
Abstract
Tuberculosis (TB) is caused by infection of Mycobacterium tuberculosis. Host genetic variability is an important determinant of the risk of developing TB in humans. Although the association between MBL2 polymorphisms and TB has been studied in various populations, the results are controversial. In this study four functional single-nucleotide polymorphisms (SNPs, H/L, X/Y, P/Q and A/B) across the MBL2 gene were genotyped by direct DNA sequencing of PCR products in a case-control population of Chinese Han origin, consisting of 1,020 patients with pulmonary TB and 1,020 controls. We found that individuals carrying variant allele at A/B (namely BB or AB genotypes) was associated with increased susceptibility to TB (odds ratios [OR] = 1.57, 95% confidence interval [CI] 1.30–1.91, P = 1.3 × 10−6). Additionally, LYPB haplotype showed a significant association with increased risk of TB (OR = 1.54, 95% CI 1.27–1.87, P = 4.2 × 10−6; global haplotype association P = 3.5 × 10−5). Furthermore, individuals bearing low- or medium- MBL expression haplotype pairs had an increased risk of TB (OR = 1.56, 95% CI 1.29–1.90, P = 1.4 × 10−6). Thus, the reduced expression of functional MBL secondary to having MBL2 variants may partially mediate the increased susceptibility to TB risk.
Similar content being viewed by others

Introduction
Tuberculosis (TB) is caused by the infection of Mycobacterium tuberculosis (Mtb). Mtb presumably infects a third of the world’s population, with China has the second-largest number of infected cases1. Despite the improved treatment during the past decades, TB remains the first leading cause of global death from infectious diseases. The host-pathogen interactions and environmental factors may contribute to TB. Furthermore, Host genetic variability is an important determinant of the risk of developing TB in humans2. Unraveling the genetic factor contributing to the pathogenesis of TB may lead to improved treatment and prevention of this disease3.
The innate immune response in human activates the T helper 1 (Th1)-type immune system and plays an important role in the host’s defense against the development of TB4. Many studies have reported the association between TB and genetic polymorphisms related to human innate immunity5. Mannose-binding lectin (MBL) is a member of the collectin family that recognizes pathogens by its carbohydrate-recognition domains6. MBL is an acute-phase serum protein, which is synthesized in the liver and circulates as oligomers complexed with MBL-associated serine proteases (MASPs)7. MASPs binds to the sugar moieties on the surface of a pathogen, and then are activated to initiate the lectin pathway of complement activation, resulting in opsonization and phagocytosis or lysis of pathogens. In addition, MBL can regulate inflammatory responses and immune activation7.
MBL is encoded by the MBL2 gene, which is located on chromosome 10. Six single-nucleotide polymorphisms (SNPs) in exon 1 (codon 52 A/D, codon 54 A/B, and codon 57 A/C) and in the promoter and 5′-untranslated regions (nt −550 H/L, nt −221 X/Y, and nt + 4 P/Q) of the MBL2 gene are associated with serum levels and/or functions of MBL. These genetic variations are associated with a wide variety of diseases, including respiratory tract infections3,8,9,10,11. However, in previously reported studies MBL2 polymorphisms have conflicting results showing protection from or susceptibility to TB5. For example, low levels of MBL (associated with variant alleles at the promoter and exon 1 regions of the MBL2 gene) were reported to protect against tuberculosis12,13, supporting the hypothesis that MBL binding can enhance the uptake of intracellular pathogens by phagocytes, and then promote infection12,14. Other investigators instead claim that protection against the disease is associated with high levels of MBL (controlled by the wild MBL2 alleles)15,16,17,18. In the Chinese Han population, no convincing evidence of association between MBL2 sequence variants and TB was observed19,20,21,22,23. For example, in a meta-analysis totally including 880 TB patients and 959 controls of Chinese origin, Shi J and colleagues reported that variants at A/B were associated with increased susceptibility to TB20. Additionally, Chen M and colleagues found that variants in Y/X was associated with increased susceptibility to TB among Chinese21,22. However, most recently Wu L reported that variants in H/L, but not Y/X, P/Q or A/B, were associated with decreased susceptibility to TB among Chinese23. Conflicting results are not unexpected in association studies for several reasons, including small sample size, marginal statistical significance, detection of genotypes, or ethnic heterogeneity.
Therefore, here we conducted a genetic association study in a large case-control population of Chinese Han origin (totally consisting of 1,020 patients with TB and 1,020 controls), to better define the association between the MBL2 SNPs and TB.
