, and p21 protein increased when treated with complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM), 6 (14 μM), 8 (10 μM) and 11 (6 μM) for 24 h. The results indicated that complexes 1–6, 8 and 11 could block the Hep-G2 cell cycle progression into S phase.

The protein levels of cell cycle protein regulators in Hep-G2 cells after treatment with complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM) and 6 (14 μM) for 24 h, respectively.
(A,C) Cell cycle protein regulators protein levels in Hep-G2 cells were analyzed by western blot. (B,D) The whole-cell extracts were prepared and analyzed by Western blot analysis using antibodies against cell cycle protein regulators proteins. The same blots were stripped and reprobed with a β-actin antibody to show equal protein loading. Western blotting bands from three independent measurements were quantified with Image J. in (B,D).
Comet Assay
To determine whether complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM) and 6 (14 μM) induced DNA damage in S phase, a comet assay at the single-cell level was used to detect DNA damage69. As shown in Fig. 15 and Table 3, treatment of Hep-G2 cells with 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM), and 6 (14 μM), especially in cells treated with complex 3, resulted in marked DNA damage as shown by the increased DNA tail size. Taken together, these results demonstrated that complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM) and 6 (14 μM) inhibited Hep-G2 cell growth via the induction of DNA damage and associated S-phase cell cycle arrest70.

Complexes 1–6 -induced DNA damage in Hep-G2 cells.
Cells were treated with complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM) and 6 (14 μM) with the indicated concentrations for 24 h and analyzed by comet assay, respectively. The length of the tail reflects the DNA damage in Hep-G2 cells.
Assessment of Changes in Δψ
Complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM) and 6 (14 μM) accumulated in the mitochondrial fraction, whereas complexes 7, 8, 10 and 11 were only accumulated into the nuclear fraction. Therefore, only complexes 1–6 were selected to study the disruption of mitochondrial function using fluorescence morphological examination and a flow cytometry assay. The Hep-G2 cells treated with complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM) and 6 (14 μM) demonstrated a change in mitochondrial membrane potential (Δψ) as evidenced by a fluorescent emission shift from orange-red to green during JC-1 staining. As shown in Fig. 16, control cells showed a weak green fluorescence and an intense red fluorescence. However, the Hep-G2 cells treated with complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM) and 6 (14 μM), especially with complex 3 (5 μM), exhibited a bright green fluorescence with a marked decrease in red fluorescence (orange-red), along with a remarkable decrease (green) in the orange-red fluorescence emission, indicating a loss of Δψ and a typical apoptotic morphology in Hep-G2 cells after 24 h. Therefore, it was concluded that complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM) and 6 (14 μM) induced Hep-G2 cell apoptosis.

Loss of Δψ in Hep-G2 cells treated with complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM) and 6 (14 μM) for 24 h, and the cells was examined by a fluorescence microscope (Nikon Te2000, 200×) with stained by JC-1.
To obtain a detailed understanding of the cytotoxicity mechanism of complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM) and 6 (14 μM)-induced Hep-G2 cell apoptosis, Rh123 (Rhodamine 123) was used to determine the change of Δψ in Hep-G2 cells. As shown in Fig. 17, upon treatment with complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM) and 6 (14 μM), the fluorescent intensity decreased in Hep-G2 cells as compared with the control cells. These results indicated that complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM) and 6 (14 μM) were able to induce Δψ disruption in Hep-G2 cells, which was in agreement with the Δψ measurements obtained by fluorescent microscopy by JC-1 staining.

Effect of complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM) and 6 (14 μM) on Δψ in Hep-G2 cells, respectively.
After treatment with complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM) and 6 (14 μM) for 24 h, the cells was examined by flow-cytometry with Rh 123 staining.
Expressions of Apoptosis Related Proteins
To demonstrate the effects of complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM) and 6 (14 μM) on cytochrome c (Cyt C), apaf-1, and bax proteins, we carried out western blot analyses on these proteins in Hep-G2 cells. As shown in Fig. 18, significant increases in Cyt C, bax, and apaf-1 levels were observed, suggesting that complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM) and 6 (14 μM) induced cell apoptosis71.

Western blot assay of apoptosis related protein levels in Hep-G2 cells treatmented with complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM) and 6 (14 μM) for 24 h, respectively.
(A,C) Apoptosis related proteins protein levels in Hep-G2 cells were analyzed by western blot. (B,D) The whole-cell extracts were prepared and analyzed by Western blot analysis using antibodies against apoptosis related proteins. The same blots were stripped and reprobed with a β-actin antibody to show equal protein loading. Western blotting bands from three independent measurements were quantified with Image J. in (B,D).
Measurement of ROS Generation
Compared with the control cells, complex 3 (5 μM) showed much higher ability than complexes 1 (8 μM), 2 (15 μM), 4 (18 μM), 5 (28 μM) and 6 (14 μM) to induce ROS generation (Fig. 19). These results are consistent with the results of the flow cytometric analysis (from left to right) in Hep-G2 cells (Fig. 20). In general, the results demonstrated that the effects of complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM), and 6 (14 μM) on the accumulation of ROS generation and finally on the induction of cell death/apoptosis72.

ROS generation assay in Hep-G2 cells was examined by a fluorescence microscope (Nikon Te2000, 200×).
(A,E) Control and (B–D,F–H) Complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM) and 6 (14 μM) treatment with Hep-G2 cells for 24 h, respectively.

ROS generation assay in Hep-G2 cells was examined by flow cytometry of Hep-G2 cells after treated with complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM) and 6 (14 μM), respectively.
1–6 Induced Ca2+ Fluctuation
It was reported that disruption of intracellular Ca2+ (calcium) homeostasis is one of the characteristic events associated with human cancer cell apoptosis and mitochondrial membrane disruption73. For this, we examined the effects of complexes 1–6 on intracellular Ca2+ mobilization in Hep-G2 cells. Figure 21 displayed that the level of intracellular Ca2+ increased steadily (from left to right) in Hep-G2 cells after treated with complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM), and 6 (14 μM), especially in complex 3 (5 μM) treated cells, whereas the values for the control cells were the lowest. These results demonstrated that Ca2+ activation was closely related to complexes 1–6 -induced cell apoptosis, which was consistent with the results of the Δψ disruption assay by JC-1 staining and Rhodamine 123 and cell apoptosis studies.

Effects of complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM) and 6 (14 μM) on Ca2+ activation level in Hep-G2 cells.
Caspase 3/8/9 Activation Assay
In order to assess the effects of 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM), and 6 (14 μM) on caspase-3, caspase-8, and caspase-9 promoter activity and to investigate the potential molecular mechanisms involved74, flow cytometric analysis was used to determine their activated expression levels. Figures 22, S89, and S90 show that the activated expression levels of caspase-3 were 15.9%, 23.4%, 27.9%, 15.0%, 16.1%, and 17.9% in cells treated with 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM) and 6 (14 μM), respectively. Similarly, the activated expression levels were 5.0%, 2.5%, 11.1%, 9.0%, 11.0%, and 16.9% for caspase-8, and 7.6%, 7.9%, 24.3%, 6.9%, 7.0% and 18.5% for caspase-9. Therefore, all the results indicated that complex 3 induced more cell apoptosis via triggering caspase-3/8/9 in Hep-G2 cells, comparing with that of complexes 1, 2 and 4–6.

Activation of caspase-3/8/9 caused by complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM) and 6 (14 μM) in Hep-G2 cells for 24 h.
The Morphological Changes of Hep-G2 Cell Apoptosis
Various experiments showed that complexes 1–6, especially 3 treated cells, were telomerase inhibitors targeting c-myc, telomeric, and bcl-2 G4s, which also caused telomeres/DNA damage, S phase arrest, and mitochondrial dysfunction, likely triggered cell apoptosis13,43,60. Thus, to further investigate the cell apoptosis or cell death mechanism, complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM), and 6 (14 μM)-treated Hep-G2 cells were stained with AO/EB and Hoechst 33258. The Hep-G2 cells stained with Hoechst 33258 showed typical apoptotic features, such as nuclear shrinkage (brightly stained) and condensation after treatment with complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM), and 6 (14 μM) for 24 h (Fig. 23), respectively. The amount of cell apoptosis significantly increased in Hep-G2 cells as the result of treatment with complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM) and 6 (14 μM). In addition, the number of apoptotic nuclei induced by complex 3 (5 μM) increased more significantly compared with that induced by complexes 1 (8 μM), 2 (15 μM), 4 (18 μM), 5 (28 μM), and 6 (14 μM). As shown in Fig. 24, the results of Hep-G2 cell apoptosis (orange) caused by complexes 1–6 agreed with cell apoptosis by AO and EB staining and double staining (Annexin V and PI).

