
Abstract
Although endometrioid endometrial cancer (EEC; comprising ~80% of all endometrial cancers diagnosed) is typically associated with favourable patient outcome, a significant portion (~20%) of women with this subtype will relapse. We hypothesised that gene expression predictors of the more aggressive non-endometrioid endometrial cancers (NEEC) could be used to predict EEC patients with poor prognosis. To explore this hypothesis, we performed meta-analysis of 12 gene expression microarray studies followed by validation using RNA-Seq data from The Cancer Genome Atlas (TCGA) and identified 1,253 genes differentially expressed between EEC and NEEC. Analysis found 121 genes were associated with poor outcome among EEC patients. Forward selection likelihood-based modelling identified a 9-gene signature associated with EEC outcome in our discovery RNA-Seq dataset which remained significant after adjustment for clinical covariates, but was not significant in a smaller RNA-Seq dataset. Our study demonstrates the value of employing meta-analysis to improve the power of gene expression microarray data, and highlight genes and molecular pathways of importance for endometrial cancer therapy.
Similar content being viewed by others


Introduction
Endometrial cancer is the most commonly diagnosed gynecological cancer in developed countries, accounting for approximately 7% of new cancer cases in women worldwide1. Unlike most other cancer in females, age-standardized rated are steadily increasing2. Endometrioid endometrial cancers (EECs) are the most commonly reported histological subtype of endometrial cancer (~80% of all new cases), are estrogen-related tumors, and generally associated with good prognosis. Conversely, non-endometrioid endometrial cancers (NEECs; commonly serous papillary or clear cell histology) are estrogen-independent, and tend to be high-grade, clinically aggressive tumors3. A subset of EEC patients (~20%) will suffer recurrent tumors, with a 5-year survival rate reduced from 75–80% to less than 10%4. Although a recent study has reported the utility of POLE mutation status for identifying women with good prognosis5, there is currently no accepted method to identify markers that predict EEC patients with poor clinical outcome. Markers to predict EEC patients with poor prognosis will identify those women requiring more extensive surgery and adjuvant therapy to improve patient outcome. Such biomarkers may be discovered by comparing “global” molecular data for poor and good outcome EEC patients, but unfortunately few public datasets have been annotated for this phenotype. We hypothesized that a comparison of all EEC patients with poor-outcome NEEC patients might provide an alternative, better powered, strategy to identify biomarkers of EEC patients with poor outcome.
Global gene expression analysis is recognized as an effective strategy for determining profiles that could be used to classify cancer tissues into clinically meaningful subgroups. For example, the classification of breast cancers into luminal A, luminal B, normal, HER2 and basal-like subtypes, and the discovery of two distinct types of B-cell lymphoma (germinal center B-cell like lymphoma and activated B-cell like lymphoma) resulted from gene expression microarray studies6,7. It is recognized that results reported from individual microarray studies often display variability8. Indeed, variability can be observed for results from endometrial cancer microarray studies. For example, in total ~1,300 genes have been reported as differentially expressed across microarray studies assessing gene expression profiles between EEC and NEEC tumors9,10,11,12,13,14,15,16, however only 160 genes were reported in more than one study and no gene was reported by more than four studies.
To overcome the discrepancy and low reproducibility of individual microarray studies of endometrial cancer, we have performed a meta-analysis of 12 microarray gene expression studies to assess genes differentially expressed between NEEC and EEC cancers, as a means to identify genes that are important for development of aggressive endometrial cancer subtypes. The differential expression of these genes was validated using an independent endometrial cancer set with RNA-Seq data from The Cancer Genome Atlas (TCGA). We then explored the hypothesis that the aggressive gene signature identified by expression profiles associated with NEEC tumors can be used to predict EEC patients with poor prognosis and used validated aggressive signature genes to construct survival prediction models for EEC patients in the TCGA cohort. Our study demonstrates the value of employing meta-analysis for gene expression microarray data, and has highlighted genes and molecular pathways of importance for endometrial cancer prognosis and therapy.
Results
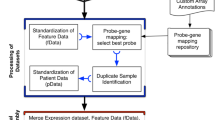
An overview of the study design can be found in Fig. 1.

