
Abstract
Breast cancer is the most common cancer in women worldwide. Despite the information provided by anatomopathological assessment and molecular markers (such as receptor expression ER, PR, HER2), breast cancer therapies and prognostics depend on the metabolic properties of tumor cells. However, metabolomics have not provided a robust and congruent biomarker yet, likely because individual metabolite contents are insufficient to encapsulate all of the alterations in metabolic fluxes. Here, we took advantage of natural 13C and 15N isotope abundance to show there are isotopic differences between healthy and cancer biopsy tissues or between healthy and malignant cultured cell lines. Isotope mass balance further suggests that these differences are mostly related to lipid metabolism, anaplerosis and urea cycle, three pathways known to be impacted in malignant cells. Our results demonstrate that the isotope signature is a good descriptor of metabolism since it integrates modifications in C partitioning and N excretion altogether. Our present study is thus a starting point to possible clinical applications such as patient screening and biopsy characterization in every cancer that is associated with metabolic changes.
Similar content being viewed by others

Introduction
Medical applications of stable isotopes are now widespread, like the well-known 13C-urea breath assay for ulcer detection1. This takes advantage of 13C-labelling and thus usually neglects differences in reaction rates between isotopic forms, because the isotopic signal used for diagnosing is far above small natural variations in 13C. By contrast, the use of isotopes at natural abundance exploits such subtle differences (referred to as isotope effects) to identify bottlenecks in metabolic pathways (rate-limiting steps) or the contribution of multiple elemental sources (mass balance), without the need to introduce expensive isotope tracers into the patient. Isotope effects in metabolism are mostly caused by enzymatic reactions that preferentially consume substrates containing either the light or the heavy isotope (isotopologues) and therefore, the natural isotope abundance in metabolites depends on metabolic fluxes and source substrates2. For example, the natural 13C abundance in respired CO2 has been used to trace diet composition and substrate changes during exercise3,4. In cancer biology, the use of natural variations in Cu and S stable isotopes in hepatocellular carcinoma has been attempted recently5. But to our knowledge, no study has looked at alterations of natural isotope abundance in breast cancer. Due to changes in primary C and N metabolism such as increased glycolysis, glutaminolysis and nucleotide synthesis6, important changes in 13C and 15N natural abundance can be anticipated.
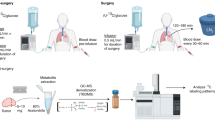
To address this question, we examined the isotopic signature of intact breast cancer biopsies (mostly from invasive ductal carcinoma, IDC) and cultured breast cancer cell lines (Supplementary Tables S1 and S2) using elemental analysis coupled to isotope ratio mass spectrometry (EA-IRMS). This technique has been recently shown to be applicable to the biochemical analysis of cancerous cell lines7.
Results
Isotope signature of breast cancer biopsies
Natural 13C and 15N abundance was found to discriminate normal and cancerous biopsies, the latter being significantly 13C-enriched by ~4‰ and tended to be (though not significantly) 15N-depleted (Fig. 1a,b). Triple negative tissues (ER- PR- HER2-) tended to be relatively 15N-depleted (Fig. 1a and Supplementary Table S1). The observed isotopic difference between cancerous and non-cancerous tissues can have been caused by either variation in chemical composition or isotope fractionations in metabolism.

Breast cancer biopsies are naturally 13C-enriched and nitrogen-rich.
(a) The natural abundance in 13C (δ13C, vs. V-PDB) and 15N (δ15N, vs. air N2) differentiates control from tumor patient tissue samples (n = 23). One sample pair of healthy (adjacent) and cancerous tissues not characterized for receptors (u and u’, respectively) and three triple negative tumor samples (t) are also shown. (b) Taken as a whole, the change in the 13C-abundance (Δδ) in cancerous tissues in tumor/adjacent tissue pairs was significant (P < 0.01, n = 5). (c) 13C-abundance (Δδ) was inversely correlated with the total lipid content (r2 = 0.48, P < 0.03). (d) Regardless of receptor expression, cancerous tissues had a higher elemental content in N (P < 0.05). In (a,c), each datum is the average of 3 sub-samples. In (d), lower case letters stand for statistical classes, P < 0.05.