The morphological changes of apoptotic cell nucleus of Hep-G2 cells induced by complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM), and 6 (14 μM) for 24 h, and the Hep-G2 cells were examined by fluorescence microscope (Nikon Te2000, 400×) by staining with Hoechst 33258.

Apoptosis of Hep-G2 cells treated with complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM), and 6 (14 μM) for 24 h, respectively, and these cells the Hep-G2 cells were examined by fluorescence microscope (Nikon Te2000, 200×) by stained with AO/EB.
Apoptosis
Cell apoptosis in Hep-G2 cells caused by complexes 1–6, 7, 8, 10 and 11 was further analyzed by flow cytometry with double staining with Annexin V and PI for visualization. 24 h treatment of the Hep-G2 cells with complexes 1 (8 μM), 2 (15 μM), 3 (5 μM), 4 (18 μM), 5 (28 μM), 6 (14 μM), 7 (16 μM), 8 (10 μM), 10 (12 μM), and 11 (6 μM) significantly induced Hep-G2 cell apoptosis (Q2 + Q3), respectively. As shown in Figs 25 and S91–S93, 3 (5 μM) caused a higher percentage of apoptotic Hep-G2 cells (Q2 + Q3, ca. 21.43%) after 24 h of treatment as compared with 10 μM 1 (ca. 19.33%), 15 μM 2 (ca. 18.61%), 18 μM 4 (ca. 13.49%), 28 μM 5 (ca. 10.60%), 14 μM 6 (ca. 15.45%), 16 μM 7 (ca. 8.30%), 10 μM 8 (ca. 10.35%), 11 μM 10 (ca. 9.90%), and 6 μM 11 (ca. 21.10%). All the results indicated that complex 3 could cause more apoptotic cell death than complexes 1, 2, 4–8, 10 and 11 could in Hep-G2 cells, and the differential induction of cell apoptosis was in the following order: 3 > 1 > 6 > 4 > 2 > 5 and 3 > 11 > 6 > 8 > 10 > 7 (or 1 > 4, 2 > 5, 3 > 6 and 11 > 8 > 10 > 7).

Populations of apoptotic Hep-G2 cells treated with complexes 1–6 were examined by FACS analysis with double staining by Annexin V and PI for visualization.
Effect on a Panel of Genes by RT-qPCR Array
A Telomeres & Telomerase PCR Array was performed to confirm whether complex 3 (5 μM) or 6 (14 μM) could down-regulate or up-regulate the telomeres/telomerase-related gene mRNA expression in the treated Hep-G2 cells75. As shown in Fig. 26 and Table S11, as compared with the untreated cells, of the 84 genes, 50 telomeres/telomerase-related gene mRNA levels more than 1.5-fold greater after treatment with complex 3 (5 μM) for 24 h (i.e., up-regulated: DCLRE1B, HSP90AA1, HSPA1L, IGF1, PAX8, SART1, SIRT6, TERF1, TERF2IP and XRCC6; down-regulated: ABL1, AKT1, ATM, BCL2, BLM, CDK2, CHEK2, EGF, EME1, KRAS, MEN1, MRE11A, MSH2, MSH3, NBN, PARP1, PIF1, PLK1, POT1, PPARG, PPP2R1A, PPP2R1B, PRKCA, PRKCB, PRKDC, PTGES3, RAD17, RAP1A, RAPGEF1, RASSF1, RB1, RTEL1, SLX4, SMAD3, SMG6, SP1, SUN1, TEP1, TP53BP1 and XRCC5). In contrast, as shown in Fig. 27 and Table S12, only 18 genes telomeres/telomerase-related mRNA levels were higher than 1.5-fold after treatment with complex 6 (14 μM) for 24 h (i.e., up-regulated: ACD, BCL2, DCLRE1C, EGF, ERCC1, MUS81, PINX1, PPARG, PURA, RAD17, RAD50, RFC1, SART1, SIRT6, SSB, TERF1 and TP53; down-regulated: PPP2R1B), which was consistent with the RT-PCR, transfection, and western blot analyses.

The mRNA expression level of telomeres/telomerase-related genes in the Hep-G2 cells after treated with complex 3 (5 μM) for 24 h.

The mRNA expression level of telomeres/telomerase-related genes in the Hep-G2 cells after treated with complex 6 (14 μM) for 24 h.
Preliminary Safety Evaluation
0.6 mL/20 g of complex 3 (5% v/v DMSO/saline) was administered by intraperitoneal injection in mice to study its acute toxicity76. Treatment of 16 mg/kg/day (qd) or two times (bid) with complex 3, KM mice (n = 6) showed similar trend (no signs of damage or peritonitis) in comparison with the control group (5% DMSO in saline, Tables S13–S15, Figs S28 and S94A). The result suggested that injection of 3 at 16 mg/kg two times a day was safe for mice; therefore, this dose was taken as the highest dose in antitumor studies. The same test method on 6 was carried out, and the results indicated that injection with 6 at 3.2 mg/kg every day was safe (Figs S94B, S95 and Tables S13–S15).
In vivo antitumor activity of 3 and 6
The in vitro cytotoxicity studies of the group-10 metal complexes suggested that the newly synthesized complexes exhibited significant cytotoxicity in several human tumor cells. To investigate the efficacy of 3 to inhibit tumor growth in vivo, a mouse model of human hepatoma (BEL-7402) xenograft was used. The tumor-bearing nude mice received treatment when the tumor size was between 100 and 160 mm3. The mice were treated with 3 at 16 and 8 mg/kg two times every day, and treatment of cisplatin with 2 mg/kg/2days as the positive control. Fig. 28A and Tables S13–S15 showed the change in tumor volume in the control group and the treatment group. The tumor volume of vehicle control group grew rapidly from day 6, and reached a mean of 1840 mm3 on day 18. In comparison, the tumor volume grew slowly in groups receiving either 3 or cisplatin, with a mean volume of 705 mm3, 873 mm3 and 566 mm3 in groups treated with 16 mg/kg, 8 mg/kg of 3 and 2 mg/kg of cisplatin (ip, q2d), respectively. The relative tumor growth rates (T/C) were 37.5%, 48.9% (p < 0.01) and 27.5% (p < 0.01), respectively. Figure 28C showed that the antitumor activity of complex 3 in BEL-7402 model was 63.1% (p < 0.001) for day 18, but it was lower than that of cisplatin (75.9%, p < 0.001). However, 6 which possesses an 8-amino moiety on the benzene of Lb, had an inhibition ratio of 27.8% (p < 0.05) at the highest dose, which was lower than that of 3 and cisplatin(Fig. S95).