Study Overview.
EEC - endometrioid endometrial cancer; NEEC - non-endometrioid endometrial cancer; TCGA - The Cancer Genome Atlas; EC-specific survival - endometrial cancer-specific survival.
Microarray Studies and Meta-Analysis
Following a literature review and repository search, twelve endometrial cancer microarray studies (Table 1) were merged and probes for 3,176 genes were extracted as being common across at least 10 studies. Principal components analysis using co-expression profiling and reproducibility estimates identified three studies as outliers (Supplementary Figure 1 and Supplementary Table 1). After considering sample size and number of probes assessed by each platform, an additional study (study 10) was removed from further analysis. The remaining eight studies were remerged, increasing the number of probes to 14,673 genes common across all studies. Genes displaying differential expression between NEEC and EEC tissue were identified for each study. Meta-analysis of individual study results found 2,053 genes (1126 upregulated, 927 downregulated) to be significantly differentially expressed between EEC and NEEC (Adjusted P-value < 0.05; Supplementary Table 2), and a consistent direction of effect observed across all eight studies.
TCGA RNA-Seq Validation
Analysis of differential expression between NEEC and EEC tissue in 317 independent samples from the TCGA Illumina GA RNA-Seq dataset validated the result for 1,581 genes from the 2,053 genes (77%) identified by microarray meta-analysis (Adjusted P-value < 0.05 and same direction of effect; Supplementary Table 2). Class prediction analysis predicted 1,253 from the 1,581 genes would be able to distinguish the subtype (EEC or NEEC) of new tumors tested using compound covariate predictor and leave-one-out cross-validation. Pathway analysis found these 1,253 genes to be enriched in pathways for cell cycle (Adjusted P-value = 5.4 × 10−7), mitotic cell cycle (Adjusted P-value = 9.34 × 10−7), progesterone-mediated oocyte maturation (Adjusted P-value = 7.9 × 10−5) and oocyte meiosis (Adjusted P-value = 2.3 × 10−4). Restricting to the 145 most significantly differentially expressed genes identified by meta-analysis (P-value < 10−19 and standardized fold change >2) was able to cluster NEEC and EEC samples in k-means cluster analysis (83.2% accuracy; Fisher’s Exact P-value < 2.2 × 10−16; Fig. 2). Similar clustering was observed in analysis of TCGA RNA-Seq data from 92 EEC and 57 NEEC tumor samples generated by HiSeq (82.6% accuracy; Fisher’s Exact P-value < 2.2 × 10−16; Supplementary Figure 2).

Gene expression patterns in endometrial cancer patients using RNA-Seq data from The Cancer Genome Atlas.
Unsupervised hierarchical clustering and heatmap showing individual expression pattern in 145 most significantly differentially expressed genes identified by microarray meta-analysis. Patient subgroup (NEEC - blue, EEC - red) is depicted by the bar across the top of the heat map. Normalized expression value is displayed by the heatmap, where blue represents upregulated genes and red represents downregulated genes. EEC - endometrioid endometrial cancer; NEEC - non-endometrioid endometrial cancer.
Functional enrichment and network analyses of the 145 most significantly differentially expressed genes
Because of computational limitations, functional enrichment analyses were restricted to the 145 most significantly differentially expressed genes identified by meta-analysis (P-value < 10−19 and standardized fold change >2). Since we have a total of 14,673 genes shared across all eight studies for differential expression analysis, we used these 14,673 genes as background for the calculation of significant P-values. In total, we found three significant functional terms: N4-(beta-N-acetylglucosaminyl)-L-asparaginase activity, Mucin type O-Glycan biosynthesis, and Walt’s disease. In our background gene list, there are only two genes (AGA and ASRGL1) related to N4-(beta-N-acetylglucosaminyl)-L-asparaginase activity. Both of these were detected in the 145 genes (GO:0003948, corrected P-value = 0.0192). For the KEGG pathway Mucin type O-Glycan biosynthesis, three genes (GALNT4, GCNT3, and ST6GALNAC1) were detected in our 145 genes (corrected P-value = 0.0456). The change of structure of mucin-type O-glycans can alter the adhesive properties of cells as well as cells’ potential to invade and metastasize in colon and breast cancers17. More interesting, we found five genes (CDKN2A, COL8A2, RASSF6, TMC4, and TMC5) from our 145 genes are associated with Walt’s disease (corrected P-value = 0.0040), which are infections in the skin caused by the human papillomavirus (HPV). In fact, the infection of HPV could precede the endometrial cancer progression18.
