
Abstract
Glucose oxidase (GO) activity is generally restricted to glucose and is susceptible to inactivation by H2O2. By comparison, the Y300A variant of gluco-oligosaccharide oxidase (GOOX) from Sarocladium strictum showed broader substrate range and higher H2O2 stability. Specifically, Y300A exhibited up to 40 times higher activity on all tested sugars except glucose, compared to GO. Moreover, fusion of the Y300A variant to a family 22 carbohydrate binding module from Clostridium thermocellum (CtCBM22A) nearly doubled its catalytic efficiency on glucose, while retaining significant activity on oligosaccharides. In the presence of 200 mM of H2O2, the recombinant CtCBM22A_Y300A retained 80% of activity on glucose and 100% of activity on cellobiose, the preferred substrate for this enzyme. By contrast, a commercial glucose oxidase reported to contain ≤0.1 units catalase/ mg protein, retained 60% activity on glucose under the same conditions. GOOX variants appear to undergo a different mechanism of inactivation, as a loss of histidine instead of methionine was observed after H2O2 incubation. The addition of CtCBM22A also promoted functional binding of the fusion enzyme to xylan, facilitating its simultaneous purification and immobilization using edible oat spelt xylan, which might benefit the usage of this enzyme preparation in food and baking applications.
Similar content being viewed by others


Introduction
Glucose oxidase (GO) belongs to the auxiliary activity family AA3_2 (www.cazy.org) and is a flavoenzyme with a tightly but non-covalently bound FAD cofactor1. The enzyme oxidizes the anomeric carbon of β-D-glucose using molecular oxygen as an electron acceptor, producing H2O2 and D-glucono-δ-lactone, which in the presence of water spontaneously hydrolyzes to gluconic acid2.
GO is one of the most widely used industrial enzymes2, and many commercial applications employ GO from Aspergillus niger (NCBI accession: AJ294936) since the properties of this enzyme have been extensively studied3. Applications of GO include use in biosensors, biofuel cells, as well as in food products4,5. Considering applications in food, GO has been used to reduce browning caused by the Maillard reaction6; the oxygen scavenging activity of GO and simultaneous production of H2O2 can also reduce food spoilage7,8. Moreover, the production of H2O2 can improve dough stability by forming cross linkages within gluten9; production of H2O2 can also oxidize water-soluble pentosans10, which then gelate and increase water uptake and softness of baked products11. The introduction of aldonic acids into dough is also expected to reduce stickiness and improve machinability11.
Despite the broad range of applications for GO, the effectiveness of GO is restricted by the narrow substrate range of this enzyme and susceptibility to H2O2 inactivation. Kleppe12 showed that less than 10 mM H2O2 that reduced 20% of enzyme activity oxidized methionine residues within the GO active site, where as 100–200 mM H2O2 reduced enzyme activity by up to 80%12. Similarly, H2O2 was reported to oxidize surface exposed methionine residues and a phenylalanine in the active site of a pyranose oxidase (Family AA3_4) from Trametes multicolor13. Commercial preparations of GO can include catalase to overcome GO inactivation by H2O2. However, this solution is not practical for applications that benefit from H2O2 production by GO.
Where broader substrate range is beneficial, alternatives to GO may be found among the oligosaccharide oxidases from family AA7 (www.cazy.org), such as the carbohydrate oxidase from Microdochium nivale (MnCO)14, and gluco-oligosaccharide oxidase from Sarocladium strictum (GOOX)15,16,17. Like GO, both MnCO and GOOX oxidize the anomeric carbon of carbohydrate substrates through an FAD reduction mechanism; however, in these cases, the FAD cofactor is bi-covalently linked through cysteine and histidine residues. Furthermore, these enzymes display a comparatively broad substrate profile, ranging from xylose, and galactose, to malto-, cello- and xylo-oligosaccharides14,16,18. Similar to MnCO, GOOX oxidizes oligosaccharides more effectively than monosaccharides16. To date, thirteen GOOX variants have been created through site-directed mutagenesis16,19, where one variant (Y300A) shows higher activity on monosaccharides (including glucose and xylose) compared to the wild-type GOOX. The Y300A variant also retains activity on oligosaccharides and displays reduced substrate inhibition19.
MnCO activity has already been reported to increase dough elasticity and consistency20 and was tested as a substitute for GO in baking applications21. Nevertheless, MnCO was inactivated by H2O2 during substrate turnover22, although the extent of H2O2 inactivation was not reported.
