
Abstract
Contemporary improvements in the type II clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 (CRISPR/Cas9) system offer a convenient way for genome editing in zebrafish. However, the low efficiencies of genome editing and germline transmission require a time-intensive and laborious screening work. Here, we reported a method based on in vitro oocyte storage by injecting oocytes in advance and incubating them in oocyte storage medium to significantly improve the efficiencies of genome editing and germline transmission by in vitro fertilization (IVF) in zebrafish. Compared to conventional methods, the prior micro-injection of zebrafish oocytes improved the efficiency of genome editing, especially for the sgRNAs with low targeting efficiency. Due to high throughputs, simplicity and flexible design, this novel strategy will provide an efficient alternative to increase the speed of generating heritable mutants in zebrafish by using CRISPR/Cas9 system.
Similar content being viewed by others

Introduction
Loss-of-function is an important approach for in vivo functional study of a gene of interest in transgenic animals. The RNA-guided clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 nuclease system has been proved to be another precise and efficient genome editing technique after zinc-finger nucleases (ZFNs) and transcription activator like effector nucleases (TALENs) in recent years1,2,3,4,5. This promising tool has been successfully applied for genome editing in many model organisms, including zebrafish, mice, dog, pig, Drosophila, C. elegans and primates, etc6,7,8,9,10,11,12. No matter which gene editing tool is used, low efficiencies of genome editing and germline mutant transmissions usually lead to labor intensive and time consuming screening work to acquire high-throughputs. For instance, zebrafish founders carried 64–81% ntl mutations by ZFNs technique, but among them only 5–32% ntl founders were resulted in heritable germline mutations13. Although TALENs have been proved to be a powerful technique for genome editing, the mutation frequency of TALENs still has room to improve14. Recently, CRISPR/Cas9 system has emerged as an efficient alternative for genome editing due to high-throughputs and broad spectrum of applications. However, the efficiency of germline transmission by CRISPR/Cas9 needs further improvements. For example, Hruscha et al. reported 50.5% mutation frequencies in zebrafish founder embryos, but only about 11% of mutations inherited to their progeny15. Varshney et al. obtained an average germline transmission rate of 28% from 162 target sites in zebrafish genome16.
To improve the efficiencies of genome editing and germline transmission, researchers explored many different approaches. Maruyama et al. promoted the homology-directed repair (HDR) and increased the genome editing efficiency of CRISPR/Cas9 system by inhibiting non-homologous end joining(NHEJ)17. Zhang et al. established a highly efficient CRISPR mutagenesis system by introducing microhomology-mediated end joining (MMEJ) with very short (approximately 35 bp) homology arms18. In addition, Yan and Schwartz used optimal promoters to drive the expression of Cas9 and sgRNAs to obtain highly efficient genome modifications19,20. In addition, Carrington et al. reported an easy fluorescent PCR-based method to pre-screen sgRNAs for target-specific activity21. Moreover, Renaud et al. also improved genome editing efficiency by using phosphorothioate-modified oligonucleotides22. Fujii et al. and Sung et al. found that Cas9 protein induced genome modifications more rapidly than Cas9 capped RNA23,24 and codon-optimized Cas9 protein improved higher knockout efficiency than original one25,26. Nevertheless, the subsequent screening for germline transmission of induced mutations could be time-consuming and labor intensive for the sgRNAs with low targeting efficiencies15,27. In 2014, Dong et al. reduced the screening time for mutants by labelling the zebrafish primordial germ cells28. However, the improvement for the specific targeting efficiency remained below expectations.
A valuable technique of ovum preservation has been successfully applied in mammals29,30,31, including humans32,33,34. Recently, the preservation of fish oocytes in vitro was also reported35,36. Goetz and his colleagues used modified cortland medium to store trout eggs for at least 2 days37. Ubilla et al. reported that in vitro storage of rainbow trout eggs in their coelomic fluid for 96 h caused a decline in the quality of eggs and reduced their capacity to develop into larvae38. The preferable preservation technology of zebrafish oocytes was established in 2008 by Seki et al., which contained 90% Leibovitz L-15 medium at pH 9.0, containing 0.5 mg mL−1 BSA and 1 ug mL−1 17α-20β-Dihydroxy-4 Pregnen-3-one (DHP). In their findings, they kept the zebrafish oocytes alive from stage III to mature in vitro, nevertheless the hatching rate remained low39,40.
