
Abstract
Respiratory syncytial virus (RSV) causes infections that range from common cold to severe lower respiratory tract infection requiring high-level medical care. Prediction of the course of disease in individual patients remains challenging at the first visit to the pediatric wards and RSV infections may rapidly progress to severe disease. In this study we investigate whether there exists a genomic signature that can accurately predict the course of RSV. We used early blood microarray transcriptome profiles from 39 hospitalized infants that were followed until recovery and of which the level of disease severity was determined retrospectively. Applying support vector machine learning on age by sex standardized transcriptomic data, an 84 gene signature was identified that discriminated hospitalized infants with eventually less severe RSV infection from infants that suffered from most severe RSV disease. This signature yielded an area under the receiver operating characteristic curve (AUC) of 0.966 using leave-one-out cross-validation on the experimental data and an AUC of 0.858 on an independent validation cohort consisting of 53 infants. A combination of the gene signature with age and sex yielded an AUC of 0.971. Thus, the presented signature may serve as the basis to develop a prognostic test to support clinical management of RSV patients.
Similar content being viewed by others

Introduction
Respiratory syncytial virus (RSV) causes infections that range from common cold to severe lower respiratory tract infection that in some instances may have a fatal outcome. Especially infants, elderly and patients with underlying chronic disorders suffer from severe RSV infections1,2. In infants, RSV is the leading cause of lower respiratory tract infections (LRTI) and is responsible for 80% of acute bronchiolitis cases3. RSV infections pose a huge burden on society in terms of disease, logistics and socio-economic sequelae. There is an unmet need for an RSV vaccine, despite considerable research efforts no licensed vaccine has been developed.
In industrialized countries, 1–5% of infants with RSV infection are hospitalized4,5,6,7. Some of these infants yet suffer from severe disease upon admittance, while others are admitted without severe symptoms since the course of bronchiolitis is highly variable and the need for supportive care cannot be predicted8,9. Several risk factors for developing severe RSV disease in infants have been identified, including preterm birth, young age, sex and environmental factors like in-house smoking10. Notwithstanding these known risk factors, current medical practice does not allow accurate prediction of whether an infant will further progress to severe RSV disease or not and could even be sent home safely. Genomic technologies have contributed to study the virus-host interaction, including virus discovery, pathogenesis studies, the design of antiviral strategies and identification of biomarkers to support clinical management of infectious diseases11,12,13,14. For RSV infections, this has supported the characterization of vaccine-induced skewed host responses upon infection15,16. Meijas et al.17 recently used blood transcriptome profiles obtained within 3 days of hospitalization to characterize the host response to RSV infection in infants compared with rhinovirus or influenza infections and identified transcriptional profiles that associate with RSV disease severity. However, a prognostic model for RSV severity based on gene expression profiles collected at admittance to the hospital has not been developed.
In this study we aim to identify and validate a gene signature that discriminates severe from less severe RSV LRTI that do not require advanced support. Such a signature together with other clinical parameters may improve the prognosis of less severe patients that could be safely sent home.
Material and Methods
Study design
Study subjects were recruited at Canisius Wilhelmina Hospital, Radboud University Medical Center, Nijmegen, and Erasmus Medical Center, Rotterdam, The Netherlands. Nasopharyngeal wash and blood samples were prospectively obtained from patients less than 2 years of age with a viral bronchiolitis. Patient enrolment occurred 7 days a week and samples were taken within 24 hours after first contact with the hospital. Seventy-three percent of all eligible bronchiolitis patients agreed to participate in the study. The major reasons for non-inclusion were parental availability to sign consent and the hesitancy for the venipuncture. Only patients with an RSV infection, as retrospectively determined by PCR were included in the study. Exclusion criteria were: immunodeficiency, systemic steroid treatment in the previous 2 weeks, blood transfusion, congenital heart and chronic lung disease. A Tempus tube (TempusTM, Applied Biosystems, Austria) and sodium heparin tube were filled with 3 ml of blood. Medical history, demographic and clinical data were collected from medical records and questionnaires. The (hospitalized) patients were followed until recovery and were retrospectively classified as: mild for children without hypoxia, moderate for patients requiring supplemental oxygen (oxygen saturations <90%, ≥10 minutes) and severe for children requiring mechanical ventilation due to apnea, exhaustion and/or respiratory failure. Recovery samples were obtained after 4–6 weeks, during home visits. Blood samples were obtained from healthy controls (HC) without underlying diseases or medication subjected to elective surgery.