Results
Population Characteristics
The selected characteristics of patients with TB and control subjects in this study are shown in Table 1.
The study consists of 1,020 patients with TB and 1,020 control subjects. The male/female ratio of patients with TB was 1.10, and the mean age was 43.2 years (±SD, 14.9) (Table 1). For the 1,020 controls, the male/female ratio was 1.02, and the mean age was 44.3 years (±SD, 15.0). Controls were comparable with cases with regard to mean age and gender (all P values > 0.05).
The genotyping results are presented in Table 2. The observed genotype frequencies for the four polymorphisms conformed to the Hardy-Weinberg equilibrium in controls (all P values > 0.05). Consistent with the previous reports11,24, the codon 52 (A/D) and 57 (A/C) variants were not observed in our ethnic northern Chinese individuals. The frequencies of variant alleles L, X, Q and B in the controls were 48.7%, 14.5%, 13.2% and 14.0%, respectively, similar to those reported in other southern or northern Chinese subpopulations11,24.
Polymorphisms and Risk of TB in this study
On the basis of unconditional logistic regression analysis with adjustment for known confounding factors including age and gender, a statistically significant association with the susceptibility to TB was observed with the condon 54 variant. Subjects homozygous and heterozygous for variant B allele (i.e. B/B plus A/B genotype) had an increased susceptibility to TB compared to those homozygous for the wild-type A allele (OR = 1.57, 95% CI 1.30–1.91, P = 1.3 × 10−6; simulation number = 10,000,000; Table 2). The association was still significant even after correction for multiple comparisons. None of the remaining three variants (H/L, Y/X and P/Q) were found to be significantly associated with susceptibility to TB. Logistic regression models including random effects obtained results similar to those obtained in models including fixed-effects (Table 2).
In addition to single SNP analysis, haplotype analysis was also performed for the MBL2 gene. We identified 5 haplotypes (HYPA, LYPB, LXPA, LYQA and LYPA) using the PHASE program, with the frequencies of these haplotypes greater than 5% in both cases and controls. The frequencies of these 5 haplotypes in controls ranged from 51.3% (HYPA haplotype) to 7.0% (LYPA haplotype), similar to those reported in other Asian populations of Japanese and Vietnamese16.
In haplotype analysis, only LYPB showed a significant association with increased risk of TB (OR = 1.54, 95% CI 1.27–1.87; P = 4.2 × 10−6; global haplotype association P = 3.5 × 10−5; simulation number = 10,000,000; Table 3). Furthermore, we grouped cases and controls by harboring haplotypes associated with high, medium, or low MBL expression25, to evaluate whether haplotype pairs associated with various levels of MBL expression were associated with susceptibility to TB risk. In 1,020 patients with TB, 618 (60.6%) were in the high expression group, 298 (29.2%) in the medium group, and 104 (10.2%) in the low group. In 1,020 controls, this distribution was 723 (70.9%), 234 (22.9%) and 63 (6.2%), respectively (OR = 1.56, 95% CI 1.29–1.90, P = 1.4 × 10−6, with adjustment for age and gender; simulation number = 10,000,000; Table 2), implicating that the individuals bearing medium or low expression haplotype pairs had an increased risk of TB and that the MBL deficiency might play a potential role in susceptibility to TB.
The associations between the four MBL2 polymorphisms and susceptibility to TB were further examined with stratification by age and gender, with no differences observed for MBL2 polymorphisms individually or in haplotype between stratums (all P values > 0.05, test for homogeneity).
Discussion
Recent studies on the genetic association between SNPs in the MBL2 gene and patients with TB of Chinese origin have generated different and even contradictory results. These studies have small sample size, usually involving about 200 or less PTB patients of Chinese origin19,20,21,22,23, which might lead to artificial associations. In addition, the four functional SNPs in the MBL2 gene is in linkage disequilibrium (LD) and more appropriate to be investigated together (in both haplotype and haplotype pairs). Moreover, marginal statistical significance reported in these association studies means that poor reproducibility of their results is not unexpected, given the concern over the possible unreliability of case-control studies26.