Complex 3 (16, 8 mg/kg/bid) and cisplatin (2 mg/kg/q2d) inhibited the growth of BEL-7402 tumor xenograft in compared with vehicle group, respectively. ***P < 0.01, **P < 0.05, p vs vehicle control.
No adverse effects were observed in the treatment. Mice treated with 3 and 6 showed favorable results without obvious losses in body weight, whereas treatment with cisplatin led to severe body weight loss (Figs 28 and S95), suggesting that 3 and 6 were safer antitumor agents and could inhibit the growth of BEL-7402 tumor in vivo comparably to cisplatin.
SARs
Based on the above described results, the cytotoxic mechanism studies of different ligand (La and Lb, Lc and Lc′) and their complexes, certain SAR (structure-activity relationships) trends emerged among the different groups substituted in oxoisoaporphine ligands, the second ligands, and the metal centers.
-
i
For the same metal center, with different ligands (La and Lb, Lc and Lc′), the in vitro antitumor activity and cellular uptake ability follow the order of 6-NH2 (La) > 8-NH2 (Lb), 7-methoxy (Lc) > 7-carbenyl (Lc′). Such SAR trends are further supported by the cytotoxic mechanism analyses, such as teleromerase activity, RT-PCR and Western blot of c-myc/hTERT/bcl-2, transfection and cell apoptosis, as well as cell cycle studies. In addition, the greater efficacy of 6-NH2 (La) compared to 8-NH2 (Lb) was also supported by telomere damage (or DNA damage) assays, cell senescence assays, a RT-qPCR array in Hep-G2 cells. Both 3 and 6 inhibited the growth of BEL-7402 xenograft in vivo.
-
ii
For the same metal center and Lc (Lc′), with different second ligand, the in vitro antitumor activity is follow the order of en > pn > Cl and DMSO.
-
iii
For the same ligand complexes, with different metal centers, the in vitro antitumor activity and cellular uptake are follow the order of Pt > Ni > Pd.
Conclusions
To incorporate a group-10 metal ion and an alkaloid into one molecule, complexes 1–14 of 6-amino-oxoisoaporphine (La), 8-amino-oxoisoaporphine (Lb), 8-chloro-oxoisoaporphine (Lc) and 10-chloro-11-amino-oxoisoaporphine (Ld) were synthesized and fully characterized. Complexes 1–14 displayed higher selectivity against Hep-G2 cells than against other selected human tumor cells, but lower toxicity in normal human cell line HL-7702. In particular, 3 showed the highest selectivity against Hep-G2 cells. In cytotoxicity studies, complexes 1–14 also displayed synergistic effects as compared with 6-amino-oxoisoaporphine (La), 8-amino-oxoisoaporphine (Lb), 8-chloro-oxoisoaporphine (Lc), 10-chloro-11-amino-oxoisoaporphine (Ld), and the free ions. Further studies demonstrated that complexes 1–6, 7, 8, 10 and 11, especially 3, were telomerase inhibitors targeting c-myc, telomeric, and bcl-2 G4s and triggered cell apoptosis and senescence, which also caused telomere/DNA damage and S phase arrest in the order 3 > 1 > 6 > 4 > 2 > 5 and 3 > 11 > 6 > 8 > 10 > 7 (or 1 > 4, 2 > 5, 3 > 6 and 11 > 8 > 10 > 7). In addition, 1–6 also caused mitochondrial dysfunction in Hep-G2 cells, which led to a significant increase in ROS, loss of Δψ, apaf-1, Ca2+ fluctuation, cytochrome c, and caspase-3/9 ratio, especially in 3 treated cells. In particular, 3 with 6-amino substituted in the ligand La, exhibited better in vivo safety profiles than that of complex 6 and cisplatin as well as significant in vivo tumor growth efficacy in BEL-7402 xenograft mouse model. These results warrant further studies of other platinum-alkaloids complexes for their anticancer activities.
Experimental Section
Synthesis and Characterization of Ligands
Synthesis and characterization of 6-amino-oxoisoaporphine (La), 8-amino-oxoisoaporphine (Lb), 8-chloro-oxoisoaporphine (Lc) and 10-chloro-11-amino-oxoisoaporphine (Ld) have been reported44,45,46. In addition, the detailed synthetic procedures for four ligands La−Ld were described in supporting information.
Data for 6-amino-oxoisoaporphine (La): The brown color product of 6-amino-oxoisoaporphine (La) was suitable for structural characterization. Yield (0.1230 g, 50.0%). 1H NMR (500 MHz, DMSO-d6): δ 7.08 (d, 1H, J = 9.5 Hz), 7.87–7.90 (m, 1H), 7.98 (t, 1H, J = 7.1Hz), 8.06 (d, 1H, J = 9.5 Hz), 8.73 (d, 1H, J = 8.2 Hz), 8.78 (d, 1H, J = 5.0 Hz), 9.21 (d, 1H, J = 8.1 Hz), 9.52 (s, Ar–NH, 1H), 12.43 (s, Ar–NH, 1H). ESI-MS m/z: 247.1 [M + H]+. Elemental analysis calcd (%) for C16H10N2O: C 78.03, H 4.09, N 11.38; found: C 78.01, H 4.14, N 11.42.
Data for 8-amino-oxoisoaporphine (Lb): The brown color product was suitable for structural characterization. Yield (0.0984 g, 40.0%). 1H NMR (600 MHz, DMSO-d6) δ 8.71 (d, J = 5.5 Hz, 1H), 8.43 (dd, J = 7.2, 0.8 Hz, 1H), 8.27 (d, J = 7.6 Hz, 1H), 8.03 (dd, J = 7.4, 0.9 Hz, 1H), 7.98 (dd, J = 8.0, 7.4 Hz, 1H), 7.92 (d, J = 5.5 Hz, 1H), 7.54–7.43 (m, 1H), 6.98 (dd, J = 8.4, 0.9 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 184.12, 153.17, 148.74, 144.08, 136.83, 134.99, 134.94, 132.46, 131.05, 129.82, 128.26, 122.17, 120.70, 119.09, 112.59, 112.55. ESI-MS m/z: 247.1 [M + H]+; IR (KBr): 3853, 3744, 3421, 3304, 3047, 2936, 1932, 1731, 1600, 1538, 1447, 1405, 1324, 1278, 1205, 1167, 1034, 934, 853, 819, 706, 606, 541, 474 cm−1. Elemental analysis calcd (%) for C16H10N2O: C 78.03, H 6.09, N 11.38; found: C 77.96, H 6.14, N 12.42.
Data for 8-chloro-oxoisoaporphine (Lc): The yellow color product (8-chloro-oxoisoaporphine (Lc)) was suitable for structural characterization. Yield (0.0191 g, 72.0%). 1H NMR (600 MHz, DMSO-d6) δ 8.74 (s, 1H), 8.67 (s, 1H), 8.34 (s, 1H), 8.28 (s, 1H), 7.92 (s, 2H), 7.69 (s, 1H), 7.64 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ 181.57, 147.24, 144.59, 139.50, 135.07, 134.69, 134.60, 134.08, 131.80, 130.41, 129.41, 128.39, 125.11, 122.25, 122.02. ESI-MS m/z: 443.9 [M + Cl + DMSO + 2CH3OH]−; IR (KBr): 3852, 3741, 3304, 3056, 1972, 1660, 1613, 1581, 1495, 1442, 1392, 1330, 1291, 1263, 1207, 1166, 1076, 1029, 953, 904, 856, 810, 737, 697, 536, 464, 416 cm−1. Elemental analysis calcd (%) for C16H8ClNO: C 72.33, H 3.03, N 5.27; found: C 72.29, H 3.10, N 5.21.