Study approval
The study protocol was approved by the Regional Committees on Research involving Human Subjects of Arnhem-Nijmegen and Rotterdam and were conducted in accordance with the principles of the Declaration of Helsinki. Written informed consent was obtained from the parents of all children prior to inclusion in the study.
Sample processing and blood transcriptome profiling
Inclusion of patients and sample collection was performed by a single MD at the hospitals. Multiplex RT-PCR was performed to test the nasopharyngeal washes on 15 different viral pathogens, as previously described18. Blood was collected in Tempus tubes for immediate stabilization of RNA and subsequently stored at −80 °C. Total RNA was isolated from each blood sample, processed, assessed, labelled and hybridized to a single Affymetrix Human Genome U133 plus 2 gene chips; and image analysis was performed in the same lab and by one technician as described in supplementary material. The raw data has been deposited in the ArrayExpress database under access number E-MTAB-5195.
Data preprocessing
Microarray data was preprocessed using R 3.1.2 19 and Bioconductor20. Upon initial quality control and VSN normalization (to render samples comparable), probeset (a combination of multiple probes) summarization was performed by median polish21,22. Unless otherwise stated, all probesets/genes present on the Affymetrix GeneChip were used for data analysis. Samples were labelled and hybridized in two batches which did not correspond to any biological variable as samples were randomly assigned to the batches. The normalized expression values were adjusted for a batch effect (see supplementary Fig. S1) using ComBat23. Additionally, we assessed confounding effects of clinical parameters age and sex on gene expression–severity relationship using “biasograms”24.
Differential expression analysis
To obtain a global view of the blood transcriptome changes in response to RSV infection (i.e. to evaluate whether whole transcriptome changes associate with severity), a principal component analysis (PCA) as an exploratory analysis was performed on the age by sex standardized data. Next, a differential expression (DE) analysis was performed on the normalized-batch-adjusted data controlling for an age by sex effect using empirical Bayes linear models25 implemented in the R package limma26. Details of the models are found in supplementary material. We controlled for multiple testing via false discovery rate (FDR) using a Benjamini and Hochberg procedure27. Gene set enrichment analysis was performed using Ingenuity pathway analysis (IPA, www.qiagen.com/ingenuity).
Identification and evaluation of prognostic biomarkers
Since we are interested in identifying RSV-infected infants that will progress to severe stage upon presentation to the hospital, we grouped mild and moderate samples and aimed to separate these samples from infants that were presented with or progressed to severe disease after hospitalization. We chose to utilized probabilistic predictors (to predict the chance of an RSV-infected infant to be severe) because in clinical applications, probabilities are more informative than absolute yes or no predictions28. Several probabilistic predictors exist in the literature and their performance depends on the type of the data they are being applied on ref. 29. Using results of ref. 29,30 and observed correlations in the data, three probabilistic classification functions that could be optimal for this data were chosen as described in supplementary material. These functions were support vector machines (SVM)31, shrunken centroids discriminant analysis (SCDA)32 and random forest (RF)33.
For each classification function, the experimental data was split into a learning set and a test set using leave-one-out cross-validation (LOOCV). Cross-validation reduces optimistic bias by ensuring that our models are evaluated on an independent dataset that was not used to constructed these models. Most probabilistic classification functions require hyper-parameters to perform variable selection among the huge number of variables (probesets). Usually, the best values for these hyper-parameters are also determined by cross-validation. Thus, the parameter(s) of the function were optimized using an inner loop of five-fold cross-validation on the learning set. Next, a prognostic model was built with the optimal parameter(s) on the entire learning set and evaluated with the test set, as described in supplementary material. The following R packages; CMA34, e107135, pamr36 and randomForest37 were utilized for class prediction. The best calibrated and refined function amongst the three functions was selected and its performance evaluated using the area under the receiver operating characteristic (ROC) curve (AUC). Finally, the transcripts that maximized the binomial log-likelihood function, with the leave-one-out cross-validated data were retained as a gene signature from the selected function as described in supplementary material.
Comparison of biomarkers to clinical parameters
Age and sex are readily available clinical parameters that have been determined to be associated to RSV disease severity38. To assess the gain attained with a genomic model over a model with these clinical parameters, and the effect of standardization, the leave-one-out cross-validated predicted probabilities of progressing to severe for all samples were transformed to genomic scores (a genomic score is single measure of the genome of a sample as predicted by a model) for models with unstandardized and standardized data. Logistic regression models (see supplementary material) were then fitted with the genomic scores and/or clinical parameters and their AUCs compared.
