
Abstract
In the present study, thirty autosomal insertion and deletion polymorphic loci were simultaneously amplified and genotyped in a multiplex system, and their allelic frequencies as well as several forensic parameters were obtained in a sample of 236 unrelated healthy Tujia individuals. All the loci were in Hardy-Weinberg equilibrium after applying a Bonferroni correction and all pair-wise loci showed no significant linkage disequilibrium. These loci were observed to be relatively informative and discriminating, quite efficient for forensic applications. Allelic frequencies of 30 loci were compared between the Tujia group and other reference populations, and the results of analysis of molecular variance indicated the Tujia group showed the least significant differences with the Shanghai Han at one locus, and the most with Central Spanish population at 22 loci. We analyzed the population genetic structure by the principal component analysis, the clustering of STRUCTURE program and a Neighbor-Joining tree, and then evaluated the genetic relationships among Tujia and other 15 populations.
Similar content being viewed by others

Introduction
Short tandem repeats (STRs) have become popular DNA markers in forensic DNA labs for more than 20 years and have been proved to possess several benefits, which make them especially suitable to identify victims, perpetrators, missing persons, and for kinship testing and population genetic analysis1,2,3,4,5. However, there were some potential limitations of STRs in forensic applications because of its relatively high mutation rate, long amplicon size, and the deficiency in the analysis of highly degraded DNA samples and complex kinship cases. In recent years, a novel genetic marker: insertion and deletion polymorphisms (InDels) dispersing through the human genome showed some advantages, such as short amplicon size, low mutation rate, and practicability of being genotyped in the present forensic DNA lab platforms6,7,8, which were useful for forensic DNA applications (Supplementary method for STR applications), population genetics9,10,11,12, and biogeographic ancestry analysis13,14,15. Population genetic and forensic validation studies have been performed using the Qiagen Investigator DIPplex® reagent including 30 autosomal InDel loci plus amelogenin locus, and population data of Chinese Han, Tibetan, Uigur, Kazak, She, Xibe and Yi populations have been reported in previous studies9,10,16,17,18. In the present study, we firstly reported the population genetic data of 30 InDels in Chinese Tujia ethnic group, evaluated their usefulness in the field of forensic sciences, analyzed the interpopulation differentiations, and retraced the genetic background of the Tujia group by the population structure construction, principal component analysis, phylogenetic tree and some other analyses.
Materials and Methods
Ethical statement and population samples
Bloodstain samples were randomly collected from 236 unrelated healthy Tujia individuals in Enshi Tujia and Miao Autonomous Prefecture of Hubei province, China. The study was conducted in accordance with the human and ethical research principles of Xi’an Jiaotong University Health Science Center and approved by the ethics committee of Xi’an Jiaotong University Health Science Center. We have obtained written informed consent from all volunteers for the purpose of research. The investigation was conducted in order to ensure that any two individuals didn’t share a common ancestry within at least three previous generations; all individuals were born and lived in the same prefecture; and their ancestors married no any other ethnic people.
DNA extraction, co-amplification and genotyping
Genomic DNA was extracted from bloodstain cards by using the Chelex® method (Solarbio, Beijing, China) according to the manufacturer’s instructions19. About 0.5–1.0 ng genomic DNA was used for amplification with a 25 ul reaction volume. PCR amplification for 30 InDel loci and Amelogenin locus was performed in a single multiplex reaction using the DIPplex Investigator reagent (Qiagen, Hilden, Germany), which was prepared on a GeneAmp® PCR System 9700 thermal cycler (Applied Biosystems, Foster City, CA, USA) under the recommended reaction condition. PCR products of all loci were separated and detected by capillary electrophoresis on the ABI 3500 Genetic Analyzer (Applied Biosystems). Genotyping of InDel loci was analyzed using the BTO 550 (Qiagen) as internal lane standard and by GeneMapper® ID software v3.2 (Applied Biosystems). Experiments were carried out according to the kit control and the ISO 17025 standard in this study.
