
Abstract
Manganese (Mn) is pivotal for plant growth and development, but little information is available regarding the strategies that evolved to improve Mn acquisition and cellular homeostasis of Mn. Using an integrated RNA-based transcriptomic and high-throughput shotgun proteomics approach, we generated a comprehensive inventory of transcripts and proteins that showed altered abundance in response to Mn deficiency in roots of the model plant Arabidopsis. A suite of 22,385 transcripts was consistently detected in three RNA-seq runs; LC-MS/MS-based iTRAQ proteomics allowed the unambiguous determination of 11,606 proteins. While high concordance between mRNA and protein expression (R = 0.87) was observed for transcript/protein pairs in which both gene products accumulated differentially upon Mn deficiency, only approximately 10% of the total alterations in the abundance of proteins could be attributed to transcription, indicating a large impact of protein-level regulation. Differentially expressed genes spanned a wide range of biological functions, including the maturation, translation, and transport of mRNAs, as well as primary and secondary metabolic processes. Metabolic analysis by UPLC-qTOF-MS revealed that the steady-state levels of several major glucosinolates were significantly altered upon Mn deficiency in both roots and leaves, possibly as a compensation for increased pathogen susceptibility under conditions of Mn deficiency.
Similar content being viewed by others

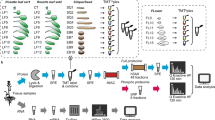

Introduction
Manganese (Mn) is an essential mineral nutrient for all organisms, playing important roles in the detoxification of reactive oxygen species and, often as a co-factor of important enzymes, in a multitude of biosynthetic pathways1. In plants, Mn is also critical for the function of photosystem II as a component of oxygen-evolving complex (OEC), which catalyzes the oxidation of water to protons and molecular oxygen. Manganese deficiency reduces plant growth, increases susceptibility to infection2,3 and induces PSII photoinhibition by destabilizing the cubane–like Mn cluster in the OEC, thereby compromising photosynthetic activity4,5. Manganese deficiency is a worldwide problem that is particularly severe in Australia, the northern parts of the United States and Canada, and in the northern parts of Europe6. In particular sandy soils, soils rich in organic matter and soils with high pH restrict the bioavailability of Mn.
The uptake of Mn across various organisms is mediated by Nramps (natural resistance-associated macrophage proteins), a group of integral membrane transporters that are highly conserved among bacteria, fungi, and animals. In Arabidopsis, NRAMP1 was shown to be critical for Mn uptake; loss of NRAMP1 function led to severe growth reduction and decreased Mn concentrations7. In support of this finding, screening of Arabidopsis accessions for reduced photosynthesis under chilling temperature identified a mutation in a conserved histidine in the NRAMP1 allele of the Hog accession as the cause for the observed phenotype8. In rice, OsNRAMP5 mediates the trans-plasma membrane transport of Mn (and Fe) from the soil9. In Arabidopsis, two other Nramp proteins, NRAMP3 and NRAMP4, are critical in remobilizing vacuolar Mn (and Fe) in leaves prior to the import into chloroplasts of mesophyll cells, and are required for functional PSII10. In addition to Nramps, several ZIP (ZRT/IRT-like Proteins) transporters were shown to transport Mn in yeast complementation assays11. The ZIP protein AtIRT1 mediates trans-plasma membrane transport of several transition metals such as Cd, Ni, Co and Mn12 and the expression level of its ortholog in barley correlates with root Mn uptake efficiency13. ZIP1 and ZIP2 have been associated with Mn translocation from the root to the shoot, but both transporters are unlikely to be involved in the uptake of Mn from the soil11. An overview of the Mn-transporting enzymes is provided in a recent review by Socha and Guerinot14.
In contrast to other soil-immobile nutrients such as phosphate and Fe for which the mechanisms that re-calibrate cellular homeostasis are relatively well explored, only fragmentary information is available regarding the responses of plants to low Mn supply. Such knowledge is of critical importance for the development of Mn-efficient germplasm with improved Mn acquisition and/or increased resource utilization efficiency. Transcriptome profiling has allowed insights into Mn deficiency-induced changes of gene activity15; however, biological processes are carried out by proteins and it is not clear how much the changes observed at the transcript level impact the proteomic readout. In fact, several studies observed only a moderate level of concordance between transcriptomics and proteomics (reviewed by Vogel and Marcotte16), indicating the necessity of integrative studies that cover disparate omics levels.
In the present study, we undertook a comprehensive quantitative profiling of gene activity in roots from plants grown on Mn-deplete media, aiming at dissecting and cataloging the responses of Arabidopsis roots to Mn deficiency. To acknowledge the generally observed moderate correlation of changes in transcript and protein abundance, we opted for an integrative transcriptomic and proteomic study. We used ultra high pressure liquid chromatography on a Q Exactive Orbitrap mass spectrometer to compile a near complete inventory of the Arabidopsis root proteome. To compare and integrate proteomic changes with alterations in the transcriptome, we investigated such changes by RNA-seq on an Illumina HiSeq 2000 platform. To validate changes observed in the activity of enzymes involved in glucosinolate metabolism at the transcript and protein level, we investigated changes in specific glucosinolates by UPLC-qTOF-MS. We identified several novel components of cellular Mn homeostasis and show that many putatively important changes in response to Mn deficiency are only evident at one of the omics levels under investigation.
Methods
Plant growth conditions
Arabidopsis (Arabidopsis thaliana (L.) Heynh) plants were grown in a growth chamber on solid medium as described by Estelle and Somerville17. Seeds of the accession Columbia (Col-0) were obtained from the Arabidopsis Biological Resource Center (Ohio State University). Seeds were surface-sterilized by immersing them in 5% (v/v) NaOCl for 5 min and 70% ethanol for 7 min, followed by four rinses in sterile water. Seeds were placed onto Petri dishes and kept for 1 d at 4 °C in the dark, before the plates were transferred to a growth chamber and grown at 21 °C under continuous illumination (50 μmol m−2 s−1; Phillips TL lamps). The medium was composed of (mM): KNO3 (5), MgSO4 (2), Ca(NO3)2 (2), KH2PO4 (2.5) and (μM): H3BO3 (70), ZnSO4 (1), CuSO4 (0.5), Na2MoO4 (0.2), CoCl2 (0.01), FeEDTA (40), 1% (w/v) MES, supplemented with sucrose (43.8 mM) and solidified with 0.8% plant cell culture tested agar (Sigma A7921). The pH was adjusted to 5.5. Plants were grown for 14 d in media supplemented with 14 μM MnCl2 (+Mn) or without Mn (−Mn).