To explore the global interaction features of the 145 most significantly differentially expressed genes, we further mapped this gene list to the human pathway-based interactome. As shown in Fig. 3, we were able to reconstruct a network of 168 genes, of which 106 (63%) were from the 145 gene list, and 570 gene-gene interactions. The majority of genes in the reconstructed map are linked to each other and the vast majority of genes (~90%) in the network are connected by less than five steps. Twenty hub genes (defined as nodes with 20 or more connections) were identified in our network, of which 13 (65%) were from the 145 gene list: AGA [33], DNM1 [32], EPHB2 [30], ATP2C2 [29], ENTPD3 [28], NUDT11 [28], ASRGL1 [27], ACSL5 [27], LOXL3 [27], EPHB1 [26], SAT1 [26], GCNT3 [23], MGST2 [22]. Interestingly, both AGA and ASRGL1, related to N4-(beta-N-acetylglucosaminyl)-L-asparaginase activity, are highly connected in the reconstructed network, which may provide clues as to how these two genes interact with other cancer genes.

Network reconstruction and mutation analysis of the 145 most significantly differentially expressed genes between EEC and NEEC.
(A) Reconstructed network using protein-protein interaction data. Genes shown in orange (n = 106) are from the 145-gene list. The remaining genes in blue (n = 62) are linker genes that bridge the 106 genes into the network. Hub genes have been denoted with red text and boxes. EEC - endometrioid endometrial cancer; NEEC - non-endometrioid endometrial cancer.
EEC-only Survival Analysis
Expression from 121 of the 1,581 genes validated as differentially expressed between NEEC and EEC associated with EC-specific survival in EEC patients at a P-value < 0.005 (Table 2). Twenty-three of these 121 genes were among the 145 most significantly differentially expressed genes (Supplementary Table 2). Using these 121 genes as input, forward selection, likelihood-based modelling selected a 9-gene signature as being associated with EC-specific survival among EEC patients. The expression of the 9-gene signature were used to construct a prognostic index for each patient (see methods), which associated with poorer survival (log-rank P-value = 2.6 × 10−4, Fig. 4A). This association remained significant after multivariate analysis, adjusting for clinical covariates, stage and grade (HR 8.2; 95% CI 1.7–40.7; P-value = 0.01). Prognostic indexes were calculated for 92 non-overlapping TCGA patients with RNA-Seq data generated using the Illumina HiSeq platform, however the difference in EC-specific survival between the high and low risk groups was non-significant in this smaller dataset (log-rank P-value = 0.16; Fig. 4B).

Kaplan-Meier plots for EC-specific survival for high- and low-risk EEC patient groups identified using 31-gene prognostic signature.
(A) 241 samples with RNA-Seq data generated by the Illumina GA platform from the TCGA. (B) 92 samples with RNA-Seq data generated by the Illumina HiSeq platform from the TCGA. EEC - endometrioid endometrial cancer; TCGA - The Cancer Genome Atlas; EC-specific survival - endometrial cancer-specific survival.
Discussion
In this study we have investigated differential gene expression between NEEC and EEC to identify 1,253 genes that are involved in aggressive disease, thus providing insight into the biological underpinnings of these two groups of endometrial cancer. By taking a meta-analysis approach, using stringent selection criteria and performing validation in a large, independent RNA-Seq data, we have minimized false positive associations and produced genes robustly associated with NEEC. The reliability of this analysis was indicated by the validation of 77% of the identified genes in RNA-Seq data from an independent set of TCGA samples. We then further explored whether genes associated with aggressive disease were associated with poor prognosis among women with EEC, identifying a 9-gene signature which was able to group EEC patients as high- or low-risk, which remained significant after adjustment for clinical features, stage and grade.