Statistical analyses
Hardy-Weinberg equilibrium (HWE), allelic frequencies and forensic statistical parameters of 30 InDels were calculated by the modified powerstat (version1.2) spreadsheet (Promega, Madison, WI, USA). Linkage disequilibrium (LD) analysis for all pair-wise InDel loci was performed using the SNPAnalyzer v2.0 (Istech, South Korea)20. Fst and p values for pairwise interpopulation comparisons were calculated based on allele frequencies of 30 InDels by analysis of molecular variance (AMOVA) performed with ARLEQUIN version 3.1 software (http://cmpg.unibe.ch/software/arlequin3). Principal component analysis (PCA) in two forms and phylogenetic reconstruction were employed in MATLAB 2007a (MathWorks Inc., USA), R statistical software v3.0.221 and genetic distance and phylogenetic analysis (DISPAN) program (http://pritch.bsd.uchicago.edu), respectively. The detailed population genetic structure was performed with the STRUCTURE program v2.2 (http://pritch.bsd.uchicago.edu.) to analyze the structure of Tujia and the other populations previously published based on the same 30 InDels.
Results and Discussion
Allele diversities within group
Probability values for Hardy-Weinberg equilibrium tests for 29 InDel loci ranged from 0.0669 (HLD40) to 0.9987 (HLD97), and p < 0.05 was only observed at the HLD88 locus (p = 0.0382). P values were adjusted after applying a Bonferroni correction for all 30 InDel loci analyzed and P > 0.00167 was considered statistically insignificant. Then, the genotype frequency data for all loci showed no deviations from HWE expectations in the sample of Tujia group. Allelic frequencies and forensic statistical parameters of 30 InDels based on the raw genotype (shown in Supplementary Table 1) were shown in Table 1. Allelic frequencies of deletion allele at the 30 InDel loci ranged from 0.0445 to 0.9089 in the group, with a mean value of 0.4939. The observed (HO) and expected heterozygosities (HE) ranged from 0.0890 (HLD118) to 0.5381(HLD92); and 0.0850 (HLD118) to 0.4985(HLD136), with a mean value of 0.4028 and 0.4073, respectively. Twenty-four InDel loci had power of discrimination (PD) values greater than 0.5, except the six loci: HLD39, HLD64, HLD81, HLD99, HLD111, and HLD118 loci. The values of the power of exclusion (PE), the matching probability (MP), the typical paternity index (TPI), and the polymorphic information content (PIC) ranged from 0.0067 to 0.2231, 0.3524 to 0.8379, 0.5488 to 1.0826 and 0.0814 to 0.3742, respectively. The lowest HO, HE, PIC, TPI, PD and PE were observed at HLD 118 locus, and this locus was also found with the lowest polymorphism in other previously studied groups10. The combined power of exclusion (CPE) and discrimination (CPD) at the 30 InDel loci in the Tujia group were 0.9860 and 0.9999999999761, respectively; combined matching probability (CMP) value of 30 InDels in the group was 2.3894 × 10−11, higher than that in our previous study which reached 1.10974 × 10−19 of 21 autosomal STRs in Tujia group11. According to our calculation, the value of CMP combining 30 InDels with 21 autosomal STRs reached 2.652 × 10−30. These data suggested that the panel of 30 InDel loci could be a valid supplement to the routine detection of autosomal STRs in forensic cases.
Linkage disequilibrium tests
Linkage disequilibrium tests of these pairwise InDels were analyzed using the SNPAnalyzer version 2.0 and obtained several indexes: LOD, r2 and |D’|. As shown in Supplementary Fig. 1, no strong linkage disequilibrium between two different InDels was observed in a total of 435 interclass correlation tests (data not shown) with the values of r2 less than 0.8, and no crimson box was coated by a thick black curve. The present LD tests suggested that 30 InDels were independent for the following statistical analyses, and also suited for forensic cases in the Tujia group.
Genetic divergences
Genetic distance is a measure method of the genetic divergence between different populations, used for understanding the origin of biodiversity and reconstructing the history of different ethnic groups22. We measured the Nei’s DA distance by examining the differences between allelic frequencies at the same set of 30 InDel loci of different populations. DA distances between the 16 groups with each other based on allelic frequencies of the 30 InDel loci were shown in Table 2. Short genetic distances were found between the Tujia group and Shanghai Han17, Guangdong Han18, South Korean23, Beijing Han9, Xibe10, She17, Tibetan9, and Yi16 groups; and further distances were observed between the Tujia group and Chinese Kazak9, Uigur9 groups; whereas the larger distances were estimated with Uruguayan24, Dane25, Central Spanish26, Basque26, and Hungarian populations27. Pairwise populations had small genetic distances, which indicated that they had close genetic relationships or shared a recent common ancestor.