The comparatively broad substrate range of Y300A along with reduced substrate inhibition make it a suitable alternative to GO particularly in applications that comprise mixed carbohydrates. Accordingly, this study directly compared the substrate profile and H2O2 inactivation of the A. niger GO and the Y300A variant of GOOX. The Y300A variant was also fused to CtCBM22A, a carbohydrate-binding module from Clostridium thermocellum, generating CtCBM22A_Y300A to facilitate enzyme purification through binding to edible oat fibre.
Materials and Methods
Materials
Glucose, xylose, oat spelt xylan (OSX), and beech wood xylan were purchased from Sigma (St. Louis, USA) while cellobiose and maltose were purchased from BioShop Inc. (Ontario, Canada). Cello-oligosaccharides and xylo-oligosaccharides (XOS) were purchased from Megazyme (Wicklow, Ireland), and mixed XOS (DP-2–7, 95% pure) were obtained from Cascade Analytical Reagents and Biochemicals (Oregon, USA). Wheat bran hemicellulose and propoxylated wheat bran hemicellulose were kindly provided by Prof. Yaman Boluk (University of Alberta, Canada). Gluconic acid was obtained from Thermo Fisher Scientific (Massachusetts, USA). Aspergillus niger glucose oxidase (Cat. no. G0543, with ≤0.1 units/mg protein catalase) and bovine liver catalase were purchased from Sigma (St. Louis, USA).
To prepare insoluble OSX, 2 g OSX was suspended in 200 mL of 50 mM Tris-HCl pH 8.0 for 48 h at room temperature and then washed three times. The washed OSX was then filtered through a 0.45-μm nylon membrane, and the dry weight of the retentate was measured.
CBM fusion synthesis
The CtCBM22A sequence with its native connecting loops (10 amino acids at the N-terminus and 7 amino acids at the C-terminus), together with a TP-rich linker (SRGGGTATPTPTPTPTP) at the C-terminus, was synthesized by NZYTech (Lisbon, Portugal) after codon optimization for Pichia pastoris expression. The synthesized CBM sequence was fused with that of Y300A in pPICZαA. The fusion also contains a His6-tag at the C-terminus to facilitate purification. DNA sequences were confirmed at the Center for Applied Genomics (the Hospital for Sick Children, Toronto). The synthetic gene was deposited in the GenBank database under the accession number of KC707634.
His-tag purification of CtCBM22A_Y300A
The confirmed plasmid was transformed into P. pastoris KM71H following the manufacturer’s instructions (Life Technologies, USA). Expression screening by immuno-colony blot, cell growth, protein expression and purification were conducted following previously reported methods23. Briefly, cells were grown at 15 °C and 250 rpm for 5 days, and 0.5% methanol was added every 24 h to induce recombinant protein expression. Culture supernatant was desalted and mixed with Ni-NTA resin (Qiagen, USA), bound proteins were then eluted with 250 mM imidazole, followed by centrifugal buffer exchange with 50 mM Tris-HCl pH 8.0.
Protein concentration was measured by the Bradford method (Bio-Rad Laboratories, USA), and verified by band densitometry on SDS-PAGE gels using known quantities of bovine serum albumin as a reference and ImageJ for image analysis (http://rsbweb.nih.gov/ij/).
OSX purification of CtCBM22A_Y300A
After five days of induction with 0.5% methanol, 250 mL P. pastoris culture supernatants comprising CtCBM22A_Y300A were harvested by centrifugation for 10 min at 2,000 × g (repeated three times), then filtered through a 0.45 μm membrane. Half of the supernatant was then exchanged to 50 mM Tris-HCl pH 8.0 using 30 kDa MWCO Vivaspin 20 centrifugal concentrators (GE healthcare, UK). Insoluble OSX was then added at 1.25% (w/v) to 40 mL filtered supernatant and buffer exchanged supernatant. Resulting solutions were mixed for 24 h at 4 °C using a 360-degree rotator set at 8 rpm. Protein-bound OSX and unbound (flow-through) fractions were then collected by vacuum-filtration using a 0.45 μm membrane. Protein-bound OSX was washed 3 times with 50 mM Tris-HCl pH 8.0 and then stored in this buffer. All fractions were kept at 4 oC.
OSX binding efficiency
The protein in 700 μL of filtered supernatant, buffer-exchanged supernatant or flow-through fractions after OSX addition was separately precipitated with 10% trichloroacetic acid. The precipitate was washed thoroughly with ice-cold acetone, and the pellet was suspended in 40 μL of SDS loading dye. The proteins in 35–40 mg wet weight of bound OSX were extracted with 40 μL of the denaturing solution (10% SDS and 10% beta-mercaptoethanol) for 10 min at 100 oC. Precipitated as well as extracted proteins were then analyzed using 10% SDS-PAGE gels.