RNA sequencing
Three independently grown batches of plants grown under Mn-sufficient and Mn-deficient conditions were used for analysis. For each sample, roots from 10 Mn-sufficient and 20 Mn-deficient plants were pooled and total RNA was extracted from roots or leaves using the RNeasy Plant Mini Kit (Qiagen), following manufacturer instructions. Nucleic acid quantity was analyzed with a NanoDrop ND-1000 UV-Vis Spectrophotometer (NanoDrop Technologies, Wilmington, USA). For RNA-seq, equal amounts of total RNA were collected and cDNA libraries for sequencing were prepared from total RNA following the manufacturer’s protocol (Illumina). The cDNA libraries were subsequently enriched by PCR amplification. The resulting cDNA libraries were subjected to sequencing on a single lane of an Illumina HiSeq 2000 machine. RNA-seq and data collection was done following the protocol of Mortazavi et al.18. The length of the cDNA library was maintained from 250 to 300 bp with a 5′-adapter of 20 bp and a 3′-adapter of 33 bp at both ends. Eventually, the fragment length range of the cDNA was 200 to 250 bp. To quantify gene expression levels, 75-mers sequences were aligned to the genomic sequence annotated in TAIR10 using the BLAT program19 and RPKM (Reads Per Kbp per Million reads) values were computed using the RACKJ (Read Analysis & Comparison Kit in Java, http://rackj.sourceforge.net/) software.
Bioinformatics
Gene clustering was performed using the MACCU software package (http://maccu.sourceforge.net/) to build co-expression clusters based on pairwise co-expression relationships of genes with Pearson coefficients greater than or equal to 0.75. In order to capture the co-expression relationships specifically in roots, Pearson coefficients were computed based on robust multi-array averaged array data derived from publically available root-specific experiments downloaded from NASCArrays (http://affymetrix.arabidopsis.info/). Visualization of the networks was performed with the Cytoscape software version 3 (http://www.cytoscape.org/).
qRT-PCR
Shoot and root samples (pooled from 10–20 seedlings from three independently grown batches of plants) were collected from 14-day-old seedlings and frozen immediately in liquid nitrogen. Total RNA was isolated using RNeasy Mini Kit (Qiagen) and treated with DNase using TURBO DNA-free Kit (Ambion) as indicated by the manufacturer. cDNA was synthesized using DNA-free RNA with Oligo-dT (20) primers and SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen). After incubation at 50 °C for 1 h and subsequently at 70 °C for 15 min, 1 μL of RNase H was added and incubated for 20 min at 37 °C. The cDNA was used as a PCR template in a 20 μL reaction system using the SYBR Green PCR Master Mix (Applied Biosystems) with programs recommended by the manufacturer in an AB QuantStudio Real Time PCR System (Applied Biosystems). Three independent replicates were performed for each sample. The ∆∆CT method was used to determine the relative amount of gene expression20, with the expression of elongation factor 1 (EF1) used as an internal control. The following primers were used: EF1A, 5′-AACTTTGATGGCGTTTGAGC-3′ and 5′-TCCGGAAACTGCATAATTGA-3′; BGLU23, 5′-CATTGGTAGCAAGCCTTTGA-3′ and 5′-ATGATCAGCGGTACCAACAG-3′; TTG1, 5′-AATGGGATTGATCCGATGTC-3′ and 5′-CACTTCACATCTGCACCTCA-3′.
ICP-OES
Fifteen to 20 shoot samples were collected from 14-day-old seedlings (yielding 0.03 to 0.05 g dry weight), washed with 10 mM CaCl2 and H2O, dried at 60 °C for 3 d, and weighed before microwave-digestion. Samples (50 mg) were digested with 5 mL of 65% HNO3 and 2 mL H2O2 in a MarsXpress microwave digestion system (CEM, Matthews, NC, USA). The volume of digested samples was adjusted to 10 mL with H2O and filtered using a 0.45 μm membrane filter. Tomato leaves (SRM-1573a) from the National Institute of Standards and Technology (Gaithersburg, MD, USA) were used as a standard. The concentration of Mn in digested samples was analyzed by inductively coupled plasma-optical emission spectrometry (ICP-OES; OPTIMA 5300, Perkin-Elmer, Wellesley, MA, USA). The Mn concentration for each sample was determined by triplicate measurements.
Protein extraction
Three independently grown batches of plants grown under Mn-sufficient and Mn-deficient conditions were used for analysis. Roots from 100 Mn-sufficient plants and 200 Mn-deficient plants (14-day-old) were pooled, ground in liquid nitrogen and suspended in 10 × volume of precooled acetone (−20 °C) containing 10% (v/v) TCA and 0.07% (v/v) 2-mercaptoethanol. Proteins were then precipitated for 2 h at −20 °C after thorough mixing. Precipitated proteins were collected by centrifuging at 35,000 g (JA-20 108 rotor; Beckman J2-HS) at 4 °C for 30 min. The supernatant was carefully removed, and the protein pellets were washed twice with cold acetone containing 0.07% (v/v) 2-mercaptoethanol and 1 mM phenylmethanesulfonyl fluoride (PMSF) and a third time with cold acetone. Protein pellets were dried and stored at −80 °C or immediately dissolved using protein extraction buffer composed of 8 M urea, 50 mM triethylammonium bicarbonate, pH 8.5, for 1 h at 6 °C under constant shaking. Protein extracts were centrifuged at 19,000 g for 20 min at 10 °C. The supernatant was then collected, and the protein concentration was determined using a protein assay kit (Pierce).
In-solution trypsin digestion and iTRAQ labeling
Total protein (100 μg) was digested and iTRAQ labeled as described elsewhere21. Subsequently, iodoacetamide was added to a final concentration of 50 mM, and the mixture was incubated for 30 min at room temperature in the dark. Then, dithiothreitol (30 mM) was added to the mixture to consume any free iodoacetamide by incubating the mixture for 1 h at room temperature in the dark. Proteins were then diluted by 50 mM Tris, pH 8.5, to reduce the urea concentration to 4 M, and digested with 0.5 μg of Lys-C (Wako) for 4 h at room temperature. After digestion, the solution was further diluted with 50 mM Tris, pH 8.0, to reduce the urea concentration to less than 1 M. The Lys-C digested protein solution was further digested with 2 μg of modified trypsin (Promega) at room temperature overnight. The resulting peptide solution was acidified with 10% trifluoroacetic acid and desalted on a C18 solid-phase extraction cartridge.