We identified 601 genes to be upregulated in EEC compared to NEEC. Unsurprisingly, given the accepted relationship of EEC with unopposed estrogen exposure, the most significantly upregulated genes included estrogen responsive genes (KIAA1324, TFF3, MLPH) and genes involved in estrogen-related processes (FOXA2, ESR1, PGR). Expression of genes involved in epithelial (Ca2+) signaling (ATP2C2, TRPM4) were also found to be highly associated with EEC, a pathway thought to be important for epithelial cancer cells19. Two other genes identified as upregulated in ECC have previously been reported to be overexpressed in EEC by numerous studies; TFF310,13,14,15 and CEACAM110,14. Both are involved in extra-cellular matrix processes and cell-adhesion pathways and have been implicated in other cancer types reviewed in refs 20 and 21.
There were 652 genes found to be upregulated in NEEC tissue compared with EEC. A number of genes are involved in cell-cycle processes, such as GPR19, CDKN2A, USP11 and MX2. GPR19 encodes for a G protein-coupled receptor and is reported to be associated with lung cancer and melanoma22. It is suggested that G protein-coupled receptors are the most “druggable” family of proteins23. The significant association of GPR19 expression in NEEC observed warrants further investigation into the utility of drugs targeting GPR19 in treatment of this disease. Defects in the mitotic spindle checkpoint genes have been implicated in aneuploidy, a well-recognized feature of NEECs, and a previous gene expression study12 found that genes involved in the regulation of the mitotic spindle checkpoint were overexpressed in NEEC. Our results are consistent with this previous study, with mitotic cell cycle pathway genes found to be enriched in pathway analyses of differentially expressed genes.
Network reconstruction identified two N4-(beta-N-acetylglucosaminyl)-L-asparaginase activity genes (AGA and ASRGL1) as hub genes. This is the first observation of the significant differential expression of these two N4-(beta-N-acetylglucosaminyl)-L-asparaginase genes, across multiple endometrial cancer expression datasets. The additional high connections in the constructed network also implicate these two genes as potentially promising biomarkers for NEEC.
The most significantly upregulated gene in NEEC was L1CAM (L1 cell adhesion molecule), a member of the immunoglobulin super family, which is involved in embryonic brain development24. L1CAM is thought to be implicated in epithelial-to-mesenchymal transition, a critical event in tumor progression25 and its expression has been reported be associated with many cancers including breast, gastric and colorectal cancers reviewed by ref. 26. Expression of L1CAM has been reported to be associated with aggressive subtypes of endometrial cancer, including NEECs27. Furthermore, L1CAM has been reported to have utility as a predictor of clinical outcome in endometrial cancer27,28. Consistent with these publications, L1CAM was found to be significantly associated with EC-specific survival among EEC patients (P-value = 8.7 × 10−4).
The 9-gene EC-specific survival signature included genes previously implicated in other cancers, particularly colorectal cancer. Reduced expression of EPHB229 and PDLIM130 are reported to be indicators of poor prognosis of colorectal cancer. Both genes appear to exhibit tissue-specific effects, with upregulation of EPHB2 reported to be associated with poor breast cancer survival31 and elevated expression of PDLIM1 reported to promote metastatic processes in breast and glioma32,33. C4BPA and NLRC3, genes involved in immune processes, are reported to be dysregulated in pancreatic34 and colorectal cancer, respectively35. FBP1 plays a role in glucose metabolism and aerobic glycolysis, and has been reported to be downregulated in hepatocellular carcinoma, colorectal, breast, gastric, and renal cancer, reviewed in ref. 36. Downregulation of FBP1 is reported to contribute to tumor progression and poor survival of hepatocellular carcinoma36 and renal cell carcinoma patients37 and has been touted as a target for therapeutic interventions for these diseases. Given the results for FBP1 expression in our study, it is conceivable that therapeutics developed targeting FBP1 may also be beneficial in the treatment of EEC.