First, there were alterations in total lipid content, which correlated to the 13C-abundance (Fig. 1c and Supplementary Figure S1). In fact, lipids are typically 13C-depleted components of human tissues8 and a systematic 13C-depletion has been observed in adipose tissue9. Here, cancerous tissues are on average less lipid-rich, reflecting the lower abundance of adipose cells in breast IDC10,11. Cancerous tissues were also enriched in nitrogenous compounds, as shown by the higher N elemental content (Fig. 1d). This effect is due to either a lower cellular N turnover rate or a higher content in protein and total amino acids including arginine (Supplementary Figure S2). Second, metabolic fluxes were modified thereby modifying the net exchange with the extracellular fluid and thus the overall (apparent) cellular isotope fractionation (see below). It is highly unlikely that the isotopic difference between biopsies was due to a confounding effect of nutritional preferences amongst patients (such as the proportion of animal proteins or sugars from C4-plants)12,13,14 because the comparison of paired normal and cancerous biopsy samples from the same patient still shows a significant difference for 13C (Fig. 1b). 13C and 15N abundance were also corrected so as to express them relative to a common elemental source (see Methods below). Moreover, female patients from which biopsies originated had the same geographical origin (western France) and thus important differences between nutritional habits or general isotope composition of body matter are improbable15.
Metabolic alterations in breast cancer biopsies
Targeted metabolomics analyses by GC-MS were then carried out to identify major metabolic alterations. Normal tissues were clearly differentiated from cancerous tissues in both unsupervised (PCA) and supervised (orthogonal partial least square-discriminant analysis, OPLS-DA) analysis (Supplementary Figures S3 and S4), with ethanolamine, methionine and arginine having the highest weight in such a differentiation (Supplementary Figures S3 and S4). Other discriminating metabolites included carbohydrates (such as fructose) and putrescine and these metabolites have also been found to be significant elsewhere16,17. While the effect on free ethanolamine reflected the general reorchestration of lipid metabolism18, the simultaneous effect on arginine and putrescine strongly suggests perturbations in arginine metabolism (urea cycle), thereby causing changes in N excretion and thus, by mass balance, in 15N-abundance and N elemental content.
Isotopic signature of cell lines
Potentially, the isotopic signature in biopsies may have been influenced by non-controlled epidemiological and clinical factors (e.g., patient age, pharmacological treatments, etc.). Therefore, we also examined different cell lines cultured under isotopically-controlled conditions: one immortalized, non-cancerous cell line (MCF10A) and six breast cancer cell lines (ZR75-1, MCF7, SKBR3, MDA-MB-231, MDA-MB-468 and Cal51, see Supplementary Table S2). The use of cultured cells further allowed us to carry out isotopic compound-specific analyses as well as targeted gene-expression, metabolomics and EA-IRMS analyses (see Methods) on the same samples.
With the exception of ZR75-1 (non-pleural origin, ER+), all other breast cancer cell lines were 15N-depleted (Fig. 2a). The 15N-abundance in DNA, proteins and total soluble metabolites paralleled that in total organic matter (Supplementary Figure S5). The 13C-abundance in non-cancerous cells was found to be lower than in cancerous cells (Fig. 2a). It did not correlate to the lipid content but rather to the 13C-abundance in lipids and loosely, to the content in C4 organic acids (Fig. 2b) which are known to be 13C-enriched19.

Cancerous cell lines are naturally 13C-enriched and 15N-depleted.
When compared to source C and N used for growth, the natural isotopic enrichment (ε13C and ε15N) differentiates the control immortalized line (MCF10A) and the breast tumor ascites-derived line (ZR75-1) from other adenocarcinoma lines (a). The isotope enrichment in lipids parallels that in total organic matter and cancerous lines contain more (13C-enriched) C4-acids (b). Despite some variations, transcript quantitation of key enzymes shows a generally lower expression in cancer cells compared to MCF10A cells of enzymes of the urea cycle, lipid synthesis or the anaplerotic pathway (gene abbreviations in Supplementary Table S5), suggesting that metabolite excretion in addition to biosynthesis is involved in the isotopic differences (c).