Desalted peptides were then labeled with iTRAQ reagents (Applied Biosystems) according to the manufacturer’s instructions. Control samples (proteins extracted from roots of control plants) were labeled with reagent 114; samples from Mn-deficient roots were labeled with reagent 116. Three independent biological experiments with two technical repeats each were performed. The reaction was allowed to proceed for 1 h at room temperature. Subsequently, treated and control peptides were combined and further fractionated offline using high-resolution strong cation-exchange chromatography (PolySulfoethyl A, 4.6 × 100 mm, 5 μm, 200-Å bead). In total, 60 fractions were collected and combined into 24 final fractions. Each final fraction was desalted on a C18 solid-phase extraction cartridge and lyophilized in a centrifugal speed vacuum concentrator. Samples were stored at −80 °C.
LC-MS/MS analysis
Liquid chromatography was performed on a Dionex UltiMate 3000 RSLCnano System coupled to a Q Exactive hybrid quadrupole-Orbitrap mass spectrometer (Thermo Scientific) equipped with a Nanospray Flex Ion Source. Peptide mixtures were loaded onto a 75 μm × 250 mm Acclaim PepMap RSLC column (Thermo Scientific) and separated using a segmented gradient in 120 min from 3 to 30% solvent B (100% acetonitrile with 0.1% formic acid) at a flow rate of 300 nL/min. Solvent A was 0.1% formic acid in water. The samples were maintained at 8 °C in the autosampler. The Orbitrap was operated in the positive ion mode with the following acquisition cycle: a full scan (m/z 350~1600) recorded in the Orbitrap analyzer at resolution R 70,000 was followed by MS/MS of the 10 most intense peptide ions with HCD of the same precursor ion. HCD collision energy was set to 30% NCE. HCD-generated ions were detected in the Orbitrap at resolution 17,500.
Database search
Two search algorithms, Mascot (version 2.4, Matrix Science) and SEQUEST, which is integrated in Proteome Discoverer software (version 1.4, Thermo Scientific), were used to simultaneously identify and quantify proteins. Searches were made against the Arabidopsis protein database (TAIR10 20110103, 27416 sequences; ftp://ftp.arabidopsis.org/home/tair/Sequences/blast_datasets/TAIR10_blastsets/ TAIR10 pep 20110103 representative gene model) concatenated with a decoy database containing the reversed sequences of the original database. The protein sequences in the database were searched with trypsin digestion at both ends and two missed cleavages allowed, fixed modifications of carbamidomethylation at Cys, iTRAQ 4plex at N-terminus and Lys, variable modifications of oxidation at Met and iTRAQ 4plex at Tyr; peptide tolerance was set to 10 ppm, and MS/MS tolerance was set to 0.05 Da. iTRAQ 4plex was chosen for quantification during the search simultaneously. The search results were passed through additional filters, peptide confidence more than 95% (P < 0.05), before exporting the data. For protein quantitation, only unique peptides were used to quantify proteins. These filters resulted in a false discovery rate of less than 5% after decoy database searches were performed. For each of the three biological repeats, spectra were combined into one file and searched. Annotated spectra of proteins and peptides identified in roots and leaves (available at ProteomeXchange Consortium; http://www.ebi.ac.uk/pride; dataset identifier PXD003309).
Statistical analysis
Proteins with significant changes in abundance upon Mn deficiency were selected using a method described by Cox and Mann22. In brief, the mean and SD from the log2 ratios of the defined 11,606 quantified proteins was calculated. Next, 95% confidence (Z score = 1.96) was used to select those proteins whose ratio was significantly different from the main distribution (mean ratio ± 1.96 × SD), protein ratios outside this range were defined as being significantly different at P < 0.05. The cutoff value for down-regulated proteins was 0.75-fold and for up-regulated proteins 1.39-fold.
Glucosinolate analysis
Glucosinolates were analyzed using a protocol described by Glauser et al.23. Samples were collected from roots and leaves of 14-day-old seedlings and frozen immediately in liquid nitrogen. Samples (200 mg) were shattered into powder with TissueLyser II (QIAGEN) and 1 mL of freshly prepared ice-cold methanol:water (70:30, v/v) with sinalbin (Applichem) as internal standard. After homogenization for 30 s with TissueLyser II, samples were incubated for 15 min at 80 °C in a block heater (BL3002, Basic Life), cooled down to room temperature and centrifuged at 3,500 g for 10 min. The supernatant was transferred to appropriate vials for UPLC-qTOF analysis. UPLC-qTOF-MS analysis was performed on an ACQUITY UPLC (Waters, Manchester, UK) and a SYNAPT G1 High Definition Mass Spectrometry (Synapt G1 HDMS) System (Waters) with an electrospray ionization interface, ion-mobility and time-of-flight system. Samples were separated on an ACQUITY CSH C18 column (2.1 × 100, 1.7 μm; Waters). The mobile phase consisted of 0.05% formic acid in 2% acetonitrile (solution A) and 0.05% formic acid in 100% acetonitrile (solution B). The elution gradient was as follows: starting with 1% B for 2 min, 1–45% B for 6 min, 45–99% B for 1 min, holding at 99% B for 1 min, 99–1% B in 0.1 min and then holding at 1% B for 0.9 min. The flow rate was 0.4 mL/min, and the column temperature was set to 25 °C. The injected volume was 2 μL. Spectra were collected in the negative ionization and V mode setting. The electrospray capillary voltage was set to 2.5 kV and the cone voltage was set to 40 V. The source and desolvation temperatures were 80 °C and 250 °C. The desolvation and cone gas flow rates were 800 L/h and 50 L/h. A lock mass calibration of sulfadimethoxin at a concentration of 0.05 mg/L in water:methanol (50:50, v/v) was introduced by HPLC pump (LC-10ATVP, Shimadzu, Japan) and split the 60 μL/min to lock spray interface. The MS range acquired was 50–1200 m/z with 0.2 sec/scan in the centroid mode. For data analysis, the acquired mass data were imported to Markerlynx (Waters) within the Masslynx software (version 4.1) for peak detection and alignment. The retention time and m/z data for each peak were determined by the software. All data were normalized to the summed total ion intensity per chromatogram.
Results
Mn deficiency-induced changes in gene activity
Plants grown on Mn-free media showed reduced growth, pronounced leaf chlorosis and significantly reduced Mn levels (Fig. 1). It is important to note that manganese-deplete medium was prepared with plant cell culture tested agar to avoid Mn contaminations introduced by other gelling agents. To identify Mn-responsive genes in Arabidopsis roots, we first surveyed transcriptional profiles of Mn-sufficient and Mn-deficient plants by RNA-seq. Experiments were performed in triplicates with more than 12 million unique reads per library (37.7 and 37.4 million unique reads for +Mn and −Mn plant, respectively). With this approach, we identified a total of 25,318 transcripts of which 22,385 were detected in all three replicates, a subset that we refer to as consistently detected transcripts (22,065 in Mn-sufficient roots and 21,866 in Mn-deficient roots; Fig. 2). To exclude noisy genes that are likely of minor importance for the processes under study, we defined biologically relevant expression based on data distribution. Only transcripts with a RPKM value larger than the square root of the median of the distribution of the RPKM values of all detected genes ( 10.95 = 3.31) were considered for further analysis. Applying this filter yielded a subset of 17,185 genes that were defined as robustly expressed. A subset of 5,173 genes was consistently detected but at low expression levels and was not further considered here (Fig. 2c).