Here, we reported a modified in vitro oocyte storage strategy to increase the efficiency of genome editing by CRISPR/Cas9 system in zebrafish. This novel technique with in vitro oocyte injection prior to their storage improved the efficiencies of gene targeting and germline transmission. Contrary to the conventional genome editing approaches, this method utilized the inherent translation system to impel the oocytes to produce Cas9 proteins in advance, which greatly increased the efficiency of genome editing. Undoubtedly, it also improved the efficiency of germline transmission. Therefore, our novel technique will increase the speed of generating heritable mutants of zebrafish using CRISPR/Cas9 system.
Materials and Methods
Ethics Statement
All experiments in this study were in accordance with the “Guide for the Care and Use of Laboratory Animals” (Eighth Edition, 2011. ILARCLS, National Research Council, Washington, D.C.) and were approved by the Animal Care and Use Committee of Chongqing in China and by the Institutional Animal Care and Use Committee of Chongqing Institute of Green and Intelligent Technology, Chinese Academy of Sciences (Approval ID: ZKCQY0068).
Zebrafish husbandry
Adult fish and embryos of AB strains were raised and maintained as described by Westerfield et al.41,42. Fish were kept in the automatic water cycle system on a 14 h light: 10 h dark period. The adult female and male zebrafish were maintained at 28 °C in separate tanks and mated once a week.
CRISPR guide RNAs design and Cas9 capped RNA synthesis
Cas9 target sites were selected in exons across the genome and designed with an online tool, ZIFIT Targeter (http://zifit.partners.org/ZiFiT/ChoiceMenu.aspx). The plasmid pXT7-Cas9 was linearized by XbaI6. Cas9 capped RNA was synthesized using Ambion mMESSAGE mMACHINE T7 Transcription Kit (Ambion, USA). The sgRNAs transcription templates were prepared by PCR with T7-targetsite-F primers and a universal reverse primer gRNA-R (Table 1)43. The sgRNAs PCR products were purified by TIANquick Midi Purification Kit (TIANGEN, China) and then transcribed with MAXIscript T7 Kit (Ambion, USA). RNAs were purified by MicroElute RNA Clean-Up Kit (OMEGA, USA). The RNA quality and concentration were analyzed by electrophoresis and the NanoDrop2000 spectrophotometer (Thermo Fisher Scientific, France). The efficiencies of sgRNAs were examined by the sgRNAs activity detection kit in vitro (Viewsolid Biotech, China).
In vitro oocyte storage
The general method of in vitro oocyte storage is adapted from Seki et al.39 and Nair et al.40. The oocyte storage medium was 90% Leibovitz’s L-15 medium with L-glutamine (Gibco) and 0.5 mg mL−1 bovine serum albumin (BSA, Sigma-Aldrich) and then pH was adjusted to 9.0 with 10N NaOH. Before oocyte collection, adult zebrafish females and males were maintained in separate spawn tanks overnight. Adult zebrafish females with swollen and elastic abdomen were chosen. Before squeezing the oocytes, zebrafish females were treated with 0.2 mg mL−1 ethyl 3-aminobenzoate methanesulfonate salt, then gently pressed the abdomen under the microscope. Oocytes were harvested from 1–2 females and scattered in the oocyte storage medium.
Oocyte micro-injection
The mature oocytes were collected for micro-injection by using a micro-injector (Eppendorf, Germany). The injected reagents were diluted in 0.2 M KCl. The final concentrations of Cas9 RNAs and sgRNAs were 300 ng μL−1 and 30~50 ng μL−1, respectively. One nL solution containing 300 pg Cas9 capped RNA and 30~50 pg sgRNAs was micro-injected into 1-cell embryo. The oocytes were arranged along the edge of a glass slide in oocyte storage medium before micro-injection (Fig. 1). For gene knock-in experiment, Cas9 capped RNAs, mc4r sgRNAs and the single-stranded mloxP donor DNAs (Table 1) were micro-injected into zebrafish oocytes. After completion of micro-injection, the oocytes were cultured in the storage medium to avoid light for 30 min.

The schematic diagram of microinjection and oocyte storage.
In vitro fertilization
Injected oocytes were fertilized after 30 min. Sperm solution was produced by shearing the testes from two AB male fish and grinding in 200~300 μL cortland solution (NaCl 7.25 g L−1; KCl 0.38 g L−1; CaCl2 0.162 g L−1; NaHCO3 1.0 g L−1; NaH2PO4 0.41 g L−1; MgSO4 0.23 g L−1; glucose 1.0 g L−1). The quality of sperm was judged by the color of the sperm solution. The color of mature sperm was milky white. The cortland solution was kept on ice for at least 1 h before use. During this experiment, the sperm solution was kept on ice all the time. The storage medium was removed from the culture dish and 20~30 μL sperm solution was added directly on the top of the oocytes, followed by a few drops of fresh water with a plastic dropper. After 1~2 min, the culture dish was washed gently with fresh water.