InDel diversities among populations
Population differentiations for 30 InDels were compared between the Tujia group and other populations previously published based on AMOVA method (p < 0.05). As shown in Table 3, the AMOVA comparison results showed significant differences between the Tujia group and Shanghai Han, Beijing Han, Guangdong Han, She, Xibe, South Korean, Tibetan, Yi, Uigur, Kazak, Uruguayan, Hungarian, Basque, Dane, Central Spanish populations at 1, 3, 3, 4, 5, 7, 8, 9, 14, 14, 20, 20, 20, 21 and 22 loci, respectively. The present results demonstrated that the HLD125, HLD99, HLD67, HLD118 loci had relatively high level of genetic variation, with the significant differentiation between Tujia group and other 9, 10, 10 and 11 populations, respectively; while the least differentiation was obtained at the HLD92, HLD101, HLD124 loci with only one pair-wise population. Therefore, allele frequency data obtained at 30 InDels are very important and necessary for forensic application research of different populations.
Principal component analyses
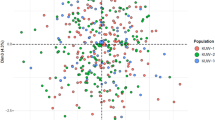
On the basis of the allelic frequencies at the same 30 InDels, PCA figures were constructed by MATLAB 2007a (MathWorks Inc., USA) and R statistical software v3.0.221 among the Tujia group and other 15 reference populations. As shown in Fig. 1a, the variance ratio contribution of the first principal component (PC) was about 77.87% of the total variation and the second accounted for 5.74%. In the PCA diagram, the 16 populations were divided into three relatively independent areas inconsistency with their languages family. Ethnic groups with similar language family basically spread closer. The results indicated that there were close relationships between the Tujia group and Chinese Han populations from different regions, as well as She and South Korean groups. Ya et al. studied the haplotypes of 17 Y-STR loci and preformed the multidimensional scaling plot which also showed the close relationship between Tujia and Han population28; and the similar result was observed in the PCA plot based on the allelic frequencies of HLA-DRB1 locus29. The relatively far genetic relationships between Tujia group and Kazak or Uigur group were observed in the PCA map constructed by mtDNA haplogroup frequencies30 and in the abovementioned HLA-DRB1 PCA plot29, respectively.

A PCA plot showing the genetic relationships.
(a) Tujia group and other 15 reference populations. (b) Tujia, central Asian, western Eurasian and other eastern Eurasian populations were analyzed at individual level.
The genetic relationships among Tujia, central Asian (Uigur and Kazak populations), western Eurasians (Hungarian, Dane, Basque and Central Spanish populations) and other eastern Eurasians (Shanghai Han, Beijing Han, Guangdong Han, She, Xibe, South Korean, Tibetan, and Yi populations) were also discerned with the aid of abovementioned InDel datasets at the individual level. Results of individual PCA were presented by the plots of the first two PCs (shown in Fig. 1b), which together accounted for 38.82% of the total variation in these populations. The first PC revealed an east-west geographic division within Eurasians. In concrete terms, all eastern Eurasians tended to cluster on the left of PCA plots, whereas western Eurasians formed a separate cluster on the right. The Tujia people were expectedly clustered within eastern Asian group.
Neighbor-joining phylogenetic reconstruction
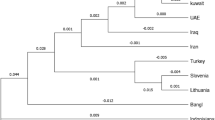
We constructed a neighbor-joining (N-J) phylogenetic tree (shown in Fig. 2). The branch in the upper-left corner contained the nine East Asian populations including Tujia group; whereas in the other branch, Dane, Basque, Central Spanish, Uruguayan, and Hungarian populations were found in the lower-left corner. The Kazak and Uigur groups were in the middle of the above two branches. In previous study, the close relationship between Tujia group and Han population was observed in the N-J dendrogram based on the allelic frequencies of HLA-A locus31. The language of Tujia belongs to Tibeto-Burman language system, without written script. Tujias lived with other nationalities like Miao and Han, and many of them can speak Mandarin Chinese and write the Chinese characters. The tight genetic relationship between the Tujia and Han population in Hubei provience was observed based on fifteen STRs, and the present and previous studies indicated that broad genetic exchanges had occurred among them in history32.

A neighbor-joining phylogenetic tree constructed to analyze phylogenetic relationships from Tujia group and 15 reference populations.