10.95 = 3.31) were considered for further analysis. Applying this filter yielded a subset of 17,185 genes that were defined as robustly expressed. A subset of 5,173 genes was consistently detected but at low expression levels and was not further considered here (Fig. 2c).

Phenotype of Mn-deficient plants.
(A) Arabidopsis plants after 12 days of growth on Mn-replete (+Mn) and Mn-deplete (−Mn) media. (B) Mn concentrations in shoots of +Mn and −Mn plants. Error bars show standard deviations from the mean of three experimental runs.

Detection of mRNAs by RNA-seq.
(a,b) Detected transcripts in three independent experiments with Mn-sufficient (+Mn) or Mn-deficient (−Mn) Arabidopsis roots. (c) Total, consistently and robustly detected transcripts in roots of both growth types.
To gain a comprehensive picture of the Mn deficiency responses, we complemented the catalog of expressed transcripts with a proteomic survey using the iTRAQ methodology. iTRAQ-labeled peptides were analyzed by liquid chromatography in combination with tandem mass spectroscopy on a Q Exactive hybrid quadrupole-Orbitrap mass spectrometer, using the Mascot and SEQUEST search algorithms to identify the proteins. With this approach, we identified a total of 22,843 proteins, of which 11,606 could be consistently and unambiguously determined in at least two out of three parallel runs. Read numbers (RNA-seq) and fold-changes (iTRAQ) for all detected transcripts and proteins are given in Supplemental Table S1.
Detection of differentially expressed transcripts and proteins
To prioritize genes with potentially high significance for the acclimation to Mn deficiency, we defined transcripts with a ∆RPKM between the two growth conditions greater than the median of the distribution of the RPKM values of all robustly expressed genes (16.3) and a P-value < 0.05 (Student’s t-test) as differentially expressed (DE transcripts). Applying these filters yielded 654 DE transcripts (Fig. 3). Since the quantification of protein abundance determined by the iTRAQ technology is defined by the ratio between Mn-deficient and Mn-sufficient plants and not by absolute values, we applied a Z-test (P < 0.05) to define differentially expressed proteins. Only proteins with abundance changed in the same direction in all three biological replicates (either increased or decreased abundance in response to Mn deficiency) were considered. These criteria yielded a subset of 338 DE proteins with significantly altered abundance upon Mn deficiency out of a total of 11,606 identified proteins that could be quantified (Fig. 3).

Detection of differentially expressed genes and proteins.
Genes that were statistically relevant (P < 0.05) and with a ∆RPKM between Mn-deficient and Mn-sufficient plants greater than the median of the RPKM values of all robustly expressed genes were referred to as differentially expressed (DE transcripts). For proteins, a change in the abundance in the same direction in all three biological replicates and a P-value < 0.05 (Z-test) was used as criteria to define differential expression (DE proteins).
Concordance of mRNA and protein abundance
A comparison of DE transcripts and DE proteins revealed a relatively small overlap of 48 genes that were differentially expressed between Mn-sufficient and Mn-deficient plants at both levels (Fig. 3; Table 1; Supplemental Table S2). Discordance of mRNA and protein expression may result from differences in mRNA/protein synthesis and stability or may be caused by other post-transcriptional processes. Notably, only 14% of the differentially expressed proteins were accompanied by a cognate transcript that was changed in the same direction, indicative of a large contribution of post-transcriptional regulation of gene activity (Fig. 3). Due to the unbiased nature of the RNA-seq technology it is unlikely that a larger fraction of differentially expressed mRNAs was not detected. As anticipated from this small overlap, transcript and protein abundance was only weakly related to each other when all detected mRNAs and proteins were considered (R = 0.19; Fig. 4a). When DE transcripts/DE proteins were used as input without considering whether the cognate partner was differentially expressed, a moderate correlation (R = 0.53) was observed (Fig. 4b). The correlation coefficient was significantly increased when only the 48 genes that constitute the overlap of the DE mRNAs/proteins were analyzed, i.e. in the population in which both partners were defined as differentially expressed (R = 0.87; Fig. 4c). Thus, it appears that the two data sets are highly discordant except for those genes that were differentially expressed at both the protein and at the transcript level. If this is the case, most (~90%) of the changes in protein abundance can be explained by changes in gene transcription. The data also allow the conclusion that most genes do not follow the anticipated pattern of parallel change of mRNA and protein abundance. In the majority of cases, the change in either transcript or protein abundance was not associated with changes of the cognate partner.

Concordance of mRNA and protein expression.
(a) Pearson correlation of all detected mRNAs and proteins. (b) Concordance calculated with DE transcripts/proteins independent of the regulation of the cognate partner. (c) Concordance of genes/protein pairs for which both partners were differentially expressed.
The ‘Manome’ is regulated at different layers
Genes that were differentially expressed between Mn-sufficient and Mn-deficient plants could be categorized into: 1) genes for which a simultaneous change in transcript and protein abundance in the same direction was observed; 2) post-transcriptionally regulated genes, i.e. genes that were only regulated at the protein level or transcript and protein were regulated in opposite directions, and 3) genes that were solely regulated at the transcriptional level without changes in the corresponding protein. Since changes in the activity of genes from the first group can be tracked at two different levels and thus have low gene expression noise, we defined these genes as being robustly Mn-regulated. From the 48 genes in this group (Supplemental Table S2), only few have been associated with Mn homeostasis. NRAMP1, the main route of entry for Mn from the soil7, was moderately up-regulated in roots of Mn-deficient plants both at the mRNA and protein level. Also CHLORIDE CHANNEL A (CLCA), mediating the transport of nitrate into the vacuole24, was up-regulated at both levels. The majority of robustly Mn-regulated genes was, however, down-regulated in response to Mn deficiency. Most of the genes with reduced transcript abundance are related to photosynthesis (PSAK, PSAN, PSAE-1, PSAD-1, PSBQ-2, PSBO-2, CAB3) or are involved in the response to hypoxia (ADH1, PDC1, PCO1, HB1, At4g27450). Also, expression of the metallothionein MT2B was reduced upon Mn deficiency.