Validation of biomarkers
For an independent validation, a subset of the Illumina RSV data of Meijas et al.17 was used. Since the experimental data and validation data were obtained using different platforms, we linked the data using gene symbols and applied cross-platform transformation (to render gene expression comparable across datasets) as described in supplementary material. The transformed data was supplied to our prognostic model for predictions of probabilities of severity. For a confirmatory analysis of how well our prognostic model performs, we built and evaluated a prediction model with the chosen function (same function used to build our prognostic model) on the entire Illumina data and compared our validation performance to the performance from this (unrestricted) data.
Results
Study subjects and sampling
Thirty-nine infants hospitalized with acute RSV bronchiolitis were included in the study. Nasopharyngeal wash and whole blood samples for mRNA profiling were collected within 24 hours upon hospital admittance. Table 1 presents the characteristics of the study subjects. As expected, patients with the most severe course of RSV bronchiolitis were significantly younger than those with a relative mild or moderate course of this disease. The variables related to disease severity; duration of oxygen, and length of stay in the hospital were highest in the severe group, with ventilation indicating the method by which oxygen was supplied. The proportion of co-infections was lower in severe patients as compared to the other severity categories. There were no differences in the occurrence of other known risk factors.
Age and sex as confounders of gene expression–severity relationship
Figure 1a,b respectively illustrate the confounding effects of sex and age on the gene expression-disease severity relationship. These figures show that whereas age is negatively correlated to severity, sex is uncorrelated to severity. Nevertheless, the high positive/negative correlations of a considerable number of transcripts to sex and age, as well as severity, indicate a confounding effect of these variables on the expression-severity relationship of these transcripts, thus warrant adjustment. Figure 1d,e illustrate the “biasograms” after an age by sex standardization. These figures show that standardization has no effect on severity correlated transcripts but as expected, transcripts that were originally correlated to age and sex become uncorrelated. A positive correlation of age to sex which signifies an age by sex interaction as a potential confounder on the gene expression-severity relationship was also observed (Fig. 1c) and eliminated after standardization (Fig. 1f).

Confounding effect of Sex, Age and Age by Sex on gene expression–severity relationship, before.
(a,b,c) and after: (d,e,f), an age by sex standardization. The blue and green lines represent the clinical variables, the cosine of the angle between the lines represents its correlation to the blue line (Sex is not correlated to Severity, Age is negatively correlated to Severity i.e. younger kids become severe and Age is positively correlated to sex i.e. females are older). The cloud of points represent the transcripts and their correlations to both variables with most transcripts uncorrelated to the variables (yellow cloud) while a considerable number (black cloud) are correlated to Severity, Sex, Age and Age *Sex. The associations between the transcripts and Sex, Age or Age *Sex are significantly eliminated after standardization while retaining that of Severity.
Global blood transcriptome profiles associate with RSV disease severity
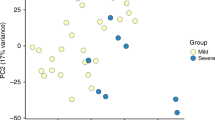
Figure 2 illustrate a PCA on the whole transcriptome and the first principal component accounts for 25% of the variance in the transcriptomes and associates with disease severity. Transcriptome profiles of HC and recovery samples group together on the first principal component and are located opposite to profiles of severe infants. The distinct groups do not form discrete clusters in the PCA but gradually shift from mild through moderate to severe, with considerable overlap. This shows that the blood mRNA profiles substantially capture the severity of lower respiratory tract RSV infection.

Global blood transcriptome profiling with principal component analysis: the first principal component (PC1) accounts for 25% of the variance in the dataset and associates with disease severity.
This can be observed as a shift from healthy controls and recovery cases (left) through mild and moderate to severe cases (right), with considerable overlap.
Number of differential gene expression relates to RSV disease severity
Table 2 presents results of differential gene expression analysis and reveals that the number of DE transcripts increases with disease severity. No DE transcript was identified between mild versus HC samples when applying a FDR of 5% and absolute fold change (FC) threshold of 2. However, 17 and 221 transcripts were DE between moderate and severe versus HC respectively. Interestingly, all transcripts that are DE in moderate class are also DE in severe class with larger FC. About 90% of these DE transcripts are up-regulated. Comparison of HC with recovery samples revealed a single down-regulated transcript while moderate versus mild yielded no DE transcript, severe versus mild or moderate yielded 178 and 49 DE transcripts respectively. Lastly, 95 transcripts were DE between severe versus combined mild/moderate samples.