Genomic DNAs isolation
Genomic DNAs were extracted by using the CTAB extraction method44. For the DNAs extraction of embryos and oocytes, one embryo and five oocytes were incubated in 40 μL and 60 μL lysis solution (100 mM Tris-Hcl pH 8.5, 0.5 M EDTA, 10% SDS, 5 M NaCl, 20 mg mL−1 Proteinase K) at 55 °C for 2 h. For adult zebrafish DNAs extraction, partial tails were lysed in 100 μL lysis solution and incubated at 55 °C for 3 h. Then, DNAs were purified by chloroform, isopropanol and 75% ethanol. Finally, 20~30 μL double distilled water was added into each sample.
Gene editing assay and sequencing analysis
The sgRNAs binding site and the primers used for T7E1 assay are shown in Table 1. PCR mixture was prepared according to the manual of TransFast Taq DNA Polymerase (Transgen Biotech, China). The PCR program was set as: 95 °C 2 min, 35 cycles of (95 °C 5 s, mc4r 52 °C, mpv17 55 °C, mstna 55 °C, mc3r 55 °C, mrap2b 60 °C, 15 s and 72 °C 10 s) and a final extension at 72 °C for 5 min (BIO-RAD, Singapore). 8.5 μL PCR products were added into 1 μL T7 endonuclease I buffer (Viewsolid Biotech, China) to perform an annealing process (95 °C for 5 min, 95 °C to 75 °C ramping at 0.1 °C s−1, 75 °C to 16 °C ramping at max rate and holding at 16 °C for 2 min) for heteroduplex formation. After annealing, products were treated with 0.5 μL T7 endonuclease I for 15 min at 37 °C and analyzed on 1.5% agarose gels. Gels were observed and analyzed by using a gel-imaging system (JUNYI, Beijing, China). To detect mloxP knock-in, larvae DNAs at 72 hpf were isolated and PCR was performed with mc4r T7E R matching the mc4r sequence and mloxP R matching the sequence of the mloxP donor sequence (Table 1). To further confirm the success of gene editing, about 2 μL above all PCR products were cloned into the pMD-19T vector (Takara, Japan) for sequencing analysis.
Statistical analysis
For statistical analysis, two-tailed unpaired Student’s t-test was performed by using SPSS software45. P values < 0.05 were considered statistically significant. Each experiment was performed independently for three times.
Results
An effective storage medium for oocyte preservation
The oocyte preservation and in vitro maturation have been investigated for many years. On the basis of previous findings, we compared the effects of three oocytes storage media: hepes-cortland medium, 90% L-15 medium at pH 9.0 with BSA and without BSA. Due to the difficulty in obtaining the coelomic fluid of zebrafish, oocyte preservation in coelomic fluid was not suitable here. Thus, we collected oocytes and stored them in three different preservation media that are mentioned above. The results showed that the mature oocytes stored in 90% L-15 medium with BSA at pH 9.0 remained stable after 2 h. The hepes-cortland medium showed almost the same effect as the 90% L-15 medium without BSA at pH 9.0 and induced the formation of a cavity between the oocyte shell and vitelline membrane (Fig. 2).

The preservation effects of three different oocyte storage media.
(A) Fresh water, (B) Hepes-cortland medium, (C) 90% L-15 medium without BSA at pH 9.0, (D) 90% L-15 medium with BSA at pH 9.0. The storage time in three oocyte storage media was 30 min, 60 min and 120 min, respectively. The cavities between the shell and vitelline membrane and the animal pole were indicated with red and blue arrows.
The hatching rates in vitro
To find a suitable storage time, the effects of storage time on hatching rates were investigated. The results showed that there was a significantly decreasing trend for oocyte hatching rates with increase in oocytes storage time in vitro (Fig. 3). The hatching rates dramatically declined, when the storage time was longer than 60 min (Fig. 3A). However, the egg hatching rates were more than 80%, if the storage time was less than 30 min (Fig. 3B). As expected, the oocytes stored in hepes-cortland medium and 90% L-15 medium without BSA failed to fertilize in vitro. In addition, a longer storage time resulted in a higher deformity rate in the 90% L-15 medium with BSA at pH 9.0 (data not shown). Our results indicated that Seki’s medium was conducive to oocytes storage, but DHP was unnecessary. However, BSA was proved as a critical component for the oocytes storage medium.