Primary metabolism in cell lines
The endometabolome (i.e., intracellular metabolome) was analyzed to gain information on primary C and N metabolism (summarized in Supplementary Figure S6a). Some metabolites such as arginine, urea and glucose were significantly different between cancer and normal cell lines. Close correlations or anti-correlations were obtained between 13C- or 15N-abundance and key metabolites or metabolic ratios such as urea/arginine and fumarate/aspartate (Fig. 3a). As such, cancer cell lines could be differentiated from the MCF10A non-cancerous cells using these ratios (Fig. 3a).

Cancerous cell lines excrete less 15N-depleted arginine (Arg).
The role of Arg metabolism (urea cycle) is further demonstrated by the differences in ratios of endometabolites since Arg appears to be more retained relative to urea in cancerous cell lines (a). The extracellular-to-intracellular relative difference (D) in Arg was found to (i) be the best D value that anti-correlates to δ15N (b) and (ii) have the largest average D value across cell lines (c). In all cases, the δ15N value of the influx (substrate consumption) was close to 0‰ while the efflux (excreted N) was 15N-depleted and Arg was even more 15N-depleted in cancerous cell lines by up to ≈7‰ (d). Therefore, the relative isotopic efflux represented by Arg excretion (i.e., the quotient of Arg isoefflux to total isoefflux) was large in ZR75-1 and MCF10A and low in other cell lines (e). Asterisks: significant differences between cell lines (*P < 0.05; **P < 0.01); dashed line: δ15N of source Gln (d); lower case letters stand for statistical classes (P < 0.05) (e). EA, ethanolamine, 2HG, 2-hydroxyglutarate; PGA, phosphoglycerate; PHB, parahydroxybenzoate.
The present results on both metabolome and isotopes suggest that three metabolic pathways are at the origin of the isotopic differences between cell lines: the urea cycle (arginine metabolism), the anaplerotic carbon fixation and lipid synthesis. Transcript levels of a selection of genes encoding key enzymes of these pathways was examined by qPCR and in most cases, these genes were less expressed in cancer cell lines (Fig. 2c). Thus, rather than substantial changes in enzyme abundance, the alteration of isotope abundance was caused by changes in metabolic fluxes and/or net exchange of metabolites (absorption, excretion and accumulation) with the extracellular medium.
Isotopic efflux in cell lines
We then examined the metabolic composition of the extracellular medium (exometabolome, Supplementary Figure S6b) to determine the preferential distribution (extracellular vs. intracellular) of metabolites and compute isotopic fluxes (isofluxes). The extracellular-to-intracellular difference and the 15N-abundance were found to be best anti-correlated with arginine, β-alanine, ethanolamine and spermidine (Supplementary Figure S6c,d). Amongst these, arginine had the highest value of the extracellular-to-intracellular difference (Fig. 3b,c) and was the most significantly different between cancer and normal cell lines (P < 0.005) and thus, appeared to be the best candidate to explain the 15N-depletion (Fig. 3d). There was little difference in the 15N-abundance of arginine itself amongst cell lines, although the non-cancerous (MCF10A) and ER lines (ZR75-1 and MCF7) synthesized 15N-enriched arginine (by a few ‰). Still, in all cases, arginine was much more 15N-depleted than the isotopic influx (isotope composition of substrates consumed by cells from supplied culture medium) or extracellular urea (Fig. 3d). As a result, arginine efflux from cells represented a very substantial part of the isotopic efflux to the extracellular medium in both ZR75-1 and MCF10A lines but was modest in others (Fig. 3e). That is, cancerous lines were naturally 15N-depleted (Fig. 3b) because of the proportionally lower excretion of 15N-depleted arginine. The higher metabolic ratio urea-to-arginine then comes as no surprise: it reflected the prevalence of arginine metabolism via the urea cycle (Fig. 3a).