Data for 10-chloro-11-amino-oxoisoaporphine (Ld): The brown color product was suitable for structural characterization. Yield (0.6826 g, 38.0%). 1H NMR (600 MHz, DMSO-d6) δ 8.80 (d, J = 5.4 Hz, 1H), 8.45 (d, J = 7.2 Hz, 1H), 8.32 (d, J = 8.1 Hz, 1H), 8.01–7.98 (m, 1H), 7.96 (d, J = 5.5 Hz, 1H), 7.53 (d, J = 9.1 Hz, 1H), 7.01 (d, J = 9.1 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 183.98, 153.26, 148.75, 143.28, 139.99, 135.07, 133.42, 132.20, 131.25, 129.36, 128.57, 122.93, 121.20, 120.82, 118.82, 114.00. ESI-MS m/z: 281.0 [M + H]+; IR (KBr): 3853, 3746, 3423, 3294, 3054, 2926, 1916, 1605, 1538, 1445, 1384, 1316, 1264, 1122, 1081, 967, 856, 823, 751, 718, 504 cm−1. Elemental analysis calcd (%) for C16H9ClN2O: C 68.46, H 3.23, N 9.98; found: C 68.50, H 3.18, N 10.02.
Synthesis of 6-Amino-Oxoisoaporphine (La) and 8-Amino-Oxoisoaporphine (Lb) Metal Complexes (1–6)
Synthesis of [Ni(La)Cl2] (1), [Pd(La)Cl2] (2), [Ni(Lb)Cl2] (4), [Pd(Lb)Cl2] (5) and [Pt(Lb)(DMSO)Cl]·H2O (6)
NiCl2·6H2O, PdCl2 or cis-Pt(DMSO)2Cl2 (5.0 mmol) was first dissolved in distilled water (2.5 mL) or CH3CN (2.5 mL) and heated to near boiling. The hot solution was added to 5.0 mmol 6-amino-oxoisoaporphine (La) or 8-amino-oxoisoaporphine (Lb) in ethanol (50.0 mL), and the mixture was stirred at reflux for 12 h, cooled overnight, filtered for about four days, and each product of 1 (clear green), 2, 5 and 6 (black) and 4 (brown) suitable for structural characterization were isolated, washed with ethanol and water or ethanol and CH3CN, similar to the synthesis of Ni(L)Cl277.
Data for 1: The green color product ([Ni(La)Cl2] (1)) was suitable for its structural characterization. Yield (0.033 g, 87%). 1H NMR (600 MHz, DMSO-d6) δ 10.18 (s, 1H), 8.73 (s, 1H), 8.49 (d, J = 55.3 Hz, 2H), 8.21 (s, 1H), 7.85 (s, 1H), 7.62 (s, 2H), 7.52 (s, 1H), 7.29 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ 182.05, 156.07, 142.76, 141.91, 136.39, 135.84, 132.89, 132.68, 129.75, 129.23, 126.43, 125.37, 124.82, 123.33, 122.19, 103.49. ESI-MS m/z: 392.92 [M−Cl + 3H2O]+; IR (KBr): 3417, 3141, 3034, 1630, 1578, 1524, 1446, 1286, 1221, 1163, 854 cm−1. Elemental analysis calcd (%) for C16H10Cl2N2ONi: C 51.13, H 2.68, N 7.45; found: C 51.10, H 2.73, N 7.48.
Data for 2: The black color product ([Pd(La)Cl2] (2)) was suitable for its structural characterization. Yield (0.034 g, 80%). 1H NMR (600 MHz, DMSO-d6) δ 10.27 (s, 1H), 9.03 (s, 1H), 8.88–8.72 (m, 1H), 8.35 (d, J = 11.9 Hz, 1H), 8.12 (d, J = 9.3 Hz, 1H), 8.02 (s, 1H), 7.91 (t, J = 5.9 Hz, 2H), 7.58 (d, J = 9.3 Hz, 1H), 7.50 (d, J = 9.8 Hz, 1H). ESI-MS m/z: 422.91 [M−Cl + 2H2O]+; IR (KBr): 3450, 3313, 2917, 1618, 1553, 1517, 1478, 1421, 1342, 1248, 1174, 848, 787 cm−1. Elemental analysis calcd (%) for C16H10Cl2N2OPd: C 45.37, H 2.38, N 6.61; found: C 45.35, H 2.41, N 6.65.
Data for 4: The brown color product ([Ni(Lb)Cl2] (4)) was suitable for its structural characterization. Yield (0.028 g, 75%). 1H NMR (600 MHz, DMSO-d6) δ 8.73 (d, J = 5.5 Hz, 1H), 8.47–8.42 (m, 1H), 8.29 (d, J = 8.2 Hz, 1H), 8.05 (d, J = 7.4 Hz, 1H), 8.02–7.97 (m, 1H), 7.94 (d, J = 5.5 Hz, 1H), 7.50 (t, J = 7.9 Hz, 1H), 6.99 (d, J = 8.3 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 184.13, 153.18, 148.76, 144.10, 136.84, 135.01, 134.96, 132.48, 131.08, 129.83, 128.28, 122.18, 120.72, 119.11, 112.59, 112.55. ESI-MS m/z: 372.31 [M−Cl + CH3OH]+; IR (KBr): 3850, 3743, 3425, 3308, 3051, 2927, 1601, 1539, 1448, 1405, 1324, 1278, 1206, 1167, 1037, 934, 820, 706, 541 cm−1. Elemental analysis calcd (%) for C16H10Cl2N2ONi: C 51.13, H 2.68, N 7.45; found: C 51.16, H 2.65, N 7.50.
Data for 5: The black color product ([Pd(Lb)Cl2] (5)) was suitable for its structural characterization. Yield (0.029 g, 68%). 1H NMR (600 MHz, DMSO-d6) δ 8.86 (d, J = 4.2 Hz, 1H), 8.25 (d, J = 5.3 Hz, 1H), 8.17 (d, J = 6.7 Hz, 1H), 8.02–7.98 (m, 1H), 7.92 (d, J = 7.9 Hz, 1H), 7.76 (d, J = 5.6 Hz, 1H), 7.63 (d, J = 8.0 Hz, 1H), 6.65 (d, J = 7.9 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 183.02, 150.81, 144.89, 142.78, 142.50, 138.40, 135.11, 133.62, 131.58, 129.97, 129.09, 121.43, 119.66 × 2, 118.80, 112.61. ESI-MS m/z: 527.45 [M + Cl + CH3OH + 2H2O]−; IR (KBr): 3854, 3745, 3321, 2923, 1597, 1514, 1391, 1276, 1168, 1115, 939, 816, 718, 649, 535, 493 cm−1. Elemental analysis calcd (%) for C16H10Cl2N2OPd: C 45.37, H 2.38, N 6.61; found: C 45.30, H 2.32, N 6.68.
Data for 6: The black color product ([Pt(Lb)(DMSO)Cl]·H2O (6)) was suitable for its structural characterization. Yield (0.0466 g, 84%). 1H NMR (600 MHz, DMSO-d6) δ 8.93 (d, J = 6.4 Hz, 1H), 8.15 (d, J = 7.2 Hz, 1H), 8.05 (d, J = 8.1 Hz, 1H), 8.02 (d, J = 8.8 Hz, 1H), 7.95–7.84 (m, 2H), 7.68 (d, J = 6.6 Hz, 1H), 6.64 (d, J = 8.8 Hz, 1H), 2.46 (s, 6 H). 13C NMR (151 MHz, DMSO-d6) δ 183.22, 161.84, 150.33, 144.22, 139.68, 135.74, 135.26, 134.34, 131.56, 130.75, 130.59, 129.42, 121.57, 120.22, 119.65, 112.74, 40.91 × 2. ESI-MS m/z: 589.11 [M + Cl]−; IR (KBr): 3853, 3746, 3441, 3311, 3067, 3007, 2919, 1598, 1565, 1509, 1467, 1405, 1340, 1282, 1237, 1196, 1125, 1023, 979, 937, 820, 699, 540, 502, 446 cm−1. Elemental analysis calcd (%) for C18H17ClN2O2PtS: C 38.89, H 3.08, N 5.04; Found: C 38.85, H 3.05, N 5.10.
Synthesis of [Pt(La)(DMSO)Cl]·H2O (3)
The brown block crystals of [Pt(La)(DMSO)Cl]·H2O (3) was prepared by treating 0.0422 g cis-Pt(DMSO)2Cl2 (0.1 mmol) and 0.1 mmol 6-amino-oxoisoaporphine (0.0246 g) in ethanol/water (20:1) under solvothermal conditions. Yield (0.0505 g, 91%). 1H NMR (500 MHz, DMSO-d6) δ 9.06 (d, J = 7.4 Hz, 1H), 8.84 (d, J = 4.7 Hz, 1H), 8.63 (d, J = 7.5 Hz, 1H), 8.27 (s, 1H), 8.01–7.96 (m, 1H), 7.92 (dd, J = 12.5, 7.1 Hz, 2H), 7.87–7.79 (m, 1H), 7.62 (d, J = 9.5 Hz, 1H), 3.17–3.20 (m, 2H), 2.54 (s, 6 H). 13C NMR (151 MHz, DMSO-d6) δ 181.87, 162.94, 156.20, 136.36, 133.00, 132.84, 130.21, 129.83, 126.64, 126.03, 124.87, 123.57, 122.52, 121.56, 103.49, 40.91×2, 40.54. ESI-MS m/z: 555.1 [M-Cl + 2H2O]+. IR (KBr): 3440, 3298, 2995, 1627, 1572, 1528, 1440, 1405, 1357, 1270, 1122, 1036, 982, 851, 787, 716 cm−1. Elemental analysis calcd (%) for C18H17ClN2O2PtS: C 38.89, H 3.08, N 5.04; Found: C 38.83, H 3.12, N 5.08.
Synthesis of 8-Chloro-Oxoisoaporphine (Lc) Platinum Complexes (7–11)
Synthesis of [Pt(Lc)(DMSO)Cl] (7)