A subset of 290 proteins was classified as chiefly or entirely post-transcriptionally regulated (Supplemental Table S2). Interestingly, in this group a more pronounced regulation of the protein abundance was observed, spanning a range from 0.5-fold down- to 6.8-fold up-regulation, while that of the robustly Mn-regulated genes was much narrower (0.41 to 1.78-fold). Several up-regulated genes in this group are related to translation (EIF4A-III, EIF4A-2, ERF1-1, EF1B, At2g45030), mRNA stability (PUM7), mRNA modification (At1g61470), or mRNA splicing (SC35, LUC7, PRP38, At2g16940, At2g29210). Splicing-related genes were regulated in a complex pattern; while the Arabidopsis ORTHOLOG OF HUMAN SPLICING FACTOR (SC35) was up-regulated upon Mn deficiency, the SC35-like splicing factors (SCLs) SCL30 and SCL30A showed reduced abundance in roots of Mn-deficient plants, suggesting Mn deficiency-induced alterations in the composition of the splicing machinery. Several translation-associated proteins were increased in abundance when plants were grown under conditions of Mn-deficiency. Notably, none of the translation-associated gene was transcriptionally regulated by Mn deficiency. Similar to what has been observed for MT2B, the metallothionein MT3 was post-transcriptionally down-regulated.
Changes in the abundance of histone-related proteins indicate that Mn deficiency also induces epigenetic modifications. For example, the H2A histone protein HTA10 showed increased abundance in Mn-deficient plants. While a specific function of HTA10 has not yet been inferred, it is of interest to note that the promoter activity of the coding gene was found to be sharply restricted to root cap cells in the root apical25. In contrast, the H2B protein HTB4 was less abundant when plants were grown on Mn-deplete media. The consequences of these changes in H2A/H2B dimer composition remain to be elucidated.
For the third and largest group of DE genes (605 genes; Supplemental Table S2), transcriptional changes were not translated into changes in protein abundance. This may be caused by a lack of reliable detection of the cognate protein or by a lack of detectable change in protein abundance. For approximately one-third of the transcripts in this group (218 mRNAs) proteins were identified and quantified, indicating that in a large fraction of the differentially expressed mRNAs transcriptional changes were disengaged from protein-level regulation. The most pronounced up-regulation was observed for the beta-glucosidase BGLU23 (PYK10), a member of myrosinase protein family that cleaves the glucose group from glucosinolates. The remaining molecule is then converted to isothiocyanate, nitrile or thiocyanate, which are the active substances that function in defense responses to herbivores. Also, the gene encoding the PYK10-binding protein PBP1, an activator of BGLU23, was strongly up-regulated upon Mn deficiency. The most strongly down-regulated transcript was the glyceraldehyde-3-phosphate dehydrogenase subunit C2 (GAPC2); transcripts of the other subunit, GAPA1, showed also reduced abundance in Mn-deficient plants. Decreased transcripts were also observed for the sucrose synthases SUS1 and SUS4 and for several genes involved in photosynthesis. These changes indicate that the aerobic sucrose catabolic pathway is dramatically down-regulated when Mn is unavailable.
Co-expression analysis reveals novel modules with putative functions in the manganese deficiency response
Functional information from differentially expressed genes can be inferred from the analysis of relationships of these genes (‘guilt by association’). To include the information of proteins that accumulate differentially upon Mn deficiency, a chimeric co-expression network was constructed from DE transcripts and DE proteins by using the MACCU software and publically available microarray experiments as a database26,27. Only DE genes that were co-expressed with a Pearson correlation coefficient greater than 0.75 were included in the network. This chimeric co-expression network comprises 366 DE transcripts and 129 DE proteins (with 33 of them regulated at both levels), which could be divided into several modules comprising transcripts/proteins of putatively related functions (Supplemental Table S3, Supplemental Fig. S1).
Sub-cluster C0 contains 105 transcripts and proteins related to photosynthesis, all of which were strongly down-regulated. At the protein level, the most strongly decreased abundance was observed for AtCg00710, a component of the photosystem II oxygen evolving core, PSBH. No corresponding transcript was detected for this protein. Due to the crucial function of Mn in PSII, we anticipated repression of genes related to photosynthesis upon Mn deficiency. It was, however, surprising that this response was evident in non-photosynthetic roots. It should be noted that the roots were exposed to light, resulting in the robust expression of PS genes in both leaves and roots, albeit with lower expression levels in the latter.
Another module in the co-expression network (C1) mainly contains down-regulated genes related to hypoxia such as HEMOGLOBIN1 (AHB1) and 3 (GLB3), ADH1, the hypoxia-responsive transcription factors ERF71 and ERF73, and genes encoding proteins involved in ethylene production such as ACO1. Also, the two sucrose synthase genes SUS1 and SUS4 were strongly down-regulated upon Mn deficiency.
The central part of the cluster C2 in this network comprises 56 nodes that mostly represent up-regulated genes (Fig. 5; Table 1). This cluster contains the Mn transporter NRAMP1 and several transcripts/proteins related to glucosinolate metabolism that were all strongly up-regulated upon Mn deficiency, including BGLU23. To gain further insights into a putative involvement of glucosinolates in the Mn deficiency response, glucosinolate-related genes were surveyed among the DE genes. A metabolic pathway depicting the role of differentially expressed proteins/transcripts in glucosinolate biosynthesis and breakdown is shown in Fig. 6a, and expression changes are given in Table 1. For two of the genes encoding enzymes related to the breakdown of glucosinolates, BGLU23 and TGG1, RNA-seq data were confirmed by qRT-PCR (Fig. 6b). Interestingly, several proteins that belong to families that participate in the pathway (e.g. glutathione transferases and glycosyl transferases) were found in this cluster, indicating a possible involvement of these transcripts/proteins in Mn deficiency-induced changes in glucosinolate metabolism.

Glucosinolate biosynthesis in Arabidopsis.
(a) RNAseq and iTRAQ results in the context of the glucosinolate biosynthesis pathway. Genes labeled in black and red increased significantly at the mRNA and protein level, respectively. (b,c) qRT-PCR results of BGLU23 and TGG1 expression levels in roots (b) and shoots (c). (d) UPLC analysis of glucosinolates in roots and leaves. Error bars represent standard deviations of three independent experiments.