The effect of Ca2+ on CtCBM22A binding to xylan was investigated by incubating the fusion enzyme with OSX overnight at 4 oC in the presence of 5 mM CaCl2; the amount of unbound and bound protein was then evaluated weekly by SDS-PAGE.
Binding affinity on soluble polymeric substrates
Binding of CtCBM22A_Y300A on soluble OSX, arabinoxylan, beech wood xylan, wheat bran hemicellulose and propoxylated wheat bran hemicellulose was evaluated by affinity gel electrophoresis24. Briefly, 0.01% of each polysaccharide was added to 7.5% (w/v) acrylamide in a Tris-glycine buffer (25 mM Tris, 250 mM glycine pH 8.3) prior to polymerization to produce native polyacrylamide gels. Protein samples (5 μg) were loaded into each gel followed by electrophoresis using ice-cold Tris-glycine buffer.
Specific activity, enzyme kinetics and thermostability measurements
The production of oxidized sugars was determined by continuously measuring the co-production of H2O2 at 37 °C for 15 min using a chromogenic horseradish peroxidase assay23. Activity measurements were performed using 16 nM of each GOOX variant and 0.5 mM substrate; kinetic measurements were performed using the following substrate concentrations: 0.05–300 mM glucose; 0.05–1,200 mM xylose; 0.05–20 mM cellobiose; 0.01–10 mM cellotriose, xylobiose and xylotriose; 0.01–4 mM of longer cello- and xylo-oligosaccharides. GraphPad Prism5 software (GraphPad Software, USA) was used for curve fitting and deriving kinetic parameters.
To compare activities of GO and GOOX variants, 24 nM of each enzyme were separately assayed in triplicate with 10 mM of xylose, maltose and cellobiose; 1 mM of glucose and cellotetraose; and 0.6 g/L of mixed XOS. GOOX enzymes were assayed in 50 mM Tris-HCl pH 8.0 while GO was assayed in 50 mM sodium acetate pH 5.0.
Thermostability tests were performed by incubating GOOX enzymes (16 nM) at temperatures between 30 and 54 °C for 1 h, and then measuring residual activity (initial rate) on 0.5 mM cellobiose.
H2O2 inactivation and stability
H2O2 inactivation of GOOX was investigated by incubating 0.1 μg of each GOOX variant (5–7 nM) at 37 °C in the dark in 250 μL solutions of 50 mM Tris-HCl (pH 8.0) containing 1 mM glucose or 0.5 mM cellobiose, and between 0–200 mM H2O2. The effect of H2O2 on GO activity was similarly determined, except that the buffer was replaced by 50 mM sodium acetate (pH 5.0) and 1 mM glucose was used. Substrate concentration was kept low to avoid any contribution from the de novo release of H2O2. Following 5-h incubation with glucose, or 2-h incubation with cellobiose, reactions were filtered through 10 K OmegaTM Nanosep centrifugal devices (Pall Corp., USA) to separate reaction products from each enzyme. To facilitate the high-performance anion-exchange chromatography (HPAEC) analyses, 100 U catalase was added to reactions containing glucose to prevent H2O2 interference, and then incubated for 1 h at 25 °C before filtration through 10 K OmegaTM Nanosep centrifugal devices.
The production of oxidized sugars was analyzed by HPAEC with pulsed amperometric detection (HPAEC-PAD) using a CarboPac PA1 (2 × 250 mm) analytical column (Dionex, USA) in an ICS5000 HPAEC-PAD system (Dionex, USA). The HPAEC-PAD samples were eluted at 0.25 mL/min using sodium acetate gradient (0–1,000 mM) in 100 mM NaOH23. Chromatograms were viewed and analyzed using Chromeleon 7.2 (Dionex, USA).
To test for stability, 16 nM GOOX enzymes and GO were also pre-incubated with 200 mM H2O2 for 30 min at 25 °C in the dark in an Eppendorf ThermoMixer with shaking (300 rpm). The H2O2 was then degraded using catalase (2 × 100 U) prior to addition of 1 mM glucose to measure residual activity. Following a 2-h incubation with glucose at 37 °C, each sample was filtered using a 10 K OmegaTM Nanosep centrifugal device prior to analysis by HPAEC-PAD.
Potential effects of residual cultivation media on H2O2 decomposition was tested in triplicate by incubating 1 μL of culture supernatant or medium with 74 μL of different H2O2 concentrations (up to 150 μM) in 96-well plates at 25 °C for 5 h in the dark. The remaining amount of H2O2 was measured using the chromogenic horseradish peroxidase assay.