In conclusion, we have used a stringent meta-analysis and validation approach to identify distinct gene expression profiles in EEC and NEEC tumors. Importantly, a 9-gene signature was associated with poorer EC-specific survival in EEC patients, indicating its utility to predict prognosis. These genes may also provide new targets for therapy or the opportunity for the repositioning of currently available drugs. Results from this study contribute to the understanding of the molecular mechanisms of endometrial cancer subtypes, and have identified avenues to develop improved methods for identifying and treating poor prognosis patients with this disease.
Materials and Methods
Acquisition of Microarray Expression Datasets
A literature review and repository search was conducted up to September 2015 to identify endometrial cancer microarray expression studies. Twenty-one endometrial cancer microarray studies were accessed from publication supplementary data, the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/), ArrayExpress (https://www.ebi.ac.uk/arrayexpress/), or by contacting the publication authors (Supplementary Table 3). Microarray data generated by TCGA were downloaded from TCGA data portal (https://tcga-data.nci.nih.gov/tcga/).
Of these 21 studies, eight were excluded as follows: four studies lacked EEC and NEEC subtype information (E-GEOD-36389, E-GEOD-21882, E-GEOD-63678 & refs 38 and 39); one study using a custom platform of which the probe annotations could not be updated40; four studies performed by the same research lab (ArrayExpress accession no: E-GEOD-14860, E-MTAB1358, E-MTAB-1007 and E-MTAB-2532) included overlapping sample sets, and, thus, only the largest study (E-MTAB-2532), was selected for inclusion in our analysis.
Expression Microarray Analysis
Analysis of microarray expression data was performed used the MetaOmics suite of packages in R41. Gene probe annotations were updated for each dataset using SOURCE (http://source-search.princeton.edu/) and expression data log transformed (by taking the logarithmic values of the signals to the base of two). Multiple probes mapping to the same gene were summarized using the inter-quartile range method, since this method is considered to be more biologically relevant than averaging probes values42. Expression data were filtered to remove the bottom 20% of unexpressed and uninformative genes (i.e. genes with low mean expression intensity values and low variation in expression intensity values) as advised by the authors of the MetaOmics packages.
Quality control measures were generated using the MetaQC package41, to identify studies which should be excluded from the meta-analysis, such as outlier studies with gene co-expression profile considered inconsistent using both unsupervised pair-wise comparisons between studies and pathway knowledge provided by curated gene sets from MSigDB (http://software.broadinstitute.org/gsea/msigdb). Other measures generated included those aimed at quantifying the reproducibility of differentially expressed genes.
Genes common across all studies were extracted and datasets merged. Differentially expressed genes were identified for each study using moderated t-tests and p-values combined using Fisher’s combined probability test. Gene expression level differences between EEC and NEEC tissue for each study were expressed as an effect size, a unit-free standardized mean difference, and combined using a random effects model. Adjustment for multiple comparisons on the combined p-values was performed using the false discovery rate procedure of Benjamini and Hochberg. All meta-analyses were performed using the MetaDE package41.
TCGA RNA-Seq data validation
RNA-Seq RSEM gene expression data (level 3 generated for 317 TCGA EEC and NEEC tissues by the Illumina GA platform and 162 TCGA EEC and NEEC by the Illumina HiSeq platform were downloaded from the cancer browser (https://genome-cancer.ucsc.edu/proj/site/hgHeatmap/). RNA-Seq data generated by the two sequencing platforms (GA and HiSeq) were treated as two separate datasets to avoid bias from batch effects. Samples that overlapped with the TCGA microarray dataset were excluded from RNA-Seq analysis. RNA-Seq data was normalized using the voom function from the package limma in R. Unsupervised hierarchical clustering was performed using the ggplot package in R. Class comparison, class prediction and KEGG pathway enrichment were performed using BRB-ArrayTools software (http://brb.nci.nih.gov/BRB-ArrayTools/index.html).
Function enrichment analysis
Functional enrichment analysis using WebGestalt (https://www.webgestalt.org/) was performed to identify potentially important gene pathways from KEGG and and gene ontology (GO). All 14,673 genes shared across all eight studies for differential expression analysis were used as background in these analyses. P-values were corrected for multiple testing by Benjamini-Hochberg adjustment and only pathways with a corrected P-value < 0.01 for any gene set were considered significant.