RSV induced blood transcriptome profiles reveal an inflammatory response
Figure 3 shows that multiple relevant categories of molecular and cellular functions are significantly enriched when comparing severe to HC samples. With “Cell-to-Cell Signaling and Interaction” top category, gene sets related to activation of several types of immune cells including lymphocytes, granulocytes and specifically neutrophils are most significantly enriched. In addition, gene sets that are involved in migration and tissue infiltration of these same activated cell types are most significantly enriched within the category “Cellular Movement” that ranks third on this figure. Finally, several high ranking molecular and cellular function categories and their underlying gene sets indicate the immune cells involved are strongly proliferating. A list of genes involved in each of these pathways is presented in supplementary Table S1. Taken together, blood transcriptome changes in RSV disease reveal a typical inflammatory response to a viral infection.

Ingenuity pathway analysis (IPA) Molecular and Cellular functions gene set analysis for severe vs healthy control contrast.
Early blood transcriptome changes to predict a severe outcome of RSV infection
To construct a predictive model, we combined mild and moderate cases as a single group and three probabilistic classification functions were chosen based on supplementary Fig. S2 and results of Jong et al. and Kim and Simon29,30. Using these functions, classifiers were built and evaluated using LOOCV on the experimental data. SVM was chosen as the best calibrated and refined as shown on supplementary Fig. S3 and henceforth considered for all analyses. The LOOCV predicted probabilities from SVM were used to evaluate its performance and are plotted on Fig. 4a against the true RSV status as retrospectively determined. This figure shows that 5 samples out of 39 were misclassified at a 50% cutoff and when applying a proposed uncertainty band of 30–70% just one false negative is witnessed. Evaluation of the clinical characteristics of the single false negative patient as well as those patients with uncertain predictions (plotted within the proposed uncertainty band) did not reveal any recognizable pattern. The false negative patient had uniquely RSV and only a single patient plotted within the uncertainty band had RSV+ other virus(es). Figure 4b presents the corresponding ROC curve from the LOOCV predicted probabilities and AUC of 0.966 demonstrates the high discriminative power of our prognostic model.

Internal validation of gene signature.
(a) samples’ predicted probabilities of being severe. (b) shows the ROC curve and the AUC for predicted probabilities. The AUC value of approximately 1 indicates how accurate our signature performs on this internal validation set. (c) shows that a genomic model from the age by sex standardized data out performs that from the unstandardized data. In addition, there is a significant difference between a model with clinical parameters and that with a genomic score and just a slight improvement when both parameters are included in a model.
Genomic biomarkers outperform clinical parameters age and sex
Figure 4c presents the ROC curves from logistic models of clinical parameters and/or genomic scores. The genomic score model from the standardized data (AUC = 0.966) outperforms that from the unstandardized data (AUC = 0.915). In addition, there is a significant difference between a model with age and sex only (AUC = 0.833) and that with a standardized (AUC = 0.966) or unstandardized genomic score (AUC = 0.915). Whereas there is a slight improvement from clinical parameters and standardized genomic score model (AUC = 0.971), there is no improvement from the clinical parameters and unstandardized genomic score (AUC = 0.911), indicating that indeed standardization completely removed an age-by-sex effect on the gene expression data.
An 84 gene expression signature predicts absence of disease progression
To extract the prognostic signature, we selected top transcripts maximizing the binomial log-likelihood function using LOOCV predicted probabilities as illustrated on supplementary Fig. S4. This figure depicts a 1-SE maximum of 95 transcripts corresponding to 84 unique genes, which are displayed on Table 3. Of the 95 transcripts constituting the prognostic signature, 81 (85.26%) were found to be significantly DE between severe and non-severe patients (FDR cutoff of 5%, supplementary Table S2). The inclusion of non-DE transcripts in the classification model is expected, since not only DE genes are instrumental in class discrimination as illustrated by the two-dimensional scenario in supplementary Fig. S5.