Total amino acid analysis
Both Y300A and CtCBM22A_Y300A (1,000 pmoles) were incubated with 200 mM H2O2 in the presence of 1 mM glucose for 5 h in the dark. The samples were then washed intensively with water using a 10 K OmegaTM Nanosep centrifugal device, and treated with 6 N HCl containing 1% phenol for 24 h at 110 °C. The amino acid composition was analyzed at the SPARC BioCentre (the Hospital for Sick Children, Toronto).
Results and Discussion
Broader substrate specificity of Y300A
The GOOX variant Y300A displays a similar substrate profile to the wild-type enzyme; however, it demonstrates higher activity on glucose16 and lower substrate inhibition19. Therefore, Y300A was chosen for the current comparative analysis with GO. The substrate preference of GO and Y300A was assayed using glucose, xylose, maltose, cellobiose, cellotetraose, and XOS. On tested mono- and oligosaccharides, GO exhibited only 2.5% activity relative to Y300A, with the exception of glucose, for which GO activity was 100% higher than Y300A (Fig. 1). The different substrate preference of Y300A and GO is likely attributed to the open active site of GOOX25 in comparison with the narrow binding pocket of GO, which is specifically suited for glucose26.

Relative activity of Y300A and GO on selected mono- and oligo-saccharides.
Each of the following substrates was assayed in triplicate for both enzymes: 1 mM of glucose and cellotetraose; 10 mM of xylose, maltose and cellobiose; and 0.6 g/L of mixed xylo-oligosaccharides (XOS). The sum of Y300A and GO activity (U/mg) towards each substrate is indicated in parentheses, and shaded bars indicate the proportion of that activity attributed to Y300A (white) versus GO (blue).
Improving glucose oxidation by Y300A
The yield of CtCBM22A_Y300A recombinantly expressed in P. pastoris KM71H was approximately 200 mg/L. Fusing CtCBM22A to Y300A also increased kcat values of the enzyme on all sugars, up to 67% in the case of cellotriose, from 670 min−1 to 1,120 min−1 (Table 1). A similar effect was previously seen when CtCBM22A was fused to the N-terminus of the wild-type GOOX23. The Km values of CtCBM22A_Y300A on monosaccharides including glucose and xylose were also decreased by around 45%, resulting in higher catalytic efficiency (Table 1). For instance, kcat/Km values of CtCBM22A_Y300A on glucose and xylose (173 and 27 mM−1 min−1, respectively) are about 100% higher than those of Y300A (Table 1), and more than 600% higher than those reported for wild-type GOOX19. As CtCBM22A does not bind xylose and glucose27, the observed improvement in Km and catalysis on monosaccharides, as well as slight thermal activation at 40 °C (Fig. 2) by CtCBM22A_Y300A, is likely due to a subtle structural change induced in the Y300A variant upon fusion to CtCBM22A, as previously proposed for GOOX23.

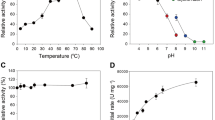
Thermostability of Y300A variants.
Enzymes (16 nM) including OSX-immobilized CtCBM22A_Y300A (or iCtCBM22A_Y300A) were incubated at temperatures between 30 and 54 °C for 1 h, and residual activity was measured using 0.5 mM cellobiose.
As expected, the catalytic efficiency of the fusion enzyme on glucose was lower than those reported for A. niger GO (i.e., 1,80028 and 2,280 mM−1 min−1 29). However, the significantly lower Km values of CtCBM22A_Y300A on glucose (5.4 mM) compared to those reported for GO (~ 3028 and ~50 mM29) may offer advantages to applications containing low glucose concentrations.
Enhanced xylan binding through CtCBM22A fusion
The presence of different xylans including OSX, arabinoxylan, beech wood xylan and wheat bran xylan significantly reduced the migration of CtCBM22A_Y300A in native polyacrylamide gels, indicating strong xylan binding by the fusion enzyme (Fig. 3). Reduced migration was more obvious on buffer-soluble oat spelt xylan and propoxylated wheat bran xylan, as shown by sharp bands at or near the loading wells (Fig. 3). There was no difference in migration between the CtCBM22A fusion of Y300A (Fig. 3) and the previously reported wild-type fusion23, suggesting that the tyrosine substitution did not impact hemicellulose binding.

Binding of the CtCBM22A_Y300A fusion on polymeric substrates.