Effects of storage time on hatching rates and embryonic development in 90% L-15 medium with BSA at pH 9.0.
(A)The relationship between the hatching rate and the storage time in vitro. The oocytes were stored in oocyte storage medium for different time and then fertilized in vitro. The hatching rates were recorded after 72 h. (B) The embryos developed normally when oocytes were stored for 30 min in vitro. The oocytes were stored in oocyte storage medium for 30 min and then fertilized in vitro. The embryonic development were observed from 0.5 h to 24 h.
Translational capacity of mature oocytes
To verify the translational capacity of mature oocytes, mCherry capped RNAs were micro-injected into mature oocytes and the oocytes were cultured in the storage medium. The red fluorescence was observed after 2 h incubation (Fig. 4). To elucidate whether the Cas9-sgRNAs complexes efficiently worked in zebrafish oocytes or not, the Cas9 capped RNAs and mc4r or mpv17 sgRNAs were micro-injected into mature oocytes and incubated in the storage medium for 30 min or more, then the PCR-based T7E1 assay was performed. However, there was no mutation detected in the oocytes genome (data not shown). Thus, we speculated that the genome DNAs of zebrafish oocytes had a high affinity between histones and DNA backbone, which prevented Cas9 protein and target sgRNAs from binding with genomic DNAs to initiate the cleavage activity of CRISPR-Cas9 complex.

The translational capacity of mature oocytes.
(A) Oocytes were stored for 2 h in vitro without injecting mCherry capped RNAs. (B) Oocytes were stored for 2 h in vitro and micro-injected with mCherry capped RNAs. (C) Fertilized eggs at 1 cell stage were micro-injected with mCherry capped RNAs after 2 h. The red fluorescence was indicated with white arrows.
Toxicity assay of Cas9 capped RNAs and sgRNAs
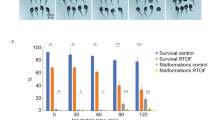
To test the toxicity of Cas9 capped RNAs and sgRNAs in the oocytes, the fertilization rate and hatching rate were statistically analyzed after injecting Cas9 capped RNAs/sgRNAs or phenol red. The results revealed that the toxicity of Cas9 capped RNAs/sgRNAs was very low, compared to phenol red injected groups. However, the fertilization and hatching rates were significantly lower than the control group (Fig. 5 and Table S1), indicating that the injection caused mechanical damage to the oocytes. In addition, there was no significant difference observed in fertilization rates between Cas9 capped RNAs/sgRNAs groups and phenol red groups, albeit their hatching rates were significantly different (Fig. 5 and Table S1). About eight percent of embryos were observed with developmental deformity and were unable to hatch properly after injecting Cas9 capped RNAs/sgRNAs.

Toxicity assay of oocytes injected with Cas9 capped RNAs and sgRNAs.
The control groups were oocytes stored in vitro for 30 min without injection. The groups of phenol red and Cas9/sgRNAs were injected with phenol red and Cas9/sgRNAs, respectively.
Improving the efficiency of gene knock-in in zebrafish founder
Although gene knock-in was successfully performed by ZFN, TALEN and Cas9 in zebrafish embryos4,46,47, the efficiency of gene knock-in was unsatisfactory. To test whether the current method of oocytes storage could enhance the efficiency of gene knock-in or not, a mloxP knock-in experiment was performed. The results were quite promising and showed that a single-stranded oligo DNA as a donor containing a mloxP site6 and 27 nt homology arms on both ends was effectively knocked into mc4r locus (Fig. 6A and Fig. S1B). The size of PCR products was 352 bp as expected (Fig. 6A). To further confirm the insertion of mloxP into mc4r locus, the PCR products were amplified from the tested embryo and sequenced. As shown in Fig. S1, mloxP were successfully knocked into mc4r locus both by the normal and oocytes storage injection. Moreover, the efficiency of gene knock-in was successfully improved by 49.6% in the oocytes storage injection groups, compared to 26% in the normal injection groups (Fig. 6B and Table S2).

The efficiencies of gene knock-in and knock-out.
(A) The schematic diagram of knocking mloxP into mc4r locus. A single-stranded oligo contained a mloxP site (blue) and 27 nt left and right homology arms (green). The primers for PCR detection were marked as black arrows. The positive fragments with mloxP were marked with red arrows. (B) The knock-in efficiencies of mloxP both in the oocytes storage injection groups and normal injection groups. The numbers of embryos for detection and knock-in in each experiment were shown in Table S4. (C) The efficiencies of genome editing of mc4r, mpv17, mstna, mc3r and mrap2b were measured by T7E1 assay and sequencing analysis. (D) The efficiencies of germline transmission of mc4r and mpv17 were also measured by T7E1 assay and sequencing analysis in zebrafish F1 generation. Three batches of F1 embryos with different numbers (17~28) were performed both in the oocytes storage injection groups and in the normal injection groups. Each experiment was repeated for three times independently.