In this study, we investigated all of the four functional SNPs in the MBL2 gene, individually or in haplotype or haplotype pairs, in a large case-control population of Chinese Han origin, totally consisting of 1,020 patients with TB and 1,020 controls. This is the first study to investigate the association between MBL2 polymorphisms with pulmonary TB susceptibility in northern Han Chinese. By genotyping all of the four functional SNPs in the MBL2 gene in the case-control population of relatively large sample size, we found that one SNP (A/B) was associated with susceptibility to TB (P = 1.3 × 10−6). Furthermore, we found that the LYPB haplotype showed a significant association with increased risk of TB in northern Han Chinese (P = 4.2 × 10−6). In addition, individuals bearing haplotype pairs indicating medium or low MBL2 expression had an increased risk of TB (P = 1.4 × 10−6). We noted that Wu et al. have reported that LYPB haplotype was associated with TB in southern Chinese23. However, frequencies of MBL2 haplotypes in Wu’s study were remarkably different from those in Chinese subjects observed in other studies. For example, frequencies of HYPA, LYPB, LXPA, LYQA and LYPA were 51.3%, 14.0%, 14.5%, 13.2% and 7.0% respectively in the controls in the present study, similar to the frequency distribution of these haplotypes in southern and northern Chinese in the 1000 genomes project. However, in the controls in Wu’s study frequencies of HYPA, LYPB, LXPA, LYQA and LYPA were 29.3%, 0.9%, 7.1%, 3.6% and 37.1% respectively. Based on the large sample size, haplotype pairs analyses and small P values, our results may be closer to the real situation of MBL2 polymorphisms in TB susceptibility.
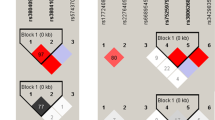
The MBL2 gene encodes a homotrimeric molecule harboring a carbohydrate recognition domain and a collagenous tail27. The variant B allele can impair the formation of a triple helix in the collagenous tail, and therefore disrupts MBL polymerization and leads to enzymatic degradation and functional deficiency of this protein28. The other SNPs, H/L, X/Y, and P/Q, also play roles in determining functional MBL concentration, among which the X allele shows the lowest transcriptional activity25,29,30. When in haplotype, these SNPs form three defective haplotypes (LYPB, LYQC and HYPD, with the latter two absent among Chinese), and five functional haplotypes with different expression levels (a low-producing LXPA haplotype, an intermediate-producing LYPA haplotype, and two high-producing haplotypes LYQA and HYPA)30. The results that B allele, LYPB haplotype, and medium- or low-expression haplotype pairs were associated with increased risk of developing TB (Tables 1, 2 and 3) suggest that MBL deficiency possibly plays a role in susceptibility to TB.
There is biological plausibility for our observed genetic associations, although the exact mechanism by which MBL deficiency is associated with increased susceptibility to TB requires further investigation. Previous studies have revealed that MBL recognizes M. tuberculosis by direct interaction, resulting in lectin pathway activation, agglutination of bacteria and enhancement of phagocytosis31,32. Additionally, the serum samples from carriers of haplotype pairs associated with high MBL levels (HYA/HYA) display significantly higher antibacterial activity of neutralization than did those associated with lower MBL levels, and the activity is mediated by MBL15. Therefore, one speculative interpretation of our results is that low serum concentrations of functional MBL caused by haplotype pairs indicating medium or low MBL2 expression might result in reduced neutralization and favor the survival of M. tuberculosis.
There are several potential limitations in the present study. The first, the patients with TB were selected from the hospital and the controls were recruited from the community population, which means that inherent selection bias cannot be completely excluded. However, by matching on age and gender and by analyzing data with further stratification, potential confounding effect might have been minimized. The second, some of the control subjects used in this study may be asymptomatic with latent TB infection. Therefore, we cannot directly specify which stage of TB, infection of Mtb or development of active disease, was more affected by MBL2. The third, although the highly significant association between MBL deficiency and increased susceptibility to TB derives from a biologically based a priori hypothesis, the initial findings presented here should be independently verified in other subpopulations of ethnic Chinese origin (e.g. southern Chinese) or of different ancestry.
In summary, our results reveal an association between genetic MBL deficiency and increased susceptibility to TB among northern Chinese, and provide supports for the importance of MBL in the pathogenesis of TB. Knowledge of genetic factors contributing to the pathogenesis of TB revealed in this study could lead to improved treatment and prevention of this disorder.
Methods
Ethics statement
Written informed consent was obtained from all participants involved in this case-control genetic association study, and the study was approved by the Medical Ethics Committee of the PLA 307 Hospital (Beijing, China) and the Fifth Hospital of Shijiazhuang (Shijiazhuang City, Hebei province, China). All the experiments were performed in accordance with the relevant guidelines and regulations.