(A) Control, no polysaccharide; (B) Arabinoxylan; (C) Soluble oat spelt xylan; (D) Beech wood xylan; (E) Wheat bran hemicellulose; (F) Propoxylated wheat bran hemicellulose. Polysaccharides (0.01%) were added to 7.5% native polyacrylamide gels, and 5 μg of CtCBM22A_Y300A were loaded in duplicate; the figure was composed from six separate gels.
Simultaneous purification and immobilization using dietary fibre
Enzyme purification at large scale is expensive and labor-intensive. Given that CtCBM22A fusion was previously shown to immobilize wild-type GOOX on xylan23, insoluble OSX was chosen for ability to immobilize and recover CtCBM22A_Y300A directly from culture supernatant while providing additional dietary fibre in food applications30.
Insoluble OSX was directly added to filtered culture supernatant containing CtCBM22A_Y300A or the supernatant that had been exchanged to 50 mM Tris buffer pH 8.0. The amount of protein that remained bound to insoluble OSX (B fraction) was then evaluated by SDS-PAGE (Fig. 4). The original amount of protein in supernatant solutions before loading (L fraction), those in the first flow-through (F fraction) and flow-through during washing (W fraction) were also analyzed. Low amounts of protein in flow-through solutions were detected, indicating that most of the protein bound to OSX (Fig. 4). Furthermore, there was a similar amount of bound protein from filtered culture supernatant and buffer-exchanged supernatant samples, suggesting that the culture supernatant did not interfere with OSX binding of CtCBM22A_Y300A. Therefore, OSX can be used to recover CtCBM22A_Y300A directly from culture supernatant without buffer-exchange, facilitating the simultaneous purification and immobilization of the enzyme.

Binding and specific activity of different CtCBM22A_Y300A preparations.
The CtCBM22A_Y300A (L fraction) from the filtered culture supernatant or the supernatant after buffer-exchange was mixed with insoluble OSX and then filtered; the amount of protein that was in the flow-through (F fraction) or in subsequent washing steps (W fraction) as well as that bound to OSX (B fraction) was analyzed. Specific activity (U/mg) of L and B fractions on cellobiose is indicated in parentheses.
Although the filtered, spent culture did not impact CtCBM22A_Y300A binding to OSX, enzyme activity towards cellobiose was reduced by approximately 50% (Fig. 4). Fresh induction media did not interfere with the assay (Fig. S1), which suggests that the reduced activity resulted in part by components released by the Pichia transformant.
Further analysis of CtCBM22A_Y300A from buffer exchanged culture medium and immobilized to OSX confirmed that immobilization did not reduce enzyme activity on cellobiose (Fig. 4). This suggests that cellobiose did not compete with OSX for CtCBM22A binding, which is consistent with the high Ka of CtCBM22A towards xylo-oligosaccharides and particularly OSX27, and with our earlier characterization of CtCBM22A_GOOX23.
Binding between CtCBM22A_Y300A and OSX remained stable in 2 M of NaCl, even after 12 weeks at 4 °C (data not shown). However, up to 10% of the protein desorbed from OSX during storage at room temperature, of which about 50% bound back to the OSX upon the addition of 5 mM CaCl2 (Fig. S2). Calcium probably stabilizes the functional form of CtCBM22A, as predicted from structural characterizations of CtCBM22A27.
H2O2 tolerance of GOOX variants and GO
The impact of H2O2 on GO activity and on activities of GOOX variants Y300A, CtCBM22A_Y300A and OSX-immobilized CtCBM22A_Y300A (iCtCBM22A_Y300A), was compared using glucose as the substrate.
Fusion of CtCBM22A to Y300A appeared to increase Y300A stability in H2O2, as CtCBM22A_Y300A and iCtCBM22A_Y300A retained approximately 80% activity after 5 h in 200 mM H2O2 (Fig. 5). The higher turnover number and better glucose binding by the fusion enzyme (Table 1) might have outweighed activity losses resulting from H2O2 inactivation. No significant difference in H2O2 tolerance was observed between Y300A and GO (Fig. 5), which displayed decreasing glucose oxidation with increasing H2O2 concentration and retained approximately 60% activity at 200 mM H2O2. It should be noted, however, that the commercial GO preparation used herein contains a very low but detectable amount of catalase (≤0.1 U/mg of GO, as reported by Sigma). An earlier report shows that GO retains approximately 20% activity after 20-min incubation with 100 mM H2O212. This difference in H2O2 tolerance of GO samples may reflect differences in activity measurement (i.e., oxygen consumption versus gluconic acid production herein) or improvement in commercial preparations of GO.

H2O2 tolerance of GOOX variants and GO.
Enzymes (0.1 μg) were incubated with 1 mM glucose and different concentrations of H2O2 (up to 200 mM). The production of gluconic acid was then quantified by HPAEC-PAD.