0.0422 g cis-Pt(DMSO)2Cl2 (0.1 mmol), 0.0265 g Lc (0.1 mmol), 1.0 mL ethanol, 0.05 mL distilled water were placed into 25 cm long Pyrex tube that was then quenched in liquid N2, before evacuated and sealed, which was heated at 80 °C for four days. Received black block crystals were used for X-ray diffraction analysis. Yield (0.0441 g, 77%). ESI-MS m/z: 529.9 [M−DMSO + Cl]−; 1H NMR (600 MHz, DMSO-d6) δ 8.82 (s, 1H), 8.71 (s, 1H), 8.56 (s, 1H), 8.26 (s, 1H), 8.15 (s, 1H), 7.78 (s, 1H), 7.37 (s, 1H), 2.53 (s, 6 H). IR (KBr): 3851, 3742, 3442, 3066, 3008, 2920, 2380, 2237, 1932, 1660, 1618, 1542, 1501, 1462, 1413, 1277, 1243, 1205, 1125, 1029, 982, 950, 823, 757, 722, 688, 581, 520, 448 cm−1. Elemental analysis calcd (%) for C18H13Cl2NO2PtS: C 37.71, H 2.29, N 2.44; found: C 37.65, H 2.34, N 2.40.
Synthesis of [Pt(Lc)(en)]Cl (8) and [Pt(Lc)(pn)]Cl (9)
5.0 mmol [Pt(Lc)(DMSO)Cl] (7) was dissolved into 20 mL anhydrous ethanol and heated to near boiling. The hot solution was added to 5.0 mmol 1,2-ethylenediamine (en) or 1,3-propanediamine (pn), and the mixture was stirred at reflux for 12 h, cooled overnight, filtered for about four days, and the black product of 8 and 9 suitable for structural characterization were isolated, washed with anhydrous ethanol, similar to the synthesis of Ni(L)Cl277.
Data for 8: The black color product ([Pt(Lc)(en)]Cl (8)) was suitable for its structural characterization. Yield (0.0499 g, 90%). 1H NMR (600 MHz, DMSO-d6) δ 8.54 (s, 1H), 8.33 (s, 2H), 8.11 (s, 1H), 8.00 (s, 1H), 7.59 (s, 1H), 7.40 (s, 1H), 6.36 (s, 4H), 5.65 (s, 4H). 13C NMR (151 MHz, DMSO-d6) δ 181.53, 160.70, 149.35, 144.78, 134.78, 134.20, 133.34, 131.91, 129.29, 128.09, 122.94, 121.78, 75.91, 48.58, 46.42, 44.49. ESI-MS m/z: 520.0 [M−Cl]+; IR (KBr): 3853, 3744, 3368, 3182, 3068, 2961, 2384, 2297, 1910, 1660, 1606, 1469, 1387, 1320, 1281, 1237, 1154, 1057, 1005, 952, 822, 764, 724, 583, 523, 445 cm−1. Elemental analysis calcd (%) for C18H15Cl2N3OPt: C 38.93, H 2.72, N 7.57; found: C 39.01, H 2.69, N 7.51.
Data for 9: The black color product ([Pt(Lc)(pn)]Cl (9)) was suitable for its structural characterization. Yield (0.0494 g, 87%). 1H NMR (600 MHz, DMSO-d6) δ 8.57 (s, 1H), 8.38 (s, 1H), 8.36 (s, 1H), 8.14 (s, 1H), 8.03 (s, 1H), 7.62 (s, 1H), 7.43 (s, 1H), 6.36 (s, 4H), 5.64 (s, 6 H). ESI-MS m/z: 534.0 [M−Cl]+; IR (KBr): 3151, 3084, 2939, 2878, 2376, 1900, 1660, 1573, 1470, 1287, 1234, 1200, 1161, 1081, 947, 818, 762, 725, 585, 521, 436 cm−1. Elemental analysis calcd (%) for C19H17Cl2N3OPt: C 40.08, H 3.01, N 7.38; found: C 40.15, H 2.94, N 3.05.
Synthesis of [Pt(Lc′)(DMSO)Cl] (10)
The brown block crystals of 0.0265 g Lc (0.1 mmol), 0.0422 g cis-Pt(DMSO)2Cl2 (0.1 mmol), 1.0 mL methanol, 0.05 mL CH3CN were placed into 25 cm long Pyrex tube that was then quenched in liquid N2, before evacuated and sealed, which was heated at 80 °C for four days. Yield (0.0541 g, 92%). ESI-MS m/z: 515.1 [M−DMSO−Cl + CH3CN]+; 1H NMR (600 MHz, DMSO-d6) δ 8.96 (s, 1H), 8.86 (s, 1H1H), 8.45 (s, 1H), 8.42 (s, 1H), 8.06 (s, 1H), 7.99 (s, 1H), 7.82 (s, 1H), 7.64 (s, 1H), 2.53 (s, 9 H). IR (KBr): 3850, 3743, 3443, 3023, 2920, 2379, 1926, 1630, 1604, 1567, 1483, 1440, 1336, 1303, 1263, 1230, 1132, 1024, 978, 854, 798, 949, 686, 595, 519, 450 cm−1. Elemental analysis calcd (%) for C19H17Cl2NO2PtS: C 38.72, H 2.91, N 2.38; found: C 38.65, H 2.97, N 2.35.
Synthesis of [Pt(Lc′)(en)]Cl (11)
5.0 mmol [Pt(Lc′)(DMSO)Cl] (10) was dissolved into 20 mL anhydrous methanol and heated to near boiling. The hot solution was added to 5.0 mmol 1,2-ethylenediamine (en), and the mixture was stirred at reflux for 12 h, cooled overnight, filtered for about four days, and the brown product of 11 suitable for structural characterization were isolated, washed with anhydrous ethanol, similar to the synthesis of Ni(L)Cl277. Yield (0.0473 g, 83%). 1H NMR (600 MHz, DMSO-d6)δ 8.13 (s, 1H), 7.82 (s, 1H), 7.62 (s, 1H), 7.48 (s, 1H), 7.43 (s, 1H), 7.26 (s, 1H), 6.87 (s, 1H), 6.11 (s, 4H), 5.38 (s, 4H), 2.58 (s, 3H). ESI-MS m/z: 534.1 [M−Cl]+; IR (KBr): 3398, 3310, 3183, 3061, 2348, 1935, 1610, 1564, 1513, 1436, 1363, 1301, 1126, 1146, 1056, 1011, 946, 850, 779, 747, 618, 589, 520, 467, 428 cm−1. Elemental analysis calcd (%) for C19H19Cl2N3OPt: C 39.94, H 3.35, N 7.35; found: C 39.87, H 3.42, N 7.32.
Synthesis of [Ni(Ld)Cl2] (12), [Pd(Ld)Cl2] (13) and [Pt(Ld)Cl2] (14)
NiCl2·6H2O, PdCl2 or cis-Pt(DMSO)2Cl2 (5.0 mmol) was first dissolved in distilled water (2.0 mL) and heated to near boiling. The hot solution was added to a hot solution of 10-chloro-11-amino-oxoisoaporphine (Lc) (5.0 mmol) in methanol and CH3CN (v/v = 30.0 mL/10.0 mL) mixture solution, and the mixture was stirred at reflux for 12 h, cooled overnight, filtered for about four days, and each product of 12 (clear brown), 13 and 14 (black) suitable for structural characterization were isolated, washed with methanol, CH3CN and water mixture solution (v/v = 15:5:1), very similar to the synthesis of Ni(L)Cl277.
Data for 12: The brown color product ([Ni(Lc)Cl2] (12)) was suitable for its structural characterization. Yield (0.029 g, 72%). 1H NMR (600 MHz, DMSO-d6) δ 8.63 (d, J = 6.5 Hz, 1H), 8.28 (d, J = 7.0 Hz, 1H), 8.15 (d, J = 8.0 Hz, 1H), 7.87–7.74 (m, 2H), 7.35 (d, J = 6.1 Hz, 1H), 6.88 (d, J = 6.2 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 184.70, 153.99, 149.44, 144.06, 140.71, 135.78, 134.26, 132.86, 132.09, 130.05, 129.33, 123.62, 122.03, 121.71, 119.51, 114.68. ESI-MS m/z: 514.29 [M + Cl + CH3OH + H2O]−; IR (KBr): 3852, 3744, 3422, 3293, 2927, 1604, 1538, 1444, 1384, 1314, 1263, 1120, 856, 823, 651, 537 cm−1. Elemental analysis calcd (%) for C16H9Cl3N2ONi: C 46.84, H 2.21, N 6.83; found: C 46.80, H 2.23, N 6.88.
Data for 13: The black color product ([Pd(Lc)Cl2] (13)) was suitable for its structural characterization. Yield (0.027 g, 60%). 1H NMR (600 MHz, DMSO-d6) δ 8.71 (d, J = 5.4 Hz, 1H), 8.35 (d, J = 7.1 Hz, 1H), 8.22 (d, J = 7.8 Hz, 1H), 7.91–7.88 (m, 1H), 7.87 (d, J = 5.4 Hz, 1H), 7.45 (d, J = 9.1 Hz, 1H), 6.95 (d, J = 9.1 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 183.60, 152.96, 148.36, 142.79, 139.63, 134.78, 133.06, 131.79, 130.94, 129.05, 128.25, 122.62, 120.92, 120.58, 118.62, 113.70. ESI-MS m/z: 587.9 [M + Cl + DMSO + H2O]−; IR (KBr): 3777, 3426, 2925, 2361, 1609, 1532, 1385, 1278, 1175, 1119, 818, 718, 479 cm−1. Elemental analysis calcd (%) for C16H9Cl3N2OPd: C 41.96, H 1.98, N 6.12; found: C 41.90, H 2.05, N 6.17.