Discussion
We showed isotopic differences between healthy and cancer tissues or cells and provided evidence that these differences are related to urea, anaplerotic C fixation and/or lipid metabolism and that the balance in nitrogen metabolism is modified in breast adenocarcinoma tissues or cells compared to normal breast tissues or cells thereby altering the natural 15N-abundance (Fig. 4). The consumption of glutamine (glutaminolysis) is a major pathway in cancerous cells20 that involves deamidation by glutaminase, thereby producing ammonia which is partly recycled and excreted in the form of urea. Here, source glutamine is naturally 15N-depleted (δ15N = −6.5‰) and 14N/15N isotope effects in both deamidation and the urea cycle21,22,23 yield 15N-depleted arginine. The efflux (excretion) of arginine does not counterbalance the N influx and this imbalance is compensated for by intracellular build-up and recycling into polyamines and β-alanine (Fig. 4) thereby causing a general 15N-depletion of organic matter. In addition to acid metabolism (C4-acids and lactate, Fig. 2b, Supplementary Table S3), the urea cycle incorporates bicarbonate (naturally 13C-enriched)24 via the action of carbamoyl phosphate synthase and thus, arginine build-up contributes to the enrichment of 13C in cells. This appears indeed to be the best explanation considering known isotope effects in metabolism6,21,25,26,27,28,29 (Supplementary Table S4). In fact, the natural 13C-enrichment cannot come from a change in total lipid content (as in tumors) since lipid content variation was small (Supplementary Figure S7) and furthermore, the isotope composition in lipids followed that in total organic matter (Fig. 2b).

Major metabolic pathways are responsible for changes in the natural 13C and 15N abundance in cancerous cultured cells.
Glutamine (Gln) is a major N and C source from which N is removed via hydrolysis (glutaminase ❶) and the urea cycle (❷). These reactions are both fractionating against 15N thereby yielding 15N-depleted urea and arginine (Arg). Therefore, build-up and recycling rather than excretion of Arg (dotted arrow) tend to deplete cancer cells of 15N. Cells are also 13C-enriched due to the fixation of bicarbonate by carbamoyl-phosphate (CP) synthesis (to feed the urea cycle ❸) and the anaplerotic pathway (❹), as well as a lower 13C content in non-structural lipids (dotted arrow). Lipids that are 13C-depleted come from the natural 13C-depletion in acetyl-CoA (Ac-CoA) (inherited from naturally 13C-depleted C-atom positions in glucose) and the isotope effect of pyruvate dehydrogenase45,46 (❺). Amine acceptors are denoted as ‘A’ and oxaloacetate (OAA) converted to aspartate (Asp) is provided as an example. 2OG, 2-oxoglutarate; Pyr, pyruvate; Lact, lactate. The potential contribution of respiration (CO2 loss) and lactate excretion to the natural 13C-abundance is described in Supplementary Table S4.
Quite noticeably, the isotopic pattern seen in tumor biopsies was similar to that found in cell lines, that is, with a tendency to be 15N-depleted and 13C-enriched (Fig. 1a). This pattern is unlikely to come from an effect of nutrition, but rather explained by the specific primary C and N metabolism in tumor tissues, as explained above (Fig. 1). The metabolic origin of the 13C-enrichment was different to that in cultured cells (a decrease in total lipid content in tumors, Supplementary Figure S7 and refs 18,30). This is not surprising since cultured cell metabolism is different from that of cells in situ. By contrast, the mechanism explaining the lower 15N-abundance was similar: a net influx of 15N-depleted nitrogen associated with any metabolism. In cancerous cultured cells, there is an increased activity of the urea cycle and non-quantitative excretion of 15N-depleted arginine. In tumors in situ, it is believed that cells are arginine-dependent to sustain polyamine metabolism31 and the auxotrophy of tumors for arginine has been exploited in the development of anticancer therapies32. Therefore cells capture naturally 15N-depleted arginine from their microenvironment, thereby leading to a 15N-depletion of cellular organic matter. Such a mechanism agrees with the well-known increase in polyamine metabolism33 and the fact that urea and glutamine (glutaminase activity) are amongst the best urinary and tumor biomarkers for breast cancer, respectively34,35. Also, differential expression of arginase, a key enzyme of the urea cycle, has been reported in breast cancer cell lines36,37.