Performance of the genomic signature retained on an independent dataset
For an independent validation, a subset of the Illumina RSV data of Meijas et al.17 was used. Since the experimental data and validation data were obtained using different platforms, we linked the data using gene symbols and applied cross-platform transformation (to render gene expression comparable across datasets) as shown on supplementary Fig. S6 and extensive described in the supplementary material. Figure 5a presents predicted probabilities of severe on the validation data using 75 of our 84 prognostic gene signature that were common in both experimental and validation datasets, while Fig. 5b presents the LOOCV predicted probabilities from SVM on the entire Illumina data. Both figures show that using the unrestricted data leads to more certain probabilities and slightly improves specificity compared to our signature. Nonetheless, Fig. 5c illustrates a large agreement between the predicted probabilities of the two models, while Fig. 5d clearly reveals that both models are alike as demonstrated by the AUCs of 0.858 and 0.856 for our signature and the unrestricted model respectively. To assess the concordance of the expression patterns of our signature on both datasets (Affymetrix and Illumina), we plotted the log2 fold changes of the common 75 genes as shown on Fig. 6. From this figure, one clearly sees that there is a huge concordance in the direction of expressions across datasets. Where there are slight differences, these differences are not significant as shown by a non-significant p-value in at least one of the datasets.

Predicted probabilities of being severe from the validation data against true RSV status using; our diagnostic signature (a) and LOOCV on unrestricted data (b). (c) illustrates the agreement of the predictions from both models, green regions are perfect agreement, blue are disagreements at a 50% cutoff and red are disagreements at a 30–70% uncertainty band. Finally, (d) presents the ROC curves and AUC from both models illustrating similar AUC values.

Log2 Fold change between Severe vs Non-Severe infants for 75 common genes in Affymetrix and Illumina datasets.
Red represent up-regulation while green represents a down-regulation and the significant FDR adjusted p-values are placed in the cells. As one can clearly see, there is a huge overlap in the direction expressions across datasets. Where there are slight differences, these differences are not significant as shown by a non-significant p-value in at least one of the datasets.
Discussion
RSV infection in infants may cause life-threatening disease. No vaccine is yet available and triage of patients is challenging since RSV infections may rapidly progress to severe disease. No reliable prognostic model to predict which RSV patient will not progress to severe disease and could be safely send home is available either. Thus, clinical care is symptom-based and a significant proportion of RSV infected infants is hospitalized for observation purposes. We have provided an 84 gene signature that discriminates hospitalized infants with less severe RSV infection from those infants with severe RSV disease. The identified signature yielded a LOOCV AUC of 0.966 on the experimental data and was independently validated with an AUC of 0.858 and might serve as a basis to develop a prognostic test for clinical management of RSV disease.
In line with epidemiological observations38 and observations of Mejias et al.17, we showed the confounding effects of age and sex on gene expression-severity relationship for RSV disease. Studies in any RSV patient cohort with a naturally occurring “skewed” distribution of age and sex can be standardized for these parameters. By adjusting for an age-by-sex effect in our analyses, we obtained age-by-sex independent results which can be effectively applied to any patient(s). The high performance of our signature on the age and sex matched validation data signifies age-by-sex independence and robustness of this signature. Fewer co-infections were observed in severe patients (Table 1). A similar trend has been described previously39. In our cohort study we did not take into account co-infections since no consistent association between the occurrence or absence of co-infections with RSV disease severity have been reported39,40,41,42,43. Furthermore, we aimed at the identification of a gene signature in a natural “real-life” cohort of patients not stratified according to age or occurrence of co-infections.
We hypothesized that changes in blood cell type distribution and/or mRNA expression changes of the circulating cells collected from peripheral blood reflect local lung host response characteristics that associate with disease severity. PCA and DE analysis indeed revealed significant changes in the transcriptome profile of whole blood. Gene set analysis further shows that relevant processes are monitored including the activation, migration and tissue infiltration of lymphocytes, granulocytes and neutrophils. Individual DE genes in severe RSV disease revealed overexpression of the neutrophil associated genes MMP8 and MMP9, which have previously been related to severe RSV disease44. ARG1 and CHI3L1 that have been linked to alternatively activated macrophages in a mouse model for vaccine enhanced RSV disease16 were also found to be strongly up-regulated. This suggests that the collected blood transcriptome profiles indeed reflect local lung host response.