Subjects and Samples
A total of 1,020 patients with pulmonary TB were recruited at the Fifth Hospital of Shijiazhuang (Shijiazhuang City, Hebei province, China), between January 2010 and January 2016. All patients with pulmonary TB were newly diagnosed based on smear or culture positive for M. tuberculosis and chest radiography. Patients with confirmed diagnosis of extrapulmonary TB were excluded. The response rate for case patients was 93%. A total of 1,020 healthy adults frequency matched to the TB patients on the basis of age and gender were recruited as control subjects during the same time period as the TB patients were collected. The response rate for controls was 91%. All participants were unrelated northern Han Chinese, and had no clinical histories of diabetes mellitus, HIV infection, or receipt of immunosuppressive therapy. The authors had access to identifying information during and after data collection.
Extraction of Genomic DNA
About 1-mL volume of peripheral blood samples was taken from each participant, and were frozen at −20 °C. Total genomic DNA was extracted from the samples using the Whole Blood DNA Extraction Kit (Tiangen Biotech, Co., Ltd, Beijing, China), according to the manufacturer’s instructions. DNA was dissolved in 0.1 × TE buffer (10 mM Tris-1 mM EDTA, pH 8.0) and stored at −20 °C.
SNP Genotyping
The promoter polymorphisms at nt −550 (H/L, rs11003125), −221 (Y/X, rs7096206), and +4 (P/Q, rs7095891) and the structural polymorphism at nt +230 (codon 54 A/B, rs1800450) of the MBL2 gene were selected for genotyping by use of PCR direct sequencing in the case-control population. The PCR primers were designed using the web-based software Primer3 (available at: http://primer3.ut.ee/). The forward primer 5′-ATGGGAGGAGGATTCAAGG-3′ and reverse primer 5′-CCTTGTGACACTGCGTGACT-3′ were used for amplifying the target region containing the MBL2 H/L and Y/X variants. The forward primer 5′-CGGTCCCATTTGTTCTCACT-3′ and reverse primer 5′-CACAAACTGCTGTGTGGAAT-3′ were used for amplifying the target region containing the MBL2 P/Q and A/B variants. Then the PCR products were sequenced by an ABI 3730 sequencer. A nucleotide was identified as a candidate polymorphism by an observer using the PolyPhred program (available at: http://droog.gs.washington.edu/polyphred/) and confirmed by another observer independently. Genotyping of all samples was performed in a blinded manner so that the case or control status was unknown. The call rates were 100% for all SNPs. The accuracy of genotyping data for each SNP was validated by masking, choosing at random, and resequencing 5% of the samples from the opposite strand.
Statistical Analyses
Comparisons of gender and age between TB patients and controls were performed using the one-sided χ2 test (SPSS software, version 10.0). Differences of mean age between TB patients and controls were analyzed using the unpaired t test (SPSS software, version 10.0). Genotype frequencies for each SNPs were determined by gene counting. The significance of deviations from the Hardy-Weinberg equilibrium was tested using the online software SNPStats (http://bioinfo.iconcologia.net/SNPstats_web). Unconditional logistic regressions under a dominant inheritance model were used due to the limited sample size, to calculate P values, Odds ratios (ORs), and 95% confidence intervals (CIs) for assessing the association between SNPs and disease risk, adjusting for age and gender (fixed effects). In additional analyses, age and gender were treated as random effects in the logistic regression to deal with the uncertainties with more flexibility. The glm-function and glmer-function in the lme4 package (version 1.1–12)33 of R (version 3.3.1)34 were used to fit the models with fixed effects and random effects, respectively. To show the robustness of the conclusions, we performed resampling statistics as previously reported35. The P values, ORs, and 95% CIs in single SNP analyses were calculated by a bootstrapping test with the number of simulations was set to 10,000,000. As to multiple comparisons, the correction factor n(m-1) (in which there are n loci with m alleles) was used to correct the significance level.
The haplotypes of the MBL2 gene of the case-control population were assigned by the PHASE program36. P values of association between haplotypes and disease risk were evaluated using the haplo.score-function37 in the haplo.stats package (version 1.7.7) of R, with the number of simulations was set to 10,000,000. ORs and 95% CIs were calculated for each haplotypes using the haplo.glm-function38 in the haplo.stats package. All P values and ORs (95% CI) were calculated by adjusting for age and gender. Analyses of haplotype pairs were similar to those of single SNPs.
Potential modification of the effect of the SNPs on TB risk was assessed for age and gender by the addition of interaction terms in the logistic model and by separate analyses of subgroups of subjects determined by these factors. A homogeneity test was used to detect the difference of ORs within each subgroups.
All tests are two-tailed unless otherwise indicated. P values less than 0.05 were considered statistically significant.