When retaining the same reaction conditions but substituting glucose for cellobiose, H2O2 inactivation was not detected for any GOOX variant (Fig. 6). The catalytic efficiency of Y300A and CtCBM22A_Y300A on cellobiose (3,400 and 4,000 min−1 mM−1, respectively) is an order of magnitude higher than that on glucose (98 and 173 min−1 mM−1, respectively) (Table 1). Presumably then, GOOX variants were able to oxidize its preferable substrate, cellobiose, before H2O2 inactivation took effect.

Cellobiose oxidation by Y300A and CtCBM22A_Y300A in the presence of different amounts of H2O2.
Relative activity of 16 nM GOOX variants on 0.5 mM cellobiose in the presence of H2O2 (0–200 mM). Activity values were derived from HPAEC-PAD quantification of reaction products.
Pre-incubation of GO and GOOX variants with 200 mM H2O2 for 30 min prior to addition of glucose did not impact enzyme activity (Fig. 7). This suggests that reduced forms of GO and GOOX are more susceptible to inactivation by H2O2, as previously reported for GO12.

Residual activity of GO, Y300A and CtCBM22A_Y300A on glucose following pre-incubation with H2O2.
After a 30-min pre-incubation with 200 mM H2O2 at 25 °C, H2O2 was removed and 1 mM glucose was added; the reaction was then incubated for 2 h at 37 °C before HPAEC-PAD analysis for the production of gluconic acid.
Apparent mode of H2O2 inactivation
The oxidation of key methionine residues in the presence of H2O2 has been proposed to cause GO inactivation12. The total amino acid analysis of GOOX variants, however, revealed significant loss of histidine in Y300A and its CtCBM22A fusion following incubation with H2O2 (Table S1). The broader active site as well as the double-covalent flavinylation in GOOX might contribute to this potential difference in H2O2 inactivation. Notably, histidine residues H70 and H138 are positioned in the GOOX active site, of which the Nδ1 atom of H70 binds to the 8α-methyl group of the isoalloxazine ring of FAD while H138 interacts with FAD via a hydrogen bond (Fig. S3). The oxidation of either these histidine residues would affect FAD binding and electrochemical properties. For instance, an H70A substitution, which led to removal of the 8α -N1-histidyl bond, retained only 0.5% catalytic efficiency and about 55% redox potential, compared with the wild-type31. Histidine residues can be oxidized by H2O2 to form a mixture of products including Asp and 2-oxo-histidine32. Since H2O2 treatment only slightly increased the (Asp + Asn) content in Y300A and CtCBM22A_Y300A samples (Table S1), 2-oxo-histidine is probably the major product of oxidation. As histidine loss was 6.7 and 9.3 mol/mol of protein in Y300A and its CtCBM22A fusion, respectively (Table S1), other histidine residues were also modified. Besides the two exposed histidine residues in the active site and six in the 6x-his tag, Y300A also has four histidine residues on the surface, and the fusion provides two additional exposed histidines. Loss of tyrosine in the H2O2-treated samples was also detected, particularly in CtCBM22A_Y300A, which has three tyrosine residues in the binding cleft out of its eleven surface-exposed tyrosine residues.
Conclusion
GOOX variants Y300A and CtCBM22A_Y300A demonstrate potential benefit in applications characterized by relatively high H2O2 concentrations and mixed sugars. Both Y300A and CtCBM22A_Y300A have broader substrate profiles than the commonly used GO, and CtCBM22A_Y300A displayed less H2O2 inactivation than GO when assayed with glucose. Furthermore, both GOOX variants showed no H2O2 inactivation on cellobiose, their favorable substrate. CtCBM22A_Y300A is the best variant of GOOX to date for glucose oxidation (as well as xylose oxidation) while retaining considerable activity on oligosaccharides. The presence of CtCBM22A also permits enzyme binding to OSX, facilitating simultaneous purification and immobilization of the enzyme, and ancillary fibre enrichment in potential food applications.
Additional Information
How to cite this article: Vuong, T. V. et al. Direct comparison of gluco-oligosaccharide oxidase variants and glucose oxidase: substrate range and H2O2 stability. Sci. Rep. 6, 37356; doi: 10.1038/srep37356 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Rando, D., Kohring, G. W. & Giffhorn, F. Production, purification and characterization of glucose oxidase from a newly isolated strain of Penicillium pinophilum. Applied Microbiology and Biotechnology 48, 34–40 (1997).