In our class prediction analysis, three functions were evaluated and the best was chosen. While it has been pointed by45,46,47,48 that selecting a minimal-error classifier leads to selection bias that should be corrected, the literature does not stipulate a selection bias when using calibration and refinement scores as evaluation measures. Nevertheless, we employed the nested cross-validation correction of selection bias46 in our model building procedure by splitting our experimental data into learning and test sets with an inner loop split on the learning set for parameter(s) optimization. Though found to contain high variance, we utilized leave-one-out cross-validation for the test set because it yields approximately an unbiased estimate of the true (expected) prediction error49 and because we were interested in the individual sample predicted probability of severe and not entirely on the expected predicted error. Nevertheless, where we were interested in the expected predicted error, as in the optimization of parameters, we utilized five-fold cross-validation as recommended by Breiman and Spector50. To validate the identified signature, an independent dataset generated on a different platform was used. Despite (i) the several sources of variability between our experimental data and the validation data that stem from - but not limited to - array platforms and different clinical cutoffs of RSV severity statuses, (ii) different time of profiling, 1–3 days after hospitalization and (iii) loss of information due to a reduction in signature because of no corresponding transcripts on Illumina platform and the aggregation of multiple transcripts to genes, our signature yielded an AUC of 0.858 that was comparable to accuracy (AUC of 0.856) when using the Illumina data (validation set) as experimental set. Cross-platform validation is rare due to lack of guidance on how this can be done reliably. We presented a cross-platform validation procedure.
The RSV patients enrolled in the study displayed varying disease severities but were all hospitalized thus representing a severe disease enriched subset of RSV infected infants. The patients enrolled however also represent a natural cohort of patients including a significant number of patients that eventually did not require extensive medical care and could have been discharged home. Since the blood samples were collected soon after hospital admission, the generated blood transcriptomes and the derived gene signature may serve as a basis for the development of a novel genomic tool to support clinical management of RSV disease including triage of patients presenting at the hospital provided that a rapid (real time) gene test can be developed. Larger transcriptome data sets are however required to construct predictive models that may also allow for discriminating mild from moderate and moderate from severe cases. Ultimately, one would like to extend the RSV biomarker program to earlier time point samples (e.g. obtained when visiting a general practitioner) and to samples collected from patients infected by other (respiratory) infectious agents or pathological conditions (comorbidities) in order to identify specific respiratory viral prognostic biomarkers. To this end a novel gene signature have to be developed using a much larger early blood sample cohort. The current results support the development of diagnostic tests for personalized medicine that not only provide information on the causative infectious agent, but also about the disease severity that may be expected.
Additional Information
How to cite this article: Jong, V. L. et al. Transcriptome assists prognosis of disease severity in respiratory syncytial virus infected infants. Sci. Rep. 6, 36603; doi: 10.1038/srep36603 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Simoes, E. a. Respiratory syncytial virus infection. Lancet 354, 847–852 (1999).
Falsey, A. R. et al. Respiratory Syncytial Virus and Other Respiratory Viral Infections in Older Adults With Moderate to Severe Influenza-like Illness. J. Infect. Dis. 1–9 doi: 10.1093/infdis/jit839 (2014).
Bush, A. & Thomson, A. H. Acute bronchiolitis. BMJ 335, 1037–1041 (2007).
Nair, H. et al. Global and regional burden of hospital admissions for severe acute lower respiratory infections in young children in 2010: a systematic analysis. Lancet 381, 1380–1390 (2013).
Hall, C. B. et al. The burden of respiratory syncytial virus infection in young children. N Engl J Med 360, 588–598 (2009).
Stockman, L. J., Curns, A. T., Anderson, L. J. & Fischer-Langley, G. Respiratory Syncytial Virus-associated Hospitalizations Among Infants and Young Children in the United States, 1997–2006. Pediatr. Infect. Dis. J. 31, 5–9 (2012).
Jain, S. et al. Community-Acquired Pneumonia Requiring Hospitalization among US Children. N. Engl. J. Med. 372, 835–845 (2015).
Adams, M. & Doull, I. Management of bronchiolitis. Paediatr. Child Health (Oxford). 19, 266–270 (2009).
Meissner, H. C. Viral Bronchiolitis in Children. N. Engl. J. Med. 374, 62–72 (2016).
Tregoning, J. S. & Schwarze, J. Respiratory viral infections in infants: causes, clinical symptoms, virology, and immunology. Clin. Microbiol. Rev. 23, 74–98 (2010).
Pulendran, B., Oh, J. Z., Nakaya, H. I., Ravindran, R. & Kazmin, D. a. Immunity to viruses: learning from successful human vaccines. Immunol. Rev. 255, 243–255 (2013).