Metabolic analysis reveals dynamic changes in glucosinolate levels in response to Mn deficiency in leaves and roots
To investigate whether Mn deficiency alters glucosinolate biosynthesis, we analyzed the concentration of several major glucosinolates by UPLC-qTOF-MS in a targeted approach. Since glucosinolates can be synthesized in both roots and shoots28, the analysis was performed in both organs. In addition, we compared the induction of BGLU23 and TGG1 by Mn deficiency in roots with the expression changes in leaves via qRT-PCR. Similar to what has been observed for roots, the expression of both genes was found to be up-regulated in leaves (Fig. 6c). Unexpectedly, in roots, all glucosinolates under investigation showed either no change or decreases in their concentration. In particular, we observed strong decreases in the concentration of the aliphatic glucosinolates 4-, 5- and 6-methylsulfinylbutylglucosinolate (4MSOB, 5MSOP and 6MSOH) (Fig. 6d). In shoots, an opposite pattern was observed, with increases in most of the compounds under investigation (Fig. 6d), including aliphatic, aromatic and indolic glucosinolates. It should be noted that the compounds analyzed here are metabolic intermediates, and increased degradation of glucosinolates could lead to relative decreases in metabolites within the pathway. Rapid degradation, secretion of biologically active compounds, and/or export of glucosinolates to aboveground plant parts could be the reason for the lack of detectable increase in the surveyed glucosinolates in roots. A re-distribution of glucosinolates from roots to leaves would explain the similar changes in transcript levels of BGLU23 and TGG1 in the two organs, which stands in contrast to the observed changes in glucosinolate levels.
Deficiencies in Mn and Fe induces antagonistically-regulated processes
A comparison with previously conducted transcriptional and proteomic surveys on roots of Fe-deficient plants28,29 showed that several genes that are induced by Fe deficiency were down-regulated in roots of Mn-deficient plants. This subset includes genes involved in Fe mobilization such as the P-type ATPase AHA2 or in the chelation of Fe (NAS1), and several genes encoding proteins with unknown function that were strongly induced by Fe deficiency. For example, IRON-RESPONSIVE PROTEIN 3 (At2g14247), dramatically up-regulated in both roots and leaves of Fe-deficient plants28,30, was strongly down-regulated by Mn deficiency. Also up-regulated in response to Fe deficiency both at the transcript and protein level but down-regulated at the protein level Mn-deficient plants was the oxygenase superfamily protein At3g12900. At3g12900 is closely co-expressed with IRT1 and the Myb-type transcription factors MYB10 and MYB72, critical regulators of Fe uptake from alkaline soils that limit the mobility of Fe31, which may indicate a role of At3g12900 in Fe acquisition from Fe pools of low availability.
Manganese deficiency was shown to increase root hair length and density, probably to compensate for the low mobility of Mn15,32. Several genes with putative or validated roles in root hair development showed increased expression under Mn-deficient conditions but decreased expression in Fe-deficient plants (DER1, AGP14, FLA9). AGP14 was previously associated with root hair elongation26 and may be critical in the formation of longer root hairs under Mn-deficient conditions. Iron deficiency does not increase root hair length33.
The most highly induced gene in Mn-deficient plants, BGLU23 (PYK10), was the most down-regulated gene in Fe-deficient plants. Also the expression of a partner of BGLU23, PBP1, followed a similar pattern. Moreover, three genes involved in glucosinolate breakdown, (CWINV1, At1g62660 and the nitrile specifier NSP1), the glucosinolate biosynthesis gene MAM3, and the plasma membrane-localized transporter ABCG34, were oppositely regulated by Mn and Fe. Together these data suggest that the acquisition responses of both nutrients are controlled in an antagonistic manner.
Discussion
Despite the importance of Mn for plant growth, limited information is available regarding the proteins involved in the acquisition of Mn from the soil solution and the mechanisms by which plants re-calibrate cellular Mn homeostasis. Also, the signaling cascades that control these processes await elucidation. Several transporters can catalyze the movement of Mn across membranes14,34, but except for the transporters of the Nramp family (AtNRAMP1, 3 and 4 in Arabidopsis;7,9,10) little is known regarding their physiological relevance for cellular Mn homeostasis under conditions of low Mn availability. In the present investigation, NRAMP1 was found to be differentially expressed between Mn-sufficient and Mn-deficient plants at both the transcript and protein level, supporting its critical importance for Mn uptake. Induction of NRAMP1 upon Mn deficiency was moderate though and not comparable with the dramatic increase in IRT1 transcript abundance upon Fe deficiency. Also of note, pronounced changes in Mn efficiency between barley genotypes were not evident in solution culture35, indicating that other factors such as root-soil or root-microbe interaction dictate Mn uptake rate from pools of low phyto-availability and thus Mn efficiency.
Concordance of mRNA and Protein Expression
Regulation of gene expression is largely decoupled from protein dynamics, a phenomenon that has been observed through all types of organisms, including plants16. Several attempts have been undertaken to estimate how much of the changes observed at the nucleotide level are translated into proteins36,37. These efforts were complicated by several factors. Firstly, in many cases transcriptomic and proteomic profiling was carried out in different laboratories, introducing a substantial bias by variations in experimental conditions. A second technical bias is introduced by the fundamental differences in extraction and detection of transcripts and proteins, which differ in their levels of sensitivity and measurement error. Moreover, proteomics have lagged behind transcriptomics in terms of coverage, leaving the majority of identified mRNAs of protein coding genes without cognate proteins in most surveys. Finally, a highly dynamic interplay between mRNA and proteins complicates all predictions from one level to another. In the present study, only for 14% of the differentially expressed proteins associated transcripts were detected that were regulated in the same direction. Considering the correlation of mRNA and protein abundance for those proteins that were detected as cognate pairs, an overall concordance of the two layers of approximately 10% can be estimated, a value that matched that estimated in a recent study on Fe-deficient plants29.
Unexpectedly, from the three groups of differentially regulated genes, the group that we defined as ‘robustly regulated by Mn’ was the smallest (48 transcripts/proteins). The second group, comprising chiefly post-transcriptionally regulated proteins, was not only much bigger (296 proteins), but also contained proteins that showed more pronounced changes in expression ratio. While the reason(s) for this observation cannot be deduced from the data at hand, it is of interest to note that in this group proteins with functions in translation, mRNA processing, and histone modifications were overrepresented, indicative of a largely unexplored regulatory layer that may have a strong impact on Mn homeostasis. This (transcriptionally) hidden layer may affect the abundance of proteins independent of their transcriptional level, thereby contributing to the gap between mRNA and protein abundance.