Bankar, S. B., Bule, M. V., Singhal, R. S. & Ananthanarayan, L. Glucose oxidase–an overview. Biotechnology Advances 27, 489–501, doi: 10.1016/j.biotechadv.2009.04.003 (2009).
Leskovac, V., Trivic, S., Wohlfahrt, G., Kandrac, J. & Pericin, D. Glucose oxidase from Aspergillus niger: the mechanism of action with molecular oxygen, quinones, and one-electron acceptors. The International Journal of Biochemistry & Cell Biology 37, 731–750, doi: 10.1016/j.biocel.2004.10.014 (2005).
Chambers, J. P., Arulanandam, B. P., Matta, L. L., Weis, A. & Valdes, J. J. Biosensor recognition elements. Current Issues in Molecular Biology 10, 1–12 (2008).
Wong, C. M., Wong, K. H. & Chen, X. D. Glucose oxidase: natural occurrence, function, properties and industrial applications. Applied Biochemistry and Biotechnology 78, 927–938, doi: 10.1007/s00253-008-1407-4 (2008).
Jiang, Z. & Ooraikul, B. Reduction of nonenzymatic browning in potato chips and french fries with glucose oxidase. Journal of Food Processing and Preservation 13, 175–186, doi: 10.1111/J.1745-4549.1989.Tb00099.X (1989).
Cruz, A. G. et al. Probiotic yogurts manufactured with increased glucose oxidase levels: Postacidification, proteolytic patterns, survival of probiotic microorganisms, production of organic acid and aroma compounds. Journal of Dairy Science 95, 2261–2269, doi: 10.3168/jds.2011-4582 (2012).
Cruz, R. S., Camilloto, G. P. & dos Santos Pires, A. C. Oxygen scavengers: An approach on food preservation. Structure and Function of Food Engineering (ed Ayman Amer Eissa ) 21–42 (InTech, 2012).
Decamps, K. et al. The bread dough stability improving effect of pyranose oxidase from Trametes multicolor and glucose oxidase from Aspergillus niger: unraveling the molecular mechanism. Journal of Agricultural and Food Chemistry 61, 7848–7854, doi: 10.1021/jf4021416 (2013).
Hanft, F. & Koehler, P. Studies on the effect of glucose oxidase in bread making. Journal of the Science of Food and Agriculture 86, 1699–1704, doi: 10.1002/jsfa.2455 (2006).
Bonet, A. et al. Glucose oxidase effect on dough rheology and bread quality: A study from macroscopic to molecular level. Food Chemistry 99, 408–415, doi: 10.1016/j.foodchem.2005.07.043 (2006).
Kleppe, K. The effect of hydrogen peroxide on glucose oxidase from Aspergillus niger. Biochemistry 5, 139–143 (1966).
Halada, P. et al. Oxidation of Phe454 in the gating segment inactivates Trametes multicolor pyranose oxidase during substrate turnover. PLOS One 11, e0148108, doi: 10.1371/journal.pone.0148108 (2016).
Kulys, J., Tetianec, L. & Schneider, P. Specificity and kinetic parameters of recombinant Microdochium nivale carbohydrate oxidase. Journal of Molecular Catalysis B: Enzymatic 13, 95–101 (2001).
Lin, S.-F., Yang, T.-Y., Inukai, T., Yamasaki, M. & Tsai, Y.-C. Purification and characterization of a novel glucooligosaccharide oxidase from Acremonium strictum T1. Biochimica et Biophysica Acta 1118, 41–47 (1991).
Foumani, M., Vuong, T. V. & Master, E. R. Altered substrate specificity of the gluco-oligosaccharide oxidase from Acremonium strictum. Biotechnology and Bioengineering 108, 2261–2269, doi: 10.1002/bit.23149 (2011).
Foumani, M., Vuong, T. V. & Master, E. R. Glucooligosaccharide oxidases from Acremonium strictum and uses thereof. WO/2012/116431, World Intellectual Property Organization (2012).
Lee, M. H. et al. Structural characterization of glucooligosaccharide oxidase from Acremonium strictum. Applied and Environmental Microbiology 71, 8881–8887, doi: 10.1128/AEM.71.12.8881-8887.2005 (2005).
Vuong, T. V. et al. Xylo- and cello-oligosaccharide oxidation by gluco-oligosaccharide oxidase from Sarocladium strictum and variants with reduced substrate inhibition. Biotechnology for Biofuels 6, 148, doi: 10.1186/1754-6834-6-148 (2013).
Schneider, P. et al. Carbohydrate oxidase and use thereof in baking. WO1999031990 A1, World Intellectual Property Organization (1999).