Additional Information
How to cite this article: Liu, C. et al. Association of Mannose-binding Lectin Polymorphisms with Tuberculosis Susceptibility among Chinese. Sci. Rep. 6, 36488; doi: 10.1038/srep36488 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Lawn, S. D. & Zumla, A. I. Tuberculosis. Lancet 378, 57–72 (2011).
Cooke, G. S. & Hill, A. V. Genetics of susceptibility to human infectious disease. Nat. Rev. Genet. 2, 967–977 (2001).
Zhang, J. et al. Polymorphisms in the interleukin 18 receptor 1 gene and tuberculosis susceptibility among Chinese. PLoS One 9, e110734 (2014).
Dorhoi, A., Reece, S. T. & Kaufmann, S. H. For better or for worse: the immune response against Mycobacterium tuberculosis balances pathology and protection. Immunol. Rev. 240, 235–251 (2011).
Azad, A. K., Sadee, W. & Schlesinger, L. S. Innate immune gene polymorphisms in tuberculosis. Infect. Immun. 80, 3343–3359 (2012).
Heitzeneder, S., Seidel, M., Forster-Waldl, E. & Heitger, A. Mannan-binding lectin deficiency-Good news, bad news, doesn’t matter? Clin. Immunol. 143, 22–38 (2012).
Ip, W. K., Takahashi, K., Ezekowitz, R. A. & Stuart, L. M. Mannose-binding lectin and innate immunity. Immunol. Rev. 230, 9–21 (2009).
Eisen, D. P. Mannose-binding lectin deficiency and respiratory tract infection. J. Innate Immun. 2, 114–122 (2010).
Chalmers, J. D. et al. Mannose-binding lectin deficiency and disease severity in non-cystic fibrosis bronchiectasis: a prospective study. Lancet Respir. Med. 1, 224–232 (2013).
Roy, S. et al. MBL genotype and risk of invasive pneumococcal disease: a case-control study. Lancet 359, 1569–1573 (2002).
Zhang, H. et al. Association between mannose-binding lectin gene polymorphisms and susceptibility to severe acute respiratory syndrome coronavirus infection. J. Infect. Dis. 192, 1355–1361 (2005).
Soborg, C. et al. Mannose-binding lectin polymorphisms in clinical tuberculosis. J. Infect. Dis. 188, 777–782 (2003).
Garred, P. et al. Mannan-binding lectin in the sub-Saharan HIV and tuberculosis epidemics. Scand J. Immunol. 46, 204–208 (1997).
Thye, T. et al. Variant G57E of mannose binding lectin associated with protection against tuberculosis caused by Mycobacterium africanum but not by M. tuberculosis. PLoS One 6, e20908 (2011).
Capparelli, R. et al. Role played by human mannose-binding lectin polymorphisms in pulmonary tuberculosis. J. Infect. Dis. 199, 666–672 (2009).
Hijikata, M. et al. Age-dependent association of mannose-binding lectin polymorphisms with the development of pulmonary tuberculosis in Viet Nam. Hum. Immunol. 75, 840–846 (2014).
Garcia-Gasalla, M. et al. Mannose-binding lectin exon 1 and promoter polymorphisms in tuberculosis disease in a Mediterranean area. Int. J. Immunogenet. 41, 306–311 (2014).
da Cruz, H. L. et al. MBL2 gene polymorphisms and susceptibility to tuberculosis in a northeastern Brazilian population. Infect. Genet. Evol. 19, 323–329 (2013).
Liu, W. et al. Sequence variations in the MBL gene and their relationship to pulmonary tuberculosis in the Chinese Han population. Int. J. Tuberc. Lung Dis. 10, 1098–1103 (2006).
Shi, J. et al. Mannose-binding lectin two gene polymorphisms and tuberculosis susceptibility in Chinese population: a meta-analysis. J. Huazhong Univ. Sci. Technolog. Med. Sci. 33, 166–171 (2013).
Chen, M. et al. Impact of MBL and MASP-2 gene polymorphism and its interaction on susceptibility to tuberculosis. BMC Infect. Dis. 15, 151 (2015).
Chen, M. et al. Impact of passive smoking, cooking with solid fuel exposure, and MBL/MASP-2 gene polymorphism upon susceptibility to tuberculosis. Int. J. Infect. Dis. 29, 1–6 (2014).
Wu, L. et al. An association study of NRAMP1, VDR, MBL and their interaction with the susceptibility to tuberculosis in a Chinese population. Int. J. Infect. Dis. 38, 129–135 (2015).