The largest group comprised genes that were solely transcriptionally regulated (605 genes). Included in this subset are: firstly, genes with noisy expression that might be beneficial in terms of system stabilization and plant fitness38; secondly, genes for which the detection or quantification of the cognate protein was obscured for technical reasons, and, thirdly, transcripts for which the cognate protein is unstable or inefficiently translated, necessitating a pronounced regulation at the transcriptional level to stabilize the proteomic readout. While a general conclusion about the relative physiological importance of these groups is difficult to be drawn from the current data set, it becomes obvious that transcription is intrinsically stochastic and information regarding the transcripts and proteins involved in a given process and their regulatory patterns can only be achieved by an integrative approach accessing different levels of gene activity. Also, it appears that only a relatively small part of Mn deficiency-induced changes in gene expression can be attributed to a regulation at the transcriptional level.
Production of glucosinolates: an integral part of the Mn acquisition strategy?
Glucosinolates are a class of sulfur- and nitrogen-containing organic compounds derived from glucose and amino acids, which are involved in plant defense. Glucosinolates are formed by an aminoacidic origin radical and a glucose moiety, linked by a thioesther bond. They occur as secondary metabolites in almost all species of the order Brassicales. The reasons for the strong regulation of glucosinolate-associated genes is not entirely clear, but the pronounced changes suggest auxiliary roles of glucosinolates in the Mn deficiency response. In roots, genes involved in the biosynthesis and degradation of glucosinolates were found to be up-regulated at the mRNA and protein level. Mn deficiency-induced up-regulation of two genes (out of two tested), BGLU23 and TGG1, was also observed in leaves, which stands in contrast to the opposite pattern of glucosinolate accumulation in the two organs. Thus, the increase in glucosinolates in leaves might have partly occurred at the expense of roots, which could export these substances to the aboveground plant parts. Glucosinolates are important for the defense against pathogens and innate immunity39,40,41. It is thus tempting to assume that the increase in the production of glucosinolates and their breakdown products is an adaptive response triggered by increased pathogen susceptibility under conditions of Mn deficiency. This stress-induced decrease in pathogen resistance might be further enhanced by the down-regulation of Fe-responsive genes, in particular those that are related to the production of phenolics, which may lead to impaired lignification42 and, as a consequence, increased disease susceptibility. This supposition is supported by the down-regulation of genes that are important for the uptake of Fe at alkaline pH, which is mainly mediated via the excretion of phenolic compounds30,43,44. While the precise role of glucosinolates needs further elucidation, we suppose that reprograming of glucosinolate metabolism in roots and leaves represents an important and integral part of the response of Arabidopsis to Mn deficiency. A plausible alternative function of glucosinolates could lie in the interaction with the rizhosphere microbiome as a chemotoxine to inhibit bacterial growth and eliminate bacterial competence for available Mn.
Conclusions
In summary, we here provide a near complete inventory of transcripts and proteins that are responsive to Mn. This catalog reveals that several regulatory processes are engaged by Mn deficiency, including changes in chromatin structure, transcription, transcript maturation and stability, as well as alterations in translation efficiency and protein stability. By comparing transcriptomic, proteomic and metabolomic data, we discovered Mn deficiency-induced changes that are hidden when only in one of these levels is interrogated. Our data further reveal massive Mn-specific changes in metabolism, which contrast similar processes responsive to Festarvation.
Additional Information
How to cite this article: Rodríguez-Celma, J. et al. Systems-wide analysis of manganese deficiency-induced changes in gene activity of Arabidopsis roots. Sci. Rep. 6, 35846; doi: 10.1038/srep35846 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Marschner, H. & Marschner, P. Marschner’s Mineral Nutrition of Higher Plants, 3 Ed., Academic Press (2012).
Hebbern, C. A., Pedas, P., Schjoerring, J. K., Knudsen, L. & Husted, S. Genotypic differences in manganese efficiency: field experiments with winter barley (Hordeum vulgare L.) Plant Soil 272, 233–244 (2005).
Nouet, C., Motte, P. & Hanikenne, M. Chloroplastic and mitochondrial metal homeostasis. Trends Plant Sci 16, 395–404 (2011).
Allen, M. D., Kropat, J., Tottey, S., Del Campo, J. A. & Merchant, S. S. Manganese deficiency in Chlamydomonas results in loss of photosystem II and MnSOD function, sensitivity to peroxides, and secondary phosphorus and iron deficiency. Plant Physiol 143, 263–277 (2007).
Husted, S. et al. Manganese deficiency leads to genotype-specific changes in fluorescence induction kinetics and state transitions. Plant Physiol 150, 825–833 (2009).
Schmidt, S. B., Jensen, P. E. & Husted, S. Manganese deficiency in plants: the impact on photosystem II. Trends Plant Sci 21, 622–632 (2016).
Cailliatte, R., Schikora, A., Briat, J. F., Mari, S. & Curie, C. High-affinity manganese uptake by the metal transporter NRAMP1 is essential for Arabidopsis growth in low manganese conditions. Plant Cell 22, 904–917 (2010).
Ihnatowicz, A. et al. Conserved histidine of metal transporter AtNRAMP1 is crucial for optimal plant growth under manganese deficiency at chilling temperatures. New Phytol 202, 1173–1183 (2014).
Ishimaru, Y. et al. (2012) Characterizing the role of rice NRAMP5 in manganese, iron and cadmium transport. Sci Rep 2 (2012).
Lanquar, V. et al. Export of vacuolar manganese by AtNRAMP3 and AtNRAMP4 is required for optimal photosynthesis and growth under manganese deficiency. Plant Physiol 152, 1986–1999 (2010).
Milner, M. J., Seamon, J., Craft, E. & Kochian, L. V. Transport properties of members of the ZIP family in plants and their role in Zn and Mn homeostasis. J Exp Bot 64, 369–381 (2013).
Korshunova, Y. O., Eide, D., Clark, W. G., Guerinot, M. L. & Pakrasi, H. B. The IRT1 protein from Arabidopsis thaliana is a metal transporter with a broad substrate range. Plant Mol Biol 40, 37–44 (1999).
Pedas, P. et al. (2008) Manganese efficiency in barley: identification and characterization of the metal ion transporter HvIRT1. Plant Physiol 148, 455–466 (2008).
Socha, A. L. & Guerinot, M. L. Mn-euvering manganese: the role of transporter gene family members in manganese uptake and mobilization in plants. Front Plant Sci 5, 106 (2014).
Yang, T. J., Perry, P. J., Ciani, S., Pandian, S. & Schmidt, W. Manganese deficiency alters the patterning and development of root hairs in Arabidopsis. J Exp Bot 59, 3453–3464 (2008).
Vogel, C. & Marcotte, E. M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet 13, 227–232 (2012).
Estelle, M. A. & Somerville, C. Auxin-resistant mutants of Arabidopsis thaliana with an altered morphology. Mol Gen Genet 206, 200–206 (1987).
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5, 621–628 (2008).