Degrand, L., Rakotozafy, L. & Nicolas, J. Activity of carbohydrate oxidases as influenced by wheat flour dough components. Food Chemistry 181, 333–338, doi: 10.1016/j.foodchem.2015.02.062 (2015).
Nordkvist, M., Nielsen, P. M. & Villadsen, J. Oxidation of lactose to lactobionic acid by a Microdochium nivale carbohydrate oxidase: Kinetics and operational stability. Biotechnology and Bioengineering 97, 694–707, doi: 10.1002/bit.21273 (2007).
Vuong, T. V. & Master, E. R. Fusion of a xylan-binding module to gluco-oligosaccharide oxidase increases activity and promotes stable immobilization. PLOS One 9, e95170, doi: 10.1371/journal.pone.0095170 (2014).
Foumani, M., Vuong, T. V., MacCormick, B. & Master, E. R. Enhanced polysaccharide binding and activity on linear beta-glucans through addition of carbohydrate-binding modules to either terminus of a glucooligosaccharide oxidase. PLOS One 10, doi: 10.1371/journal.pone.0125398 (2015).
Huang, C. H. et al. Crystal structure of glucooligosaccharide oxidase from Acremonium strictum: a novel flavinylation of 6-S-cysteinyl, 8alpha-N1-histidyl FAD. The Journal of Biological Chemistry 280, 38831–38838, doi: 10.1074/jbc.M506078200 (2005).
Hecht, H. J., Kalisz, H. M., Hendle, J., Schmid, R. D. & Schomburg, D. Crystal structure of glucose oxidase from Aspergillus niger refined at 2.3 Å resolution. Journal of Molecular Biology 229, 153–172 (1993).
Charnock, S. J. et al. The X6 “thermostabilizing” domains of xylanases are carbohydrate-binding modules: structure and biochemistry of the Clostridium thermocellum X6b domain. Biochemistry 39, 5013–5021 (2000).
Kalisz, H. M., Hecht, H. J., Schomburg, D. & Schmid, R. D. Effects of carbohydrate depletion on the structure, stability and activity of glucose oxidase from Aspergillus niger. Biochimica et Biophysica Acta 1080, 138–142 (1991).
Courjean, O. & Mano, N. Recombinant glucose oxidase from Penicillium amagasakiense for efficient bioelectrochemical applications in physiological conditions. Journal of Biotechnology 151, 122–129, doi: 10.1016/j.jbiotec.2010.10.077 (2011).
Eastwood, M. A. The physiological effect of dietary fiber: an update. Annual Review of Nutrition 12, 19–35, doi: 10.1146/annurev.nu.12.070192.000315 (1992).
Huang, C. H. et al. Functional roles of the 6-S-cysteinyl, 8alpha-N1-histidyl FAD in glucooligosaccharide oxidase from Acremonium strictum. The Journal of Biological Chemistry 283, 30990–30996, doi: 10.1074/jbc.M804331200 (2008).
Uchida, K. & Kawakishi, S. Identification of oxidized histidine generated at the active site of Cu,Zn-superoxide dismutase exposed to H2O2 . Selective generation of 2-oxo-histidine at the histidine 118. The Journal of Biological Chemistry 269, 2405–2410 (1994).
Acknowledgements
Funding for this research was provided by the Government of Ontario for the project “Forest FAB: Applied Genomics for Functionalized Fibre and Biochemicals” (ORF-RE-05-005), and by the Natural Sciences and Engineering Research Council of Canada for the Strategic Network Grant “Industrial Biocatalysis Network”.
Author information
Authors and Affiliations
Contributions
E.R.M. coordinated the study; T.V.V., M.F., B.M., and R.K. conducted the experiments; T.V.V., M.F., and E.R.M. wrote the manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Vuong, T., Foumani, M., MacCormick, B. et al. Direct comparison of gluco-oligosaccharide oxidase variants and glucose oxidase: substrate range and H2O2 stability. Sci Rep 6, 37356 (2016). https://doi.org/10.1038/srep37356
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep37356
This article is cited by
-
Glucose oxidase and metal catalysts combined tumor synergistic therapy: mechanism, advance and nanodelivery system
Journal of Nanobiotechnology (2023)
-
Insights into the genome and secretome of Fusarium metavorans DSM105788 by cultivation on agro-residual biomass and synthetic nutrient sources
Biotechnology for Biofuels (2021)
-
Enzymatic production of 4-O-methyl d-glucaric acid from hardwood xylan
Biotechnology for Biofuels (2020)
-
Novel redox-active enzymes for ligninolytic applications revealed from multiomics analyses of Peniophora sp. CBMAI 1063, a laccase hyper-producer strain
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