Network Analysis
Recent advances in high-throughput technologies have generated data for protein-protein interaction (PPI). This huge data have stimulated pathway reconstruction for improving the systems-level understanding of specific cellular events. However, most of PPI data derived from mass-spectrum and yeast-two-hybrid technologies are only physical interaction, which may not really exist in vivo. Additionally, the physical interaction-based PPI network tends to a highly skewed degree distribution, which may not represent the global interactome involving basic cellular processes. To avoid the inaccuracy, a non-redundant pathway-based human interactome was built based on the PPIs in PathCommons43. These PPIs are derived from human-curated pathway databases, including HumanCyc, the NCI signaling pathway database, Reactome, and KEGG pathway. The final human pathway-based interactome contains 3629 genes and 36034 interacting edges. Using a module searching method as previously described44, we extracted a subnetwork from all human pathway-based interactomes. This algorithm mapped all interesting input genes to the human interactome, and then it generated a sub-network with the shortest paths between input genes and other genes. Network visualization was performed using Cytoscape 2.845.
Survival analysis
Validated genes were used in survival prediction analyses of 241 EEC patients from TCGA with Illumina GA RNA-Seq and outcome data available, using the survival package in R. Gene expression was grouped using the auto-cutoff method as described in ref. 46. Briefly, each percentile of expression between the first and third quartiles was computed and best performing threshold was used as the cut-off in the Cox proportional hazards model. Forward selection, likelihood-based modelling to identify the 9-gene prognostic signature from all genes associated with EC outcome was performed using the rbserv package in R. Prognostic indexes using the 9-gene signature were calculated for each patient by subtracting the sum of the normalised expression values of genes with lower expression in EEC compared to NEEC (PDLIM1, FBP1, NLRC3, ST6GALNAC1, C4BPA) from the sum of expression values of genes with higher expression (PPP2R3A, TRIM46, EPH2, PRRG1). Indexes were grouped into low- and high-risk group using the auto-cutoff method as described above. Kaplan-Meier survival curves were and differences between groups assessed using log-rank test. Multivariate analyses of other clinical features were performed using Cox proportional hazard models. Endpoint for endometrial cancer specific survival (EC-specific survival) was defined as time from diagnosis until death with endometrial tumor present. Results were then tested in 92 EEC patients from TCGA with Illumina HiSeq RNA-Seq and outcome data available.
Additional Information
How to cite this article: O’Mara, T. A. et al. Meta-analysis of gene expression studies in endometrial cancer identifies gene expression profiles associated with aggressive disease and patient outcome. Sci. Rep. 6, 36677; doi: 10.1038/srep36677 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Ferlay, J. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 136, E359–386 (2015).
AIHW. Cancer incidence projections: Australia, 2011 to 2020. (ed. AIHW) (Aust Gov, Canberra, 2012).
Matias-Guiu, X. et al. Molecular pathology of endometrial hyperplasia and carcinoma. Hum Pathol 32, 569–577 (2001).
Gottwald, L. et al. Long-term survival of endometrioid endometrial cancer patients. Arch Med Sci 6, 937–944 (2010).
McConechy, M. K. et al. Endometrial Carcinomas with POLE Exonuclease Domain Mutations Have a Favorable Prognosis. Clin Cancer Res 22, 2865–2873 (2016).
Perou, C. M. et al. Distinctive gene expression patterns in human mammary epithelial cells and breast cancers. Proc Natl Acad Sci USA 96, 9212–9217 (1999).
Alizadeh, A. A. et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 403, 503–511 (2000).
Draghici, S., Khatri, P., Eklund, A. C. & Szallasi, Z. Reliability and reproducibility issues in DNA microarray measurements. Trends Genet 22, 101–109 (2006).
Kharma, B. et al. STAT1 drives tumor progression in serous papillary endometrial cancer. Cancer Res 74, 6519–6530 (2014).
Maxwell, G. L. et al. Microarray analysis of endometrial carcinomas and mixed mullerian tumors reveals distinct gene expression profiles associated with different histologic types of uterine cancer. Clin Cancer Res 11, 4056–4066 (2005).
Mhawech-Fauceglia, P. et al. Microarray analysis reveals distinct gene expression profiles among different tumor histology, stage and disease outcomes in endometrial adenocarcinoma. PLoS One 5, e15415 (2010).