Enhancing the efficiency of gene knock-out in zebrafish founder
Zebrafish mc4r, mpv17, mstna, mc3r and mrap2b founders were selected to evaluate the efficiency of genome editing by using this novel technique based on in vitro oocyte injection and storage. Five sgRNAs were designed, which targeted mc4r, mpv17, mstna, mc3r and mrap2b, respectively. The results showed that 5 target genes were efficiently knocked out by using this technique (Figs S2 and S3). The mutation rates of those 5 genes (mc4r, mpv17, mstna, mc3r and mrap2b) were 94.4%, 88.9%, 91.1%, 90.0% and 93.3%, respectively. Compared to the conventional 1-cell micro-injection, this technique significantly improved the efficiency of genome editing in zebrafish founder. Micro-injection of sgRNAs and Cas9 capped RNAs into 1-cell embryos by using a CRISPR/Cas9 system achieved 86.7%, 18.9%, 32.2%, 33.3% and 40.7% mutation rates for mc4r, mpv17, mstna, mc3r and mrap2b, respectively (Fig. 6C and Table S3).
Improving the efficiencies of germline transmission in the offspring
To test the efficiencies of germline transmission, mc4r and mpv17 genome DNAs were extracted from F1 generation and T7E1 assay and sequencing analysis were performed. Results showed that the efficiencies of germline transmission for mc4r and mpv17 mutations were 96.7% and 91% (Fig. 6D and Table S4), which were significantly higher than the common CRISPR/Cas9 system (70% and 35.2%).
Discussion
Here, a novel technique based on in vitro oocyte injection was developed and optimized to improve the efficiencies of genome editing and germline transmission in zebrafish by using a CRISPR/Cas9 system. Previously, Nakajima and Yaoita reported that the gene editing activity of TALEN was improved by injecting TALEN capped RNAs into the oocytes of Xenopus laevis48. However, little is known about translational capacity of mature oocytes in zebrafish by using a CRISPR/Cas9 system.
Chang et al. reported that the CRISPR/Cas9 nuclease generated site-specific cleavage (~35%) by editing three genes in zebrafish6. Hwang et al. reported that the mutation frequencies for ten genes induced by CRISPR/Cas9 nuclease were low in zebrafish and the highest mutation was only 59.4%49. The low efficiencies of genome editing and germline transmission are the common issues confronted with many researchers. To solve these problems, researchers developed and evaluated different approaches. In this study, the rates of genome editing and germline transmission for all the replicates were more than 90%, which indicated that the micro-injection of Cas9 capped RNAs into oocytes could reduce the time for Cas9 translation and subsequently improve the probability of mutations transferred to the next generation.
Our findings are consistent with the results reported by Fujii et al.23. Gagnon et al. revealed that the direct injection of Cas9 protein and sgRNAs complex into zebrafish eggs strikingly increased the mutagenic activity, but the efficiency of germline transmission was not discussed50. Our current findings are entirely different from the results using the direct injection of Cas9 protein/sgRNAs complex. Since the commercial Cas9 protein purified from bacteria lacked post-translational modifications, Cas9 proteins antecedently expressed in oocytes exhibited higher enzyme activity than the Cas9 protein derived from bacteria. Thus, this novel technique based on in vitro oocyte injection can improve the efficiencies of knock-out, knock-in and germline transmission by using a CRISPR/Cas9 system. It’s worth mentioning that an increasing efficiency of knocking mloxP into mc4r locus was achieved in this study, but whether the knocking-in efficiency was correlated with that of germline transmission deserves further studies.
The choices of effective storage media and appropriate storage time were the critical factors to affect the probability of the successful oocytes storage in vitro. The oocytes of freshwater fish usually develop the cavity between the shell and vitelline membrane due to osmotic phenomenon. In this study, the cavity forming effects of three oocytes storage media were compared. However, 90% L-15 medium at pH 9.0 with BSA was the only medium for inhibiting the formation of cavity. The cavity between the oocyte shell and vitelline membrane can hinder the oocytes’ fertilization, which was confirmed by hepes-cortland medium and 90% L-15 medium without BSA. Generally, the hatching rates of oocytes are closely associated with the storage time in vitro. Seki et al. obtained 12% hatching rates by making the zebrafish oocytes matured in vitro from stage III35. In this study, relatively longer storage time was resulted in higher oocytes deformity rates. Therefore, 30 min storage time was appropriate to improve the hatching rate and the translation of Cas9 protein in zebrafish oocytes.