Kent, W. J. BLAT-the BLAST-like alignment tool. Genome Res 12, 656–664 (2002).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 (2001).
Lan, P. et al. iTRAQ protein profile analysis of Arabidopsis roots reveals new aspects critical for iron homeostasis. Plant Physiol 155, 821–834 (2011).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26, 1367–1372 (2008).
Glauser, G., Schweizer, F., Turlings, T. C. & Reymond, P. Rapid profiling of intact glucosinolates in Arabidopsis leaves by UHPLC-QTOFMS using a charged surface hybrid column. Phytochem Analysis 23, 520–528 (2012).
De Angeli, A. et al. ATP binding to the C terminus of the Arabidopsis thaliana nitrate/proton antiporter, AtCLCa, regulates nitrate transport into plant vacuoles. J Biol Chem 284, 26526–26532 (2009).
Yi, H., Sardesai, N., Fujinuma, T., Chan, C. W., Veena & Gelvin, S. B. Constitutive expression exposes functional redundancy between the Arabidopsis histone H2A gene HTA1 and other H2A gene family members. Plant Cell 18, 1575–1589 (2006).
Lin, W. D. et al. Coexpression-based clustering of Arabidopsis root genes predicts functional modules in early phosphate deficiency signaling. Plant Physiol 155, 1383–1402 (2011).
Rodríguez-Celma, J. et al. The transcriptional response of Arabidopsis leaves to Fe deficiency. Front Plant Sci 4, 276 (2013).
Andersen, T. G. et al. Integration of biosynthesis and long-distance transport establish organ-specific glucosinolate profiles in vegetative Arabidopsis. Plant Cell 25, 3133–3145 (2013).
Pan, I. C., Tsai, H. H., Wen, T. N., Buckhout, T. J. & Schmidt, W. Post-transcriptional coordination of the Arabidopsis iron deficiency response is partially dependent on the E3 Ligases RGLG1 and RGLG2. Mol Cell Proteomics 14, 2733–2752 (2015).
Rodríguez-Celma, J. et al. Mutually exclusive alterations in secondary metabolism are critical for the uptake of insoluble iron compounds by Arabidopsis and Medicago truncatula. Plant Physiol 162, 1473–1485 (2013).
Palmer, C. M., Hindt, M. N., Schmidt, H., Clemens, S. & Guerinot, M. L. MYB10 and MYB72 are required for growth under iron-limiting conditions. Plos Genet. 9, e1003953 (2013).
Ma, Z., Bielenberg, D. G., Brown, K. M. & Lynch, J. P. Regulation of root hair density by phosphorus availability in Arabidopsis thaliana. Plant Cell Environ 24, 459–467 (2001).
Müller, M. & Schmidt, W. Environmentally induced plasticity of root hair development in Arabidopsis. Plant Physiol 134, 409–419 (2004).
Schneider, A. et al. The Evolutionarily Conserved protein photosynthesis affected mutant71 is required for efficient manganese uptake at the thylakoid membrane in Arabidopsis. Plant Cell 28, 892–910 (2016).
Huang, C., Webb, M. J. & Graham, R. D. Manganese efficiency is expressed in barley growing in soil system but not in solution culture. J. Plant. Nutr. 17, 83–95 (1994).
Abreu, R. D., Penalva, L. O., Marcotte, E. M. & Vogel, C. Global signatures of protein and mRNA expression levels. Mol Biosyst 5, 1512–1526 (2009).
Vogel, C. Protein expression under pressure. Science 342, 1052–1053 (2013).
Pilpel, Y. Noise in biological systems: pros, cons, and mechanisms of control. Method Microbiol 759, 407–425 (2011).
Schlaeppi, K., Abou-Mansour, E., Buchala, A. J. & Mauch, F. Disease resistance of Arabidopsis to Phytophthora brassicae is established by the sequential action of indole glucosinolates and camalexin. Plant J 62, 840–851 (2010).
Clay, N. K., Adio, A. M., Denoux, C., Jander, G. & Ausubel, F. M. Glucosinolate metabolites required for an Arabidopsis innate immune response. Science 323, 95–101 (2009).
Buxdorf, K., Yaffe, H., Barda, O. & Levy, M. The effects of glucosinolates and their breakdown products on necrotrophic fungi. PloS One 8, e70771 (2013).
Marschner, H. Mineral nutrition of higher plants. Academic Press, London (1995).
Fourcroy, P. et al. Involvement of the ABCG37 transporter in secretion of scopoletin and derivatives by Arabidopsis roots in response to iron deficiency. New Phytol 201, 155–167 (2014).
Schmid, N. B. et al. Feruloyl-CoA 6′-Hydroxylase1-dependent coumarins mediate iron acquisition from alkaline substrates in arabidopsis. Plant Physiol 164, 160–172 (2014).
Acknowledgements
We thank Thomas J. Buckhout (Humboldt University, Germany) for valuable suggestions and critical comments on the manuscript. Proteomic mass spectrometry analyses and in-gel digestion was performed by the Proteomics Core Laboratory sponsored by the Institute of Plant and Microbial Biology (IPMB) and the Agricultural Biotechnology Research Center (ABRC), Academia Sinica. Libraries for RNA-seq analysis were constructed by the Affymetrix Gene Expression Service Laboratory (http://ipmb.sinica.edu.tw/affy/) supported by Academia Sinica. We acknowledge the Small Molecule Metabolomics Core Facility, sponsored by IPMB and the Scientific Instrument Center, Academia Sinica, for providing technical assistance and support with data acquisition and analysis of Synapt HDMS experiments. We further thank Wen-Dar Lin from the Bioinformatics Core Lab at IPMB for bioinformatics support and the laboratory of Dr. Kuo-Chen Yeh (ABRC) for experimental and technical help with ICP-OES analysis. The study was supported by a bilateral grant from the Ministry of Science and Technology (MoST) and the Agence Nationale Recherche (ANR) to C.C. and W.S. (grant Nr. 101-2923-B-001-001-MY3). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD003309.
Author information
Authors and Affiliations
Contributions
J.R.-C. contributed to the design of the study, carried out experiments, interpreted data and contributed to the manuscript. Y.-H.T. and Y.-C.W. conducted experiments and data analysis. T.-N.W. analyzed data. C.C. and W.S. conceived and designed the study. W.S. participated in the analysis and interpretation of the data and wrote the manuscript. All authors read and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Rodríguez-Celma, J., Tsai, YH., Wen, TN. et al. Systems-wide analysis of manganese deficiency-induced changes in gene activity of Arabidopsis roots. Sci Rep 6, 35846 (2016). https://doi.org/10.1038/srep35846
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep35846
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