Moreno-Bueno, G. et al. Differential gene expression profile in endometrioid and nonendometrioid endometrial carcinoma: STK15 is frequently overexpressed and amplified in nonendometrioid carcinomas. Cancer Res 63, 5697–5702 (2003).
Risinger, J. I. et al. Microarray analysis reveals distinct gene expression profiles among different histologic types of endometrial cancer. Cancer Res 63, 6–11 (2003).
Shedden, K. A. et al. Histologic type, organ of origin, and Wnt pathway status: effect on gene expression in ovarian and uterine carcinomas. Clin Cancer Res 11, 2123–2131 (2005).
Sung, C. O. & Sohn, I. The expression pattern of 19 genes predicts the histology of endometrial carcinoma. Sci Rep 4, 5174 (2014).
Zorn, K. K. et al. Gene expression profiles of serous, endometrioid, and clear cell subtypes of ovarian and endometrial cancer. Clin Cancer Res 11, 6422–6430 (2005).
Brockhausen, I. Mucin-type O-glycans in human colon and breast cancer: glycodynamics and functions. EMBO Rep 7, 599–604 (2006).
Giatromanolaki, A., Sivridis, E., Papazoglou, D., Koukourakis, M. I. & Maltezos, E. Human papillomavirus in endometrial adenocarcinomas: infectious agent or a mere “passenger”? Infect Dis Obstet Gynecol 2007, 60549 (2007).
Kohn, K. W., Zeeberg, B. M., Reinhold, W. C. & Pommier, Y. Gene expression correlations in human cancer cell lines define molecular interaction networks for epithelial phenotype. PLoS One 9, e99269 (2014).
Beauchemin, N. & Arabzadeh, A. Carcinoembryonic antigen-related cell adhesion molecules (CEACAMs) in cancer progression and metastasis. Cancer Metastasis Rev 32, 643–671 (2013).
May, F. E. The potential of trefoil proteins as biomarkers in human cancer. Biomark Med 6, 301–304 (2012).
Kastner, S. et al. Expression of G protein-coupled receptor 19 in human lung cancer cells is triggered by entry into S-phase and supports G(2)-M cell-cycle progression. Mol Cancer Res 10, 1343–1358 (2012).
Dorsam, R. T. & Gutkind, J. S. G-protein-coupled receptors and cancer. Nat Rev Cancer 7, 79–94 (2007).
Brummendorf, T., Kenwrick, S. & Rathjen, F. G. Neural cell recognition molecule L1: from cell biology to human hereditary brain malformations. Curr Opin Neurobiol 8, 87–97 (1998).
Colas, E. et al. The EMT signaling pathways in endometrial carcinoma. Clin Transl Oncol 14, 715–720 (2012).
Altevogt, P., Doberstein, K. & Fogel, M. L1CAM in human cancer. Int J Cancer 138, 1565–1576 (2016).
Dellinger, T. H. et al. L1CAM is an independent predictor of poor survival in endometrial cancer - An analysis of The Cancer Genome Atlas (TCGA). Gynecol Oncol (2016).
Geels, Y. P. et al. L1CAM Expression is Related to Non-Endometrioid Histology, and Prognostic for Poor Outcome in Endometrioid Endometrial Carcinoma. Pathol Oncol Res (2016).
Zhang, X. EphB2: a signature of colorectal cancer stem cells to predict relapse. Protein Cell 2, 347–348 (2011).
Chen, H. N. et al. PDLIM1 Stabilizes the E-Cadherin/beta-Catenin Complex to Prevent Epithelial-Mesenchymal Transition and Metastatic Potential of Colorectal Cancer Cells. Cancer Res 76, 1122–1134 (2016).
Husa, A. M., Magic, Z., Larsson, M., Fornander, T. & Perez-Tenorio, G. EPH/ephrin profile and EPHB2 expression predicts patient survival in breast cancer. Oncotarget 7, 21362–21380 (2016).
Ahn, B. Y. et al. Glioma invasion mediated by the p75 neurotrophin receptor (p75(NTR)/CD271) requires regulated interaction with PDLIM1. Oncogene 35, 1411–1422 (2016).