Data for 14: The black color product ([Pt(Ld)Cl2] (14)) was suitable for its structural characterization. Yield (0.0447 g, 82%). 1H NMR (600 MHz, DMSO-d6) δ 8.72 (d, J = 5.4 Hz, 1H), 8.35 (d, J = 6.6 Hz, 1H), 8.22 (d, J = 8.1 Hz, 1H), 7.90 (t, J = 7.8 Hz, 1H), 7.87 (d, J = 6.2 Hz, 1H), 7.45 (d, J = 9.1 Hz, 2H), 6.95 (d, J = 9.0 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 183.76, 153.11, 148.52, 139.79, 138.89, 134.92, 133.21, 131.95, 131.09, 129.19, 128.40, 122.77, 121.07, 120.73, 118.77, 113.86. ESI-MS m/z: 585.2 [M-Cl + CH3CN + CH3OH]+; IR (KBr): 3854, 3746, 3433, 3068, 2921, 1614, 1570, 1512, 1387, 1334, 1125, 1025, 820, 706, 535 cm−1. Elemental analysis calcd (%) for C16H9Cl3N2OPt: C 35.15, H 1.66, N 5.12; Found: C 35.11, H 1.62, N 5.18.
Materials and Methods
Abbreviations, DNA oligomers and cell lines used in this work are listed in Table 4. The X-Ray crystallography structures of La, complexes 3, 7 and 10 were solved by Sheldrick method78. The antitumor mechanism of complexes 1–14 were similar to that reported by Chen43,79,80. The TRAP-silver staining assay of complexes 1–6, 8 and 11 were performed as reported by Neidle and Reed31. In addition, the comet assay of complexes 1–6 were performed as Hofer and co-workers reported81. Animal used, acute toxicity studies and antitumor activity toward BEL-7402 in vivo of complex 3 (16 and 8 mg/kg, two time a day,17 days (ip)), complex 6 (3.2 and 1.6 mg/kg/ day, 17 days) and cisplatin (2 mg/kg/2days), were similar to that reported by Chen43,80. BEL-7402 xenograft mouse models were purchased from Beijing HFK Bioscience Co., Ltd (Beijing, China, approval No. SCXK 2014-004). The animal procedures were approved by the Institute of Radiation Medicine Chinese Academy of Medical Sciences (Tian Jin, China, approval No. SYXK 2014-0002). And all of the experimental procedures were carried out in accordance with the NIH Guidelines for the Care and Use of Laboratory Animals. Animal experiments were approved by the Animal Care and Use Committee of Institute of Radiation Medicine Chinese Academy of Medical Sciences. In addition, statistical analysis and abbreviations used have been reported43,80.
Additional Information
How to cite this article: Qin, Q.-P. et al. Preparation of 6/8/11-Amino/Chloro-Oxoisoaporphine and Group-10 Metal Complexes and Evaluation of Their in Vitro and in Vivo Antitumor Activity. Sci. Rep. 6, 37644; doi: 10.1038/srep37644 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Han, H. & Hurley, L. H. G-Quadruplex DNA: a potential target for anti-cancer drug design. Trends Pharmacol. Sci. 21, 136–142 (2000).
Amrane, S. et al. Topology of a NDA G-qudruplex structure formed in the HIV-1 promoter: a potential target for anti-HIV drug development. J. Am. Chem. Soc. 136, 5249–5252 (2014).
Paritala, H. & Firestine, S. M. Characterization of insulin ILPR sequences for their ability to adopt a G-quadruplex structure. Nucleos. Nucleot. Nucl. 29, 81–90 (2010).
Phan, A. T., Kuryavyi, V., Gaw, H. Y. & Patel, D. J. Small-molecule interaction with a five-guanine-tract G-quadruplex structure from the human MYC promoter. Nat. Chem. Biol. 1, 167–173 (2005).
Phan, A. T., Kuryavyi, V., Burge, S., Neidle, S. & Patel, D. J. Structure of an unprecedented G-quadruplex scaffold in the human c-kit promoter. J. Am. Chem. Soc. 129, 4386–4392 (2007).
Shirude, P. S., Okoumus, B., Ying, L., Ha, T. & Balasubramanian, S. Single-molecule conformational analysis of G-quadruplex formation in the promoter DNA duplex of the proto-oncogene C-Kit. J. Am. Chem. Soc. 129, 7484–7485 (2007).
Todd, A. K., Haider, S. M., Parkinson, G. N. & Neidle, S. Sequence occurrence and structural uniqueness of a G-quadruplex in the human c-kit promoter. Nucleic Acids Res. 35, 5799–5808 (2007).
Dai, J. et al. An intramolecular G-quadruplex structure with mixed parallel/antiparallel G-strands formed in the human BCL-2 promoter region in solution. J. Am. Chem. Soc. 128, 1096–1098 (2006).
Dexheimer, T. S., Sun, D. & Hurley, L. H. Deconvoluting the structural and drug-recognition omplexity of the G-quadruplex-forming region upstream of the bcl-2 P1 promoter. J. Am. Chem. Soc. 128, 5404–5415 (2006).
Yuan, L. et al. Existence of G-quadruplex structures in promoter region of oncogenes confirmed bu G-quadruplex DNA cross-lingkong strategy. Scietific Reports 3, 1811 (2013).
Flores, I., Evan, G. & Blasco, M. A. Genetic analysis of myc and telomerase interactions in vivo. Mol. Cell. Biol. 26, 6130–6138 (2006).
Sampedro Camarena, F., Cano Serral, G. & Sampedro Santalo, F. Telomerase and telomere dynamics in ageing and cancer: current status and future directions. Clin. Transl. Oncol. 9, 145–154 (2007).
Wang, X.-D. et al. Turning off transcription of the bcl-2 gene by stabilizing the bcl-2 promoter quadruplex with quindoline derivatives. J. Med. Chem. 53, 4390–4398 (2010).
Balasubramanian, S. H., Hurley, L. H. & Neidle, S. Targeting G-quadruplexes in gene promoters: a novel anticancer strategy? Nat. Rev. Drug Discovery 10, 261–275 (2011).
Maji, B. & Bhattacharya, S. Advances in the molecular dsign of potential anticancer agents via targeting of human telomeric DNA. Chem. Commun. 50, 6422–6438 (2014).
Muller, S. & Rodriguez, R. G-quadruplex interacting samll molecules and drugs: from bench toward bedside. Expert. Rev. Clin. Pharmacol. 7, 663–679 (2014).
March and, A. et al. Ligand-induced conformational changes with cation ejection upon binding to human telomeric DNA G-quadruplexes. J. Am. Chem. Soc. 137, 750–756 (2015).
Yu, Z., Han, M. & Cowan, J. A. Toward the design of a catalytic metallodrug: selective cleavage of G-quadruplex telomeric DNA by an anticancer copper-acridine-ATCUN complex. Angew. Chem. Int. Ed. 54, 1901–1905 (2015).
Wu, R. A. & Collins, K. Sequence specificity of human telomerase. Proc. Natl. Acad. Sci. USA 111, 11234–11235 (2014).
Sekaran, V., Soares, J. & Jarstfer, M. B. Telomere maintenance as a targte for drug discovery. J. Med. Chem. 57, 521–538 (2014).
Ruden, M. & Puri, N. Novel anticancer therapeutics targeting telomerase. Cancer Treatment Rev. 39, 444–456 (2013).