Sekaly, R. & Pulendran, B. Systems biology in understanding HIV pathogenesis and guiding vaccine development. Curr. Opin. HIV AIDS 1–3 doi: 10.1097/COH.0b013e32834e0667 (2011).
van de Weg, C. a. M. et al. Time since Onset of Disease and Individual Clinical Markers Associate with Transcriptional Changes in Uncomplicated Dengue. PLoS Negl. Trop. Dis. 9, e0003522 (2015).
Zhai, Y. et al. Host Transcriptional Response to Influenza and Other Acute Respiratory Viral Infections – A Prospective Cohort Study. PLOS Pathog. 11, e1004869 (2015).
Schuurhof, A. et al. Gene expression differences in lungs of mice during secondary immune responses to respiratory syncytial virus infection. J. Virol. 84, 9584–9594 (2010).
van Diepen, A. et al. Host Proteome Correlates of Vaccine-Mediated Enhanced Disease in a Mouse Model of Respiratory Syncytial Virus Infection. J. Virol. JVI.03630–14 doi: 10.1128/JVI.03630-14 (2015).
Mejias, A. et al. Whole Blood Gene Expression Profiles to Assess Pathogenesis and Disease Severity in Infants with Respiratory Syncytial Virus Infection. PLoS Med. 10, e1001549 (2013).
Templeton, K. E., Scheltinga, S. a, Beersma, M. F. C., Kroes, A. C. M. & Claas, E. C. J. Rapid and sensitive method using multiplex real-time PCR for diagnosis of infections by influenza a and influenza B viruses, respiratory syncytial virus, and parainfluenza viruses 1, 2, 3, and 4. J Clin Microbiol 42, 1564–1569 (2004).
R. Core Team . R: A language and environment for statistical computing. http://www.R-project.org/ (2014).
Gentleman, R. C. et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5, R80 (2004).
Gautier, L., Cope, L., Bolstad, B. M. & Irizarry, R. A. Affy - Analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20, 307–315 (2004).
Huber, W., von Heydebreck, A., Sültmann, H., Poustka, A. & Vingron, M. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics 18 Suppl 1, S96–S104 (2002).
Johnson, W. E., Li, C. & Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics, doi: 10.1093/biostatistics/kxj037 (2007).
Krzystanek, M., Szallasi, Z. & Eklund, A. C. Biasogram: Visualization of Confounding Technical Bias in Gene Expression Data. PLoS One doi: 10.1371/journal.pone.0061872 (2013).
Smyth, G. K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 3, Article 3 (2004).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. 1–13 doi: 10.1093/nar/gkv007 (2015).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. 57, 289–300 (1995).
Pepe, M. S. Evaluating technologies for classification and prediction in medicine. Stat. Med. 24, 3687–3696 (2005).
Jong, V. L., Novianti, P. W., Roes, K. C. B. & Eijkemans, M. J. C. Exploring homogeneity of correlation structures of gene expression datasets within and between etiological disease categories. Stat. Appl. Genet. Mol. Biol. doi: 10.1515/sagmb-2014-0003 (2014).
Kim, K. I. & Simon, R. Probabilistic classifiers with high-dimensional data. Biostatistics doi: 10.1093/biostatistics/kxq069 (2011).
Bernhard Schölkopf & Alexander J. Smola . Learning with Kernels. (MIT Press, 2001).
Tibshirani, R., Hastie, T., Narasimhan, B. & Chu, G. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc. Natl. Acad. Sci. USA doi: 10.1073/pnas.082099299 (2002).
Breiman, L. Random forests. Mach. Learn. 5–32 doi: 10.1023/A:1010933404324 (2001).
Slawski, M., Daumer, M. & Boulesteix, A.-L. CMA: a comprehensive Bioconductor package for supervised classification with high dimensional data. BMC Bioinformatics doi: 10.1186/1471-2105-9-439 (2008).
Meyer, D., Dimitriadou, E., Hornik, K., Weingessel, A. & Leisch, F. Misc Functions of the Department of Statistics (e1071). R Packag. version 1.6-4 (2014).
Hastie, T., Tibshirani, R., Narasimhan, B. & Chu, G. pamr: Pam: prediction analysis for microarrays. R Packag. version 1.55 (2014).
Liaw, A. & Wiener, M. Classification and Regression by randomForest. R News 2, 18–22 (2002).