In conclusion, a novel technique based on in vitro oocyte injection and storage was developed and evaluated in this study. The combination of these two techniques was confirmed to improve the efficiencies of genome editing and germline transmission in zebrafish by using a CRISPR/Cas9 system. This novel strategy will reduce the unnecessary labor intensive screening and will significantly increase the speed of generating heritable mutants in zebrafish.
Additional Information
How to cite this article: Xie, S.-L. et al. A novel technique based on in vitro oocyte injection to improve CRISPR/Cas9 gene editing in zebrafish. Sci. Rep. 6, 34555; doi: 10.1038/srep34555 (2016).
References
Ran, F. A. et al. Genome engineering using the CRISPR-Cas9 system. Nature protocols 8, 2281–2308, doi: 10.1038/nprot.2013.143 (2013).
Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., Hsu, P. D., Wu, X., Jiang, W., Marraffini, L. A. & Zhang, F. Multiplex genome engineering using CRISPR/Cas systems. Scientific reports 339, 23, doi: 10.1126/science.1231143 (2013).
Remy, S. et al. Zinc-finger nucleases: a powerful tool for genetic engineering of animals. Transgenic research 19, 363–371, doi: 10.1007/s11248-009-9323-7 (2010).
Bedell, V. M. et al. In vivo genome editing using a high-efficiency TALEN system. Nature 491, 114–118, doi: 10.1038/nature11537 (2012).
Feng, Y., Zhang, S. & Huang, X. A robust TALENs system for highly efficient mammalian genome editing. Scientific reports 4, 3632, doi: 10.1038/srep03632 (2014).
Chang, N. et al. Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos. Cell research 23, 465–472, doi: 10.1038/cr.2013.45 (2013).
Terao, M. et al. Utilization of the CRISPR/Cas9 system for the efficient production of mutant mice using crRNA/tracrRNA with Cas9 nickase and FokI-dCas9. Experimental animals/Japanese Association for Laboratory Animal Science, doi: 10.1538/expanim.15-0116 (2016).
Zou, Q. et al. Generation of gene-target dogs using CRISPR/Cas9 system. Journal of molecular cell biology 7, 580–583, doi: 10.1093/jmcb/mjv061 (2015).
Su, Y. H., Lin, T. Y., Huang, C. L., Tu, C. F. & Chuang, C. K. Construction of a CRISPR-Cas9 System for Pig Genome Targeting. Animal biotechnology 26, 279–288, doi: 10.1080/10495398.2015.1027774 (2015).
Lee, J. S. et al. RNA-guided genome editing in Drosophila with the purified Cas9 protein. G3 4, 1291–1295, doi: 10.1534/g3.114.012179 (2014).
Friedland, A. E. et al. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nature methods 10, 741–743, doi: 10.1038/nmeth.2532 (2013).
Niu, Y. et al. Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell 156, 836–843, doi: 10.1016/j.cell.2014.01.027 (2014).
Doyon, Y. et al. Heritable targeted gene disruption in zebrafish using designed zinc-finger nucleases. Nature biotechnology 26, 702–708, doi: 10.1038/nbt1409 (2008).
Bedell, V. M. et al. In vivo genome editing using a high-efficiency TALEN system. Nature 491, 114–118, doi: 10.1038/nature11537 (2012).
Hruscha, A. et al. Efficient CRISPR/Cas9 genome editing with low off-target effects in zebrafish. Development 140, 4982–4987, doi: 10.1242/dev.099085 (2013).
Varshney, G. K. et al. High-throughput gene targeting and phenotyping in zebrafish using CRISPR/Cas9. Genome research 25, 1030–1042, doi: 10.1101/gr.186379.114 (2015).
Maruyama, T. et al. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nature biotechnology 33, 538–542, doi: 10.1038/nbt.3190 (2015).
Zhang, C., Meng, X., Wei, X. & Lu, L. Highly efficient CRISPR mutagenesis by microhomology-mediated end joining in Aspergillus fumigatus. Fungal Genetics and Biology 86, 47–57, doi: 10.1016/j.fgb.2015.12.007 (2016).
Yan, L. et al. High-Efficiency Genome Editing in Arabidopsis Using YAO Promoter-Driven CRISPR/Cas9 System. Molecular Plant 8, 1820–1823, doi: 10.1016/j.molp.2015.10.004 (2015).