Garoufits, A., Hadjikakou, S. K. & Hadjiliadis, N. Palladium coordination compounds as anti-viral, anti-fugal, anti-microbial and anti-tumor agents. Coord. Chem. Rev. 253, 1384–1397 (2009).
Shimazaki, Y. & Yamauchi, O. Group-10 metal complexes of biological molecules and related ligands: Structural and functional properties. Chem. & Biodiversity 9, 1635–1658 (2012).
Zhao, C., Ren, J., Xu, Y. & Qu, X. Targeting human telomeric higher-order DNA: Dimeric G-quadruplex units serve as preferred binding site. J. Am. Chem. Soc. 135, 18786–18789 (2013).
Bianco, S., Musetti, C., Krapcho, A. P., Palumbo, M. & Sissi, C. Ni2+ and Cu2+ complexes of a phenanthroline-based ligand bind to G-quadruplexes at non-overlapping sites. Chem. Commun. 49, 8057–8059 (2013).
Terenzi, A. et al. Selective G-quadruplex stabilizers: Schiff-base metal complexes with anticancer activity. RSC Adv. 4, 33245–33256 (2014).
Le, F. et al. Stablization of G-quadruplex DNA and antitumor activity by different structures of nickel(II) complexes. Inorg. Chem. Commun. 38, 20–27 (2013).
Campbell, N. H. et al. Molecular basis of strucure-activity relationships between salphen metal complexes and human telomeric DNA quadruplexes. J. Med. Chem. 55, 209–222 (2012).
Largy, E. et al. Tridentate N-donor palladium(II) complexes as efficient coordinating quadruplex DNA binders. Chem. -Eur. J. 17, 13274–13283 (2011).
Donzello, M. P. et al. Tetra-2,3-pyrazinoporphyrazines with externally appended pyridine rings. 9. Novel heterobimetallic macrocycles and related hydrosoluble hexacations as potentially active photo/chemotherapeutic anticancer agents. Inorg. Chem. 50, 7391–7402 (2011).
Reed, J. E., Neidle, S. & Vilar, R. Stabilisation of human telomeric quadruplex DNA and inhibition of telomerase by a platinum-phenanthroline complex. Chem. Commun. (42), 4366–4368 (2007).
Ma, D.-L., Che, C.-M. & Yan, S.-C. Platinum(II) complexes with dipyridophenazine ligands as human telomerase inhibitors and luminescent probes for G-quadruplex DNA. J. Am. Chem. Soc. 131, 1835–1846 (2009).
Zhu, J. et al. DNA cross-linking patterns induced by an antitumor-active trnuclear platinum complexes and comparison with its dinuclear analogue. Chem.-Eur. J. 15, 5245–5253 (2009).
Xu, C.-X. et al. Stabilization of human telomerci G-quadruplex and inhibition of telomerase activity by propeller-shape trinuclear PtII complexes. Chem. -Asian J. 9, 2519–2526 (2014).
Pickard, A. et al. Redesgning the DNA-targeted chromophore in platinum-acridine anticancer agents: a structure-activity relationship study. Chem.-Eur. J. 20, 16174–16187 (2014).
Zheng, X.-H., Zhong, Y.-F., Tan, C.-P., Ji, L.-N. & Mao, Z.-W. Pt(II) squares as selective and effective human telomeric G-quadruplex binders and potential cancer therapeutics. Dalton Trans. 42, 11807–11812 (2012).
Wang, J. et al. A Pt(II)-Dip complex stabilizes parallel c-myc G-quadruplex. Chem. Commun. 49, 4758–4760 (2013).
Xu, Y. Chemistry in human telomere biology: structure, function and targeting of telomere DNA/RNA. Chem. Soc. Rev. 40, 2719–2740 (2011).
Sekaran, V., Soares, J. & Jarstfer, M. B. Telomere maintenance as a target for drug discovery. J. Med. Chem. 57, 521–538 (2014).
Georgiades, S. N., Abd Karim, N. H., Suntharalingam, K. & Vilar, R. Interaction of metal complexes with G-quadruplex DNA. Angew. Chem. Int. Ed. 49, 4020–4034 (2010).
Xiong, Y.-X., Huang, Z.-S. & Tan, J.-H. Targeting G-quadruplex nucleic acids with heterocyclic alkaloids and their derivatives. Eur. J. Med. Chem. 97, 538–551 (2015).
Li, Y.-L., Qin, Q.-P., Liu, Y.-C., Chen, Z.-F. & Liang, H. A platinum(II) complex of liriodenine from traditional Chinese medicine (TCM): Cell cycle arrest, cell apoptosis induction and telomerase inhibition activity via G-quadruplex DNA stabilization. J. Inorg. Biochem. 137, 12–21 (2014).
Chen, Z.-F. et al. Stabilization of G-quadruplex DNA, inhibition of telomerase activity and tumor cell apoptosis of organoplatinum(II) complexes with oxoisoaporphine. J. Med. Chem. 58, 2159–2179 (2015).
Tang, H., Wang, Z.-Y., Zhong, S.-M. & Qin, J.-K. Synthesis and antitumor activity of 6-substituted 1-azabenzanthrone. J. Guangxi Normal University 30, 78–82 (2012).
Tang, H. 8-Substituted oxoisoaporphine derivatives as acetylcholinesterase inhibitor and their preparation. Faming Zhuanli Shenqing (2014), CN 103923009 A 20140716.
Tang, H. 11-substituted oxoisoaporphine derivative, its synthesis method and application. Faming Zhuanli Shenqing (2014), CN 103923010 A 20140716.
Fryer, R. I. et al. Structure-activity relationship studies at the benzodiazepine receptor (BZR): a comparison of the substituent effects of pyrazoloquinolinone analogs. J. Med. Chem. 36, 1669–1673 (1993).
Neu, H. C. Quinolone antimicrobial agents. Annu. Rev. Med. 43, 465–486 (1992).
Yan, Y. K., Melchart, M., Habtemariam, A. & Sadler, P. J. Organometallic chemistry, biology and medicine: ruthenium arene anticancer complexes. Chem. Commun. (38), 4764–4776 (2005).
Monchaud, D. et al. Ligands playing musical chairs with G-quadruplex DNA: A rapid and simple displacement assay for identifying selective G-quadruplex binders. Biochimie 90, 1207–1223 (2008).
Dai, J. et al. An intramolecular G-quadruplex structure with mixed parallel/antiparallel G-strands formed in the human BCL-2 promoter region in solution. J. Am. Chem. Soc. 128, 1096–1098 (2006).
Seenisamy, J. et al. The dynamic character of the G-quadruplex element in the c-MYC promoter and modification by TMPyP4. J. Am. Chem. Soc. 126, 8702–8709 (2004).
Smogorzewska, A. et al. Control of human telomere length by TRF1 and TRF2. Mol. Cell. Biol. 20, 1659–1668 (2000).
Smucker, E. J. & Turchi, J. J. TRF1 inhibits telomere C-strand DNA synthesis in vitro. Biochemistry 40, 2426–2432 (2001).
Karlseder, J., Smogorzewska, A. & de Lange, T. Senescence induced by altered telomere state, not telomere loss. Science 295, 2446–2449 (2002).
Takai, H., Smogorzewska, A. & de Lange, T. DNA damage foci at dysfunctional telomeres. Curr. Biol. 13, 1549–1556 (2003).
Celli, G. B. & de Lange, T. DNA processing is not required for ATM mediated telomere damage response after TRF2 deletion. Nat. Cell. Biol. 7, 712–718 (2005).
Fradet-Turcotte, A. et al. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 499, 50–54 (2013).