Liu, Z. et al. PDZ and LIM domain protein 1(PDLIM1)/CLP36 promotes breast cancer cell migration, invasion and metastasis through interaction with alpha-actinin. Oncogene 34, 1300–1311 (2015).
Sogawa, K. et al. Identification of a novel serum biomarker for pancreatic cancer, C4b-binding protein alpha-chain (C4BPA) by quantitative proteomic analysis using tandem mass tags. Br J Cancer (2016).
Liu, R. et al. Expression profile of innate immune receptors, NLRs and AIM2, in human colorectal cancer: correlation with cancer stages and inflammasome components. Oncotarget 6, 33456–33469 (2015).
Hirata, H. et al. Decreased Expression of Fructose-1,6-bisphosphatase Associates with Glucose Metabolism and Tumor Progression in Hepatocellular Carcinoma. Cancer Res 76, 3265–3276 (2016).
Li, B. et al. Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature 513, 251–255 (2014).
Mutter, G. L. et al. Global expression changes of constitutive and hormonally regulated genes during endometrial neoplastic transformation. Gynecol Oncol 83, 177–185 (2001).
Huvila, J. et al. Gene expression profiling of endometrial adenocarcinomas reveals increased apolipoprotein E expression in poorly differentiated tumors. Int J Gynecol Cancer 19, 1226–1231 (2009).
Saidi, S. A. et al. Independent component analysis of microarray data in the study of endometrial cancer. Oncogene 23, 6677–6683 (2004).
Wang, X. et al. An R package suite for microarray meta-analysis in quality control, differentially expressed gene analysis and pathway enrichment detection. Bioinformatics 28, 2534–2536 (2012).
Hahne, F., Huber, W., Gentleman, R. & Falcon, S. Bioconductor Case Studies (Use R!) (Springer, 2008).
Cerami, E. G. et al. Pathway Commons, a web resource for biological pathway data. Nucleic Acids Res 39, D685–690 (2011).
Zhao, M., Liu, Y. & O’Mara, T. A. ECGene: A Literature-Based Knowledgebase of Endometrial Cancer Genes. Hum Mutat 37, 337–343 (2016).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504 (2003).
Mihaly, Z. et al. A meta-analysis of gene expression-based biomarkers predicting outcome after tamoxifen treatment in breast cancer. Breast Cancer Res Treat 140, 219–232 (2013).
Acknowledgements
We gratefully acknowledge the TCGA endometrial cancer consortium for providing samples, tissues, data processing, and making data and results available. We thank Dr Dylan Glubb for critical review of the manuscript. TOM is supported by a CJ Martin Early Career Fellowship from the National Health and Medical Research Council (NHMRC), Australia; MZ is supported by a University of the Sunshine Coast research start-up fellowship; ABS is supported by an NHMRC Senior Research Fellowship.
Author information
Authors and Affiliations
Contributions
T.O’M. and M.Z. performed the analyses; T.O’M. designed the study and prepared the manuscript with support from A.B.S. All authors reviewed and approved the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
O’Mara, T., Zhao, M. & Spurdle, A. Meta-analysis of gene expression studies in endometrial cancer identifies gene expression profiles associated with aggressive disease and patient outcome. Sci Rep 6, 36677 (2016). https://doi.org/10.1038/srep36677
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep36677
This article is cited by
-
BUB1, BUB1B, CCNA2, and CDCA8, along with miR-524-5p, as clinically relevant biomarkers for the diagnosis and treatment of endometrial carcinoma
BMC Cancer (2023)
-
Identification of a prognostic gene signature of colon cancer using integrated bioinformatics analysis
World Journal of Surgical Oncology (2021)
-
Transgenic mice overexpressing the LH receptor in the female reproductive system spontaneously develop endometrial tumour masses
Scientific Reports (2021)
-
Integrated analysis of immune-related genes in endometrial carcinoma
Cancer Cell International (2020)
-
Whole transcriptome signature for prognostic prediction (WTSPP): application of whole transcriptome signature for prognostic prediction in cancer
Laboratory Investigation (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