Schwartz, C. M., Hussain, M. S., Blenner, M. & Wheeldon, I. Synthetic RNA Polymerase III Promoters Facilitate High-Efficiency CRISPR-Cas9-Mediated Genome Editing in Yarrowia lipolytica. ACS synthetic biology, doi: 10.1021/acssynbio.5b00162 (2016).
Carrington, B., Varshney, G. K., Burgess, S. M. & Sood, R. CRISPR-STAT: an easy and reliable PCR-based method to evaluate target-specific sgRNA activity. Nucleic acids research 43, e157, doi: 10.1093/nar/gkv802 (2015).
Renaud, J.-B. et al. Improved Genome Editing Efficiency and Flexibility Using Modified Oligonucleotides with TALEN and CRISPR-Cas9 Nucleases. Cell Reports 14, 2263–2272, doi: 10.1016/j.celrep.2016.02.018 (2016).
Fujii, H. et al. Efficient Multiple Genome Modifications Induced by the crRNAs, tracrRNA and Cas9 Protein Complex in Zebrafish. PloS one 10, e0128319, doi: 10.1371/journal.pone.0128319 (2015).
Sung, Y. H. et al. Highly efficient gene knockout in mice and zebrafish with RNA-guided endonucleases. Genome research 24, 125–131, doi: 10.1101/gr.163394.113 (2014).
Fenghua, Z. et al. A comparison of the knockout efficiencies of two codon-optimized Cas9 coding sequences in zebrafish embryos. Yi chuan = Hereditas/Zhongguo yi chuan xue hui bian ji 38, 144–154, doi: 10.16288/j.yczz.15-452 (2016).
Jao, L., Wente, S. R. & Chen, W. Efficient multiplex biallelic zebrafish genome editing using a CRISPR nuclease system. Proceedings of the National Academy of Sciences of the United States of America 110, 13904 (2013).
Hwang, W. Y. et al. Heritable and precise zebrafish genome editing using a CRISPR-Cas system. PloS one 8, e68708, doi: 10.1371/journal.pone.0068708 (2013).
Dong, Z., Dong, X., Jia, W., Cao, S. & Zhao, Q. Improving the efficiency for generation of genome-edited zebrafish by labeling primordial germ cells. The international journal of biochemistry & cell biology 55, 329–334, doi: 10.1016/j.biocel.2014.08.020 (2014).
Sakamoto, A. et al. Effect of modification of ovary preservation solution by adding glucose on the maturation and development of pig oocytes after prolonged storage. The Journal of reproduction and development 52, 669–674 (2006).
Suttirojpattana, T. et al. The effect of temperature during liquid storage of in vitro-matured bovine oocytes on subsequent embryo development. Theriogenology 85, 509–518.e501, doi: 10.1016/j.theriogenology.2015.09.033 (2016).
Lee, S. H. & Park, C. K. Effect of magnetized extender on sperm membrane integrity and development of oocytes in vitro fertilized with liquid storage boar semen. Animal reproduction science 154, 86–94, doi: 10.1016/j.anireprosci.2014.12.015 (2015).
Cobo, A., Garrido, N., Pellicer, A. & Remohi, J. Six years’ experience in ovum donation using vitrified oocytes: report of cumulative outcomes, impact of storage time and development of a predictive model for oocyte survival rate. Fertility and sterility 104, 1426–1434.e1421–1428, doi: 10.1016/j.fertnstert.2015.08.020 (2015).
De Munck, N., Belva, F., Van de Velde, H., Verheyen, G. & Stoop, D. Closed oocyte vitrification and storage in an oocyte donation programme: obstetric and neonatal outcome. Human reproduction (Oxford, England), doi: 10.1093/humrep/dew029 (2016).
Urquiza, M. F. et al. Successful live birth from oocytes after more than 14 years of cryopreservation. Journal of assisted reproduction and genetics 31, 1553–1555, doi: 10.1007/s10815-014-0318-9 (2014).
Yasui, G. S. et al. Improvement of gamete quality and its short-term storage: an approach for biotechnology in laboratory fish. Animal : an international journal of animal bioscience 9, 464–470, doi: 10.1017/s1751731114002511 (2015).
Sanches, E. A. et al. Storage of Steindachneridion parahybae oocytes at different temperatures. Animal reproduction science 151, 262–268, doi: 10.1016/j.anireprosci.2014.09.022 (2014).
Goetz, F. W. & Coffman, M. A. Storage of unfertilized eggs of rainbow trout (Oncorhynchus mykiss) in artificial media. Aquaculture 184, 267–276 (2000).