Berger, T. M., Aebi, C., Duppenthaler, a. & Stocker, M. Prospective population-based study of rsv-related intermediate care and intensive care unit admissions in Switzerland over a 4-year period (2001–2005). Infection 37, 109–116 (2009).
Brand, H. K. et al. Infection with multiple viruses is not associated with increased disease severity in children with bronchiolitis. Pediatr. Pulmonol. 47, 393–400 (2012).
Hasegawa, K. et al. Multicenter study of viral etiology and relapse in hospitalized children with bronchiolitis. Pediatr. Infect. Dis. J. 33, 809–813 (2014).
Mansbach, J. M. et al. Prospective multicenter study of the viral etiology of bronchiolitis in the emergency department. Acad. Emerg. Med. 15, 111–118 (2008).
Chorazy, M. L. et al. Polymicrobial acute respiratory infections in a hospital-based pediatric population. Pediatr. Infect. Dis. J. 32, 460–466 (2013).
Martin, E. T., Kuypers, J., Wald, A. & Englund, J. A. Multiple versus single virus respiratory infections: viral load and clinical disease severity in hospitalized children. Influenza Other Respi. Viruses 6, 71–77 (2012).
Brand, K. et al. Use of MMP-8 and MMP-9 to Assess Disease Severity in Children With Viral Lower Respiratory Tract Infections. J. Med. Virol. 84, 1471–1480 (2012).
Varma, S. & Simon, R. Bias in error estimation when using cross-validation for model selection. BMC Bioinformatics 7, 91 (2006).
Tibshirani, R. J. & Tibshirani, R. A bias correction for the minimum error rate in cross-validation. The Annals of Applied Statistics 3, 822–829 (2009).
Bernau, C., Augustin, T. & Boulesteix, A. L. Correcting the Optimal Resampling-Based Error Rate by Estimating the Error Rate of Wrapper Algorithms. Biometrics 69, 693–702 (2013).
Ding, Y. et al. Bias correction for selecting the minimal-error classifier from many machine learning models. Bioinformatics btu520– (2014).
Hastie, T., Tibshirani, R. & Friedman, J. The Elements of Statistical Learning: Data Mining, Inference, and Prediction. (Springer, 2009).
Breiman, L. & Spector, P. Submodel Selection and Evaluation in Regression. The X-Random Case. Int. Stat. Rev. 60, 291–319 (1992).
Acknowledgements
We are grateful for the parents and children that participated in this study, the physicians and nurses from the participating hospitals and Mariëtte Las from Pediatric Drug Research Centre, Nijmegen. We wish to thank Mediq Tefa for providing the Cheiron Dynamic II apparatus during the study. This study was financially supported by the VIRGO consortium, which is funded by the Netherlands Genomics Initiative and by the Dutch Government (FES0908).
Author information
Authors and Affiliations
Contributions
V.L.J. performed data analysis and wrote the manuscript, I.M.L.A. collected the data and contributed in writing the manuscript, H.J.v.d.H. contributed in data analysis and writing the manuscript, J.J. contributed in data collection, F.Z. performed gene expression profiling, A.Z. contributed in data analysis, E.S. contributed in data collection, M.A.B. contributed in data analysis, H.K.B. contributed in data collection, W.F.J.v.I.J. performed gene expression profiling, M.I.d.J. contributed in data collection, P.L.F. contributed in data collection, R.d.G. designed the study and contributed in data collection, A.D.M.E.O. designed the study and contributed in data analysis, M.J.E. performed data analysis and contributed in writing the manuscript, G.F. collected the data and contributed in writing the manuscript, and A.C.A. designed the study, performed gene expression profiling, contributed in data analysis and wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Jong, V., Ahout, I., van den Ham, HJ. et al. Transcriptome assists prognosis of disease severity in respiratory syncytial virus infected infants. Sci Rep 6, 36603 (2016). https://doi.org/10.1038/srep36603
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep36603
This article is cited by
-
Airway gene-expression classifiers for respiratory syncytial virus (RSV) disease severity in infants
BMC Medical Genomics (2021)
-
Gene signature of children with severe respiratory syncytial virus infection
Pediatric Research (2021)
-
Haemophilus is overrepresented in the nasopharynx of infants hospitalized with RSV infection and associated with increased viral load and enhanced mucosal CXCL8 responses
Microbiome (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