Ip, W. K., To, Y. F., Cheng, S. K. & Lau, Y. L. Serum mannose-binding lectin levels and mbl2 gene polymorphisms in different age and gender groups of southern Chinese adults. Scand J. Immunol. 59, 310–314 (2004).
Madsen, H. O. et al. Interplay between promoter and structural gene variants control basal serum level of mannan-binding protein. J. Immunol. 155, 3013–3020 (1995).
Wacholder, S., Chanock, S., Garcia-Closas, M., El Ghormli, L. & Rothman, N. Assessing the probability that a positive report is false: an approach for molecular epidemiology studies. J. Natl. Cancer Inst. 96, 434–442 (2004).
Sastry, K. et al. The human mannose-binding protein gene. Exon structure reveals its evolutionary relationship to a human pulmonary surfactant gene and localization to chromosome 10. J. Exp. Med. 170, 1175–1189 (1989).
Lipscombe, R. J. et al. High frequencies in African and non-African populations of independent mutations in the mannose binding protein gene. Hum. Mol. Genet. 1, 709–715 (1992).
Zhao, J., Lin, G., Zhang, W. H., Ge, M. & Zhang, Y. Contribution of CD14-159C/T polymorphism to tuberculosis susceptibility: a meta-analysis. Int. J. Tuberc. Lung Dis. 17, 1472–1478 (2013).
Madsen, H. O., Satz, M. L., Hogh, B., Svejgaard, A. & Garred, P. Different molecular events result in low protein levels of mannan-binding lectin in populations from southeast Africa and South America. J. Immunol. 161, 3169–3175 (1998).
Bartlomiejczyk, M. A., Swierzko, A. S., Brzostek, A., Dziadek, J. & Cedzynski, M. Interaction of lectin pathway of complement-activating pattern recognition molecules with mycobacteria. Clin. Exp. Immunol. 178, 310–319 (2014).
Garred, P., Harboe, M., Oettinger, T., Koch, C. & Svejgaard, A. Dual role of mannan-binding protein in infections: another case of heterosis? Eur. J. Immunogenet. 21, 125–131 (1994).
Bates, D., Maechler, M., Bolker, B. & Walker, S. Fitting Linear Mixed-Effects Models Using lme4. J. Stat. Softw. 67, 1–48 (2015).
R Development Core Team. A Language and Environment for Statistical Computing. Vienna, Austria: the R Foundation for Statistical Computing. (2012).
Li, J. et al. Identification of high-quality cancer prognostic markers and metastasis network modules. Nat. Commun. 1, 34 (2010).
Stephens, M., Smith, N. J. & Donnelly, P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 68, 978–989 (2001).
Schaid, D. J., Rowland, C. M., Tines, D. E., Jacobson, R. M. & Poland, G. A. Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am. J. Hum. Genet. 70, 425–434 (2002).
Lake, S. L. et al. Estimation and tests of haplotype-environment interaction when linkage phase is ambiguous. Hum. Hered. 55, 56–65 (2003).
Acknowledgements
We thank everyone who provided blood samples and consent for genetic analysis. In addition, we thank all of the clinicians, nurses and study coordinators for their contributions to the work. The study was funded by grants from the Army Scientific Research Foundation (CWS11J323), Collaborative Innovation Center of Infectious Diseases (PXM2014_014226_000011 and PXM2015_014226_000058), and Beijing Municipal Administration of Hospitals Clinical Medicine Development of Special Funding Support (ZYLX201304). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
L.Z., X.W., H.L. and W.D. conceived and designed the study. C.L., T.H. and Y.R. performed genotyping. Y.Z., Z.Z., D.M. and Y.W. helped to analyze the data. Z.M., Y.W. and H.W. were responsible for recruitment of the case-control samples. F.D. helped to prepare samples. L.Z., H.L., W.D., C.L., T.H., F.D. and Y.R. conducted samples selection and data management, performed the statistical analyses, interpreted the results, and drafted the manuscript. All authors reviewed the manuscript. L.Z., X.W., H.L. and W.D. approved the final version of the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Liu, C., He, T., Rong, Y. et al. Association of Mannose-binding Lectin Polymorphisms with Tuberculosis Susceptibility among Chinese. Sci Rep 6, 36488 (2016). https://doi.org/10.1038/srep36488
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep36488
This article is cited by
-
Mannose-binding lectin (MBL) deficiency and tuberculosis infection in patients with ankylosing spondylitis
Clinical Rheumatology (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