Ubilla, A., Valdebenito, I., Arias, M. E. & Risopatron, J. Viability and DNA fragmentation of rainbow trout embryos (Oncorhynchus mykiss) obtained from eggs stored at 4 degrees C. Theriogenology 85, 1499–1506, doi: 10.1016/j.theriogenology.2016.01.012 (2016).
Seki, S. et al. Development of a reliable in vitro maturation system for zebrafish oocytes. Reproduction 135, 285–292, doi: 10.1530/REP-07-0416 (2008).
Nair, S., Lindeman, R. E. & Pelegri, F. In vitro oocyte culture-based manipulation of zebrafish maternal genes. Developmental dynamics : an official publication of the American Association of Anatomists 242, 44–52, doi: 10.1002/dvdy.23894 (2013).
Kimmel, C. B., Ballard, W. W., Kimmel, S. R., Ullmann, B. & Schilling, T. F. Stages of embryonic development of the zebrafish. Developmental dynamics: an official publication of the American Association of Anatomists 203, 253–310, doi: 10.1002/aja.1002030302 (1995).
Westerfield, W. M. The zebrafish book: a guide for the laboratory use of zebrafish (Danio rerio). (University of Oregon press, 2000).
Wang, J. W. et al. CRISPR/Cas9 nuclease cleavage combined with Gibson assembly for seamless cloning. BioTechniques 58, 161–170, doi: 10.2144/000114261 (2015).
Borges, A. et al. CTAB methods for DNA extraction of sweetpotato for microsatellite analysis. Scientia Agricola 66, 529–534 (2009).
Starkings, S. Quantitative Data Analysis with IBM SPSS 17, 18 & 19: A Guide for Social Scientists by Alan Bryman and Duncan Cramer. International Statistical Review 80, 334–335 (2012).
Chen, F. et al. High-frequency genome editing using ssDNA oligonucleotides with zinc-finger nucleases. Nature methods 8, 753–755, doi: 10.1038/nmeth.1653 (2011).
Kimura, Y., Hisano, Y., Kawahara, A. & Higashijima, S. Efficient generation of knock-in transgenic zebrafish carrying reporter/driver genes by CRISPR/Cas9-mediated genome engineering. Scientific reports 4, 6545, doi: 10.1038/srep06545 (2014).
Nakajima, K. & Yaoita, Y. Highly efficient gene knockout by injection of TALEN mRNAs into oocytes and host transfer in Xenopus laevis. Biology open 4, 180–185, doi: 10.1242/bio.201410009 (2015).
Hwang, W. Y. et al. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nature biotechnology 31, 227–229, doi: 10.1038/nbt.2501 (2013).
Gagnon, J. A. et al. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PloS one 9, e98186, doi: 10.1371/journal.pone.0098186 (2014).
Acknowledgements
The authors are grateful for supports from the Hundred Talents Program of Chinese Academy of Sciences, the Key Application and Development Program of Chongqing Science and Technology Commission and Beibei District, Chongqing (Grant No. cstc2014yykfC20004 and 2015-07), the Scientific Research Starting Foundation for Returned Overseas Chinese Scholars from Ministry of Education and the National Technology Foundation for Selected Overseas Chinese Scholar of Chongqing Human Resources and Social Security Bureau, China.
Author information
Authors and Affiliations
Contributions
D.S.P. and J.X.Z. conceived and designed the experiments. S.L.X., W.P.B. and C.W. performed the experiments. S.L.X. and D.S.P. analyzed the data. D.S.P., S.L.X. and M.J. wrote the main manuscript text. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Xie, SL., Bian, WP., Wang, C. et al. A novel technique based on in vitro oocyte injection to improve CRISPR/Cas9 gene editing in zebrafish. Sci Rep 6, 34555 (2016). https://doi.org/10.1038/srep34555
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep34555
This article is cited by
-
Improved Growth and High Inheritance of Melanocortin-4 Receptor (mc4r) Mutation in CRISPR/Cas-9 Gene-Edited Channel Catfish, Ictalurus punctatus
Marine Biotechnology (2022)
-
Improving CRISPR/Cas9 mutagenesis efficiency by delaying the early development of zebrafish embryos
Scientific Reports (2020)
-
Full-color carbon dots with multiple red-emission tuning: on/off sensors, in vitro and in vivo multicolor bioimaging
Journal of Materials Science (2019)
-
Non-viral and viral delivery systems for CRISPR-Cas9 technology in the biomedical field
Science China Life Sciences (2017)
-
In vivo characterization of hair and skin derived carbon quantum dots with high quantum yield as long-term bioprobes in zebrafish
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