Population STRUCTURE analyses
The STRUCTURE program was used to evaluate the genetic structure of Tujia and other 15 populations. As shown in Fig. 3, at K = 2, three clusters were highly visible and easily distinguishable basically by red, green and mixture of the two. When K = 2–7 (in Supplementary Fig. 2), the STRUCTURE analyses revealed three major clusters: the first subpopulation of Dane, Basque, Central Spanish, Uruguayan, and Hungarian populations, the second of Kazak and Uigur; the last one of nine East Asian populations including Tujia group. The results presented here were similar to that of the PCA plot and N-J tree. With the increase of K values, no further population structures were obtained. We should, just as a precaution, study more ancestry informative InDels in the future in order to subdivide the genetic structure of different ethnic groups in China, and to infer the population origin and ancestral components of an unknown individual.

Population STRUCTURE analysis of 16 populations at K = 2, which revealed three major clusters.
Conclusion
In summary, the population data here indicated the 30 InDels had high diversities within the studied group and genetic differentiations among different populations; and could be a useful supplement to the routine detection of autosomal STRs in forensic cases. The PCA plot, N-J tree and STRUCTURE analyses suggested the close relationships between Tujia and Han population in different regions. More ancestry informative InDels and SNPs should be selected and validated to clarify the Tujia ancestral origin.
Additional Information
How to cite this article: Shen, C. et al. A 30-InDel Assay for Genetic Variation and Population Structure Analysis of Chinese Tujia Group. Sci. Rep. 6, 36842; doi: 10.1038/srep36842 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Meng, H. T. et al. Genetic diversities of 20 novel autosomal STRs in Chinese Xibe ethnic group and its genetic relationships with neighboring populations. Gene. 557, 222–228 (2015).
Yuan, J. Y. et al. Genetic profile characterization and population study of 21 autosomal STR in Chinese Kazak ethnic minority group. Electrophoresis. 35, 503–510 (2014).
Liu, W. J. et al. 24 Y-chromosomal STR polymorphisms for Chinese Uigur ethnic group and its phylogenic analysis with other Chinese groups. Electrophoresis. 36, 626–632 (2014).
Meng, H. T. et al. Diversity study of 12 X-chromosomal STR loci in Hui ethnic from China. Electrophoresis. 35, 2001–2007 (2014).
Zhu, B. et al. Developmental validation of the AGCU 21+1 STR kit A novel multiplex assay for forensic application. Electrophoresis. 36, 271–276 (2015).
LaRue, B. L. et al. Characterization of 114 insertion/deletion (INDEL) polymorphisms, and selection for a global INDEL panel for human identification. Leg Med (Tokyo). 16, 26–32 (2014).
Li, C. et al. Selection of 29 highly informative InDel markers for human identification and paternity analysis in Chinese Han population by the SNPlex genotyping system. Mol Biol Rep. 39, 3143–3152 (2012).
Weber, J. L. et al. Human diallelic insertion/deletion polymorphisms. Am J Hum Genet. 71, 854–862 (2002).
Wei, Y. L., Qin, C. J., Dong, H., Jia, J. & Li, C. X. A validation study of a multiplex INDEL assay for forensic use in four Chinese populations. Forensic Sci Int Genet. 9, e22–e25 (2014).
Meng, H. T. et al. Genetic polymorphism analyses of 30 InDels in Chinese Xibe ethnic group and its population genetic differentiations with other groups. Sci Rep. 5, 8260 (2015).
Yuan, G. L. et al. Genetic data provided by 21 autosomal STR loci from Chinese Tujia ethnic group. Mol Biol Rep. 39, 10265–10271 (2012).
Montgomery, S. B. et al. The origin, evolution, and functional impact of short insertion-deletion variants identified in 179 human genomes. Genome Res. 23, 749–761 (2013).
Zaumsegel, D., Rothschild, M. A. & Schneider, P. M. A 21 marker insertion deletion polymorphism panel to study biogeographic ancestry. Forensic Sci Int Genet. 7, 305–312 (2013).
Francez, P. A., Ribeiro-Rodrigues, E. M. & dos Santos, S. E. Allelic frequencies and statistical data obtained from 48 AIM INDEL loci in an admixed population from the Brazilian Amazon. Forensic Sci Int Genet. 6, 132–135 (2012).