Ou, T.-M. et al. Inhibition of cell proliferation by quindoline derivative (SYUIQ-05) through its preferential interaction with c-myc promoter G-quadruplex. J. Med. Chem. 54, 5671–9679 (2011).
Postel, E. H., Berberich, S. J., Flint, S. J. & Ferrone, C. A. Human c-myc transcription factor PuF identified as nm23-2 nucleoside diphosphate kinase, a candidate suppressor of tumor metastasis. Science 261, 478–480 (1993).
Chalfie, M., Tu, Y., Euskirchen, G., Ward, W. W. & Prasher, D. C. Green fluorescent protein as a marker for gene expression. Science 263, 802–805 (1994).
He, T.-C. et al. Identification of c-MYC as a target of the APC pathway. Science 281, 1509–1512 (1998).
Ou, T.-M. et al. G-quadruplexes: Targets in anticancer drug design. ChemMedChem 3, 690–713 (2008).
Redon, S., Reichenbach, P. & Lingner, J. The non-coding RNA TERRA is a natural ligand and direct inhibitor of human telomerase. Nucleic Acids Res. 38, 5797–5806 (2010).
Taggart, A. K. P., Teng, S.-C. & Zakian, V. A. Est1p as a cell cycle-regulated activator of telomere-bound telomerase. Science 297, 1023–1026 (2002).
Diede, S. J. & Gottschling, D. E. Telomerase-mediated telomere addition in vivo requires DNA primase and DNA polymerases α and δ. Cell 99, 723–733 (1999).
Park, J. W. et al. Chemopreventive agent resveratrol, a natural product derived from grapes, reversibly inhibits progression through S and G2 phases of the cell cycle in U937 cells. Cancer lett. 163, 43–49 (2001).
Busino, L. et al. Degradation of Cdc25A by β-TrCP during S phase and in response to DNA damage. Nature 426, 87–91 (2003).
Bartek, J., Lukas, C. & Lukas, J. Checking on DNA damage in S phase. Nature Rev. Mol. Cell Biol. 5, 792–804 (2004).
Wang, B., Matsuoka, S. H., Carpenter, P. B. & Elledge, S. J. 53BP1, a mediator of the DNA damage checkpoint. Science 298, 1435–1438 (2002).
Kerr, J. F. R., Wyllie, A. H. & Currie, A. R. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 26, 239–257 (1972).
Hu, X.-Z., Xu, Y., Hu, D.-C., Hui, Y. & Yang, F.-X. Apoptosis induction on human hepatoma cells Hep G2 of decabrominated diphenyl ether (PBDE-209). Toxicol Lett. 171, 19–28 (2007).
Laver, D. R. Ca2+ stores regulate ryanodine receptor Ca2+ release channels via luminal and cytosolic Ca2+ sites. Biophys. J. 92, 3541–3555 (2007).
Chai, J. et al. Structural and biochemical basis of apoptotic activation by Smac/DIABLO. Nature 406, 855–862 (2000).
Lin, Y.-C. et al. Genome dynamics of the huaman embryonic kidney 293 lineage in response to cell biology manipulations. Nat. Commun. 5, 4767, doi: 10.1038/ncomms5767 (2014).
Diehl, K. H. et al. A good practice guide to the administration of substances and removal of blood, including routes and volumes. J. Appl. Toxlcol. 21, 15–23 (2001).
Zou, H.-H. et al. Preparation of 4-([2,2′:6′,2″-terpyridin]-4′-yl)-N, N-diethylaniline NiII and PtII complexes and exploration of their in vitro cytotoxic activities. Eur. J. Med. Chem. 108, 1–12 (2016).
G M Sheldrick Team SHELXTL-97, Program for refinement of crystal structures, University of Göttingen, Germany (1997).
Qin, Q.-P. et al. Synthesis of a platinum (II) complex with 2-(4-methoxy-phenyl) imidazo [4,5-f]-[1,10] phenanthrolin and study of its antitumor activity. Eur. J. Med. Chem. 89, 77–87 (2015).
Chen, Z.-F. et al. Water-soluble ruthenium(II) complexes with chiral 4- (2,3-dihydroxypropyl)-formamide oxoaporphine (FOA): In vitro and in vivo anticancer activity by stabilization of G-Quadruplex DNA, inhibition of telomerase activity, and induction of tumor cell apoptosis. J. Med. Chem. 58, 4771–4789 (2015).
Hofer, M. et al. Two new faces of amifostine: protector from DNA damage in normal cells and inhibitor of DNA repair in cancer cells. J. Med. Chem. 59, 3003–3017 (2016).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants 21271051, 21431001, 81473102), IRT_16R15, the NSF of Guangxi Province of China (Grant 2016GXNSFGA380005), CMEMR2012-A22. Innovation Project of Guangxi Graduate Education (YCBZ2015024) as well as “BAGUI Scholar” program of Guangxi Province of China. Microarray experiments of telomeres/telomerase-related genes were performed by KangCheng Bio-tech, Shanghai, China.
Author information
Authors and Affiliations
Contributions
Q.P.Q. and Z.F.C. conceived and designed the experiments, Q.P.Q., Z.Z.W. and G.A.Y. synthesized chemical compounds, Q.P.Q. and T.M. carried out comet assay, cytotoxicity assay, uptake of metal complexes in Hep-G2 cells, the morphological changes of Hep-G2 cell apoptosis, SA-β-Gal assay, cell cycle and apoptosis analysis, determination of caspase-3/8/9 activity by flow cytometric analysis, measurement of ROS generation, RNA extraction, Ca2+ Fluctuation, RT-PCR, Western blot, TRAP-silver staining assay, effect on a panel of genes by RT-qPCR array, and transfection assay of complexes 1–14, and selective binding for G4 and duplex DNA by complexes 1–14 (including CD spectroscopy, FID assays, and FRET assays) and the effect of complexes 3 and 6 on the protein levels of telomeres/telomerase-related genes in Hep-G2 cells. Q.P.Q., Z.Z.W. and G.A.Y. contributed to some synthesis with assistance from Y.C.L., J.L.Q. contributed to maximum tolerated dose (MTD) analysis, acute toxicity studies and antitumor activity toward BEL-7402 models in vivo of complexes 3 and 6. Z.F.C. and H.L. provided effective directions during the research process. Q.P.Q., Z.F.C. and J.L.Q. analyzed data, discussed results, wrote and commented on the paper. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Qin, QP., Qin, JL., Meng, T. et al. Preparation of 6/8/11-Amino/Chloro-Oxoisoaporphine and Group-10 Metal Complexes and Evaluation of Their in Vitro and in Vivo Antitumor Activity. Sci Rep 6, 37644 (2016). https://doi.org/10.1038/srep37644
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep37644
This article is cited by
-
4-Hydroxyl-oxoisoaporphine, one small molecule as theranostic agent for simultaneous fluorescence imaging and photodynamic therapy as type II photosensitizer
Photochemical & Photobiological Sciences (2021)
-
Oxoaporphine Metal Complexes (CoII, NiII, ZnII) with High Antitumor Activity by Inducing Mitochondria-Mediated Apoptosis and S-phase Arrest in HepG2
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