Romanini, C. et al. Ancestry informative markers: inference of ancestry in aged bone samples using an autosomal AIM-Indel multiplex. Forensic Sci Int Genet. 16, 58–63 (2015).
Zhang, Y. D. et al. Forensic evaluation and population genetic study of 30 insertion/deletion polymorphisms in a Chinese Yi group. Electrophoresis. 36, 1196–1201 (2015).
Wang, Z. et al. Population genetics of 30 insertion-deletion polymorphisms in two Chinese populations using Qiagen Investigator® DIPplex kit. Forensic Sci Int Genet. 11, e12–e14 (2014).
Hong, L. et al. Genetic polymorphisms of 30 Indel loci in Guangdong Han population. J SUN Yat-sen Univ (Med Sci). 34, 299–304 (2013).
Walsh, P. S., Metzger, D. A. & Higuchi, R. Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques 10, 506–513 (1991).
Yoo, J., Lee, Y., Kim, Y., Rha, S. Y. & Kim, Y. SNPAnalyzer 2.0: a web-based integrated workbench for linkage disequilibrium analysis and association analysis. BMC Bioinformatics. 9, 290 (2008).
Core, R. R: A language and environment for statistical computing. R Foundation for Statistical Computing. Vienna (2013).
Nei, M. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics. 89, 583–590 (1978).
Seong, K. M. et al. Population genetics of insertion-deletion polymorphisms in South Koreans using Investigator DIPplex kit. Forensic Sci Int Genet. 8, 80–83 (2014).
Saiz, M. et al. Allelic frequencies and statistical data from 30 INDEL loci in Uruguayan population. Forensic Sci Int Genet. 9, e27–e29 (2014).
Friis, S. L. et al. Typing of 30 insertion/deletions in Danes using the first commercial indel kit–Mentype® DIPplex. Forensic Sci Int Genet. 6, e72–e74 (2012).
Martín, P. et al. Population genetic data of 30 autosomal indels in Central Spain and the Basque Country populations. Forensic Sci Int Genet. 7, e27–e30 (2013).
Kis, Z. et al. Genome deletion and insertion polymorphisms (DIPs) in the Hungarian population. Forensic Sci Int Genet. 6, e125–e126 (2012).
Yang, Y. R., Jing, Y. T., Zhang, G. D., Fang, X. D. & Yan, J. W. Genetic analysis of 17 Y-chromosomal STR loci of Chinese Tujia ethnic group residing in Youyang Region of Southern China. Leg Med (Tokyo). 16, 173–175 (2014).
Qiu, X. P. et al. Genetic diversity of HLA-DRB1 alleles in the Tujia population of Wufeng, Hubei Province, China. Genet Mol Res. 13, 4756–4765 (2014).
Li, B. et al. Genetic pol ymorphism of mitochondrial DNA in Dong, Gelao, Tujia, and Yi ethnic populations from Guizhou, China. J Genet Genomics. 34, 800–810 (2007).
Zhang, L. et al. Distribution of HLA-A, -B and -DRB1 genes and haplotypes in the Tujia population living in the Wufeng Region of Hubei Province, China. PLoS One. 7, e38774 (2012).
Zou, J., Zhu, Y. Y., Shen, G. & Yang, J. k. Genetic structure and phylogeny of the Tujia and the Han populations in Hubei Province. China revealed by 15 autosomal short tandem repeats loci polymorphisms. Fudan Univ J Med Sci. 38, 204–210 (2011).
Acknowledgements
This project was supported by the National Natural Science Foundation of China (NSFC, No. 81471824, 81525015, 81373248), and Shaanxi Science and Technology Co-ordination and Innovation Project (2015KTCL03-03). Thank Professor Shaoqing Wen for helping us to draw the Figure 1b.
Author information
Authors and Affiliations
Contributions
C.S. and B.Z. performed the data acquisition and wrote the main manuscript text, X.B. designed the research, T.Y., Z.L., Y.Z., B.W. and X.B. did the data processing and the manuscript modification, J.Y., F.T. and B.Z. prepared the figures. All authors reviewed the manuscript. All authors read and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Shen, C., Zhu, B., Yao, T. et al. A 30-InDel Assay for Genetic Variation and Population Structure Analysis of Chinese Tujia Group. Sci Rep 6, 36842 (2016). https://doi.org/10.1038/srep36842
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep36842
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