The natural abundance in 13C and 15N thus reflects primary metabolism of cancer cells and thus, offers potential opportunities for development of biomarkers for clinical research. Consistently, healthy and cancerous samples were found to fall in corresponding regions of the isotopic landscape (Fig. 1a) and normal and cancerous cell lines could be differentiated (Fig. 2a). The ability of natural 13C- or 15N-abundance to further differentiate tumor grade and severity remains to be explored. In addition, there is a potential use the natural isotope abundance to distinguish between different breast cancer subtypes, as they have been shown to have different metabolic phenotypes38. Also, isotopic measurements would be best adapted to clinical settings if measured in plasma metabolites rather than in biopsies and thus, future work on both the accuracy and applicability of such plasma isotopic markers is required. For example, 15N natural abundance in plasma arginine could be exploited as a marker provided the metabolic activity of the tumor has a detectable effect on the total circulating arginine pool. That isotopic signature of tumorous breast tissues is due to alterations in major pathways of primary metabolism highlights the potential relevance of our findings to other cancer types which also display changes in metabolic fluxes.
Methods
Human Tissues
Fresh human mammary samples were obtained from chemotherapy naive patients with invasive carcinoma after surgical resection at the Institut de Cancérologie de l’Ouest – René Gauducheau, Nantes, France. Informed consent was obtained from patients to use their surgical specimens and clinicopathological data for research purposes, as required by the French Committee for the Protection of Human Subjects (CCPPRB). Ouest IV – Nantes CCPPRB approved use of tumor tissues for this study (6 May 2013: no. 357/2013). The collection of tumors was approved by the French Minister of higher education and research (no. AC-2008-141). This study did not require additional ethical approval. Amongst 17 breast cancer patients, 12 breast cancer samples and 5 pairs of breast cancer and adjacent non-cancerous tissues were obtained. One normal breast sample was obtained from reduction mammoplasty. For tumors, hormonal receptors, histological type and grade information were available, except that a paired of tissue #22/#19 (Supplementary Table S1, u and u’ in Fig. 1a) did not have receptor status available.
Cultured cells
Six human breast cancer cell lines and one non-cancerous cell line (Supplementary Table S2) were used and grown as monolayer cultures (107 cells/75 m2 flask). The cancer cells were grown in no-glucose no-glutamine DMEM supplemented with 10% FBS and 1% 10,000 units/ml penicillin, 10,000 units/ml streptomycin at 37 °C in a humidified atmosphere of 5% CO2. The non-cancerous cell line (MCF10A) was grown in DMEM/F-12 (no-glucose no-glutamine) supplemented with 5% horse serum, 0.01% EGF (Epidermal Growth Factor), 0.005% cholera toxin, 0.1% insulin, 0.005% hydrocortisone and 1% HEPES. L-glutamine and D-glucose (4 and 25 mM, respectively) were added separately to culture mediums to ensure these nutrients were isotopically identical for all cells. The same cells were grown several times to test the biological variability. As soon as cells were confluent (~107 cells), cell counting and extraction were done using triplicate cell culture flasks. One flask was used for cell counting, after harvesting with trypsin, using trypan blue staining and microscopic hemocytometry. For other analyses carried out on the other flasks, cells were then prepared as an aqueous (isotopes) or methanolic (metabolomics) extract without using trypsin as explained below.
Extraction and separation of studied fractions
Human tissues
Human fresh tissues were lyophilised. Lipids were extracted from the dried human tissues (10 mg) with hexane (1 mL). After centrifugation (4000 rpm, 15 min, 4 °C), the pellet was kept for metabolomics and the lipids were recovered by evaporation of hexane under a stream of N2 gas at 40 °C.
Cultured cells
After culture, cells were washed twice with PBS and 3 mL of cold water was added to the flask. Cells were detached from the flask using a cell scraper. The lysed cell preparation was kept at −20 °C for 48 h and then lyophilized. Different fractions (lipids, total soluble fraction (TSF), protein and DNA) were extracted and separated from the dry cell material. To obtain a lipid fraction, lyophilized cells (40 mg) were ground and extracted twice under agitation with 2 mL cyclohexane for 5 min at 20 °C. After centrifugation (4000 rpm, 5 min, 20 °C), the lipid fraction (4 mL of cyclohexane) was dried under a stream of N2 gas at 40 °C and the dry residue stored at −20 °C until required. To obtain a total soluble fraction, the pellet (insoluble in cyclohexane) obtained after centrifugation were ground and extracted twice with 2 mL methanol/water (80:20 v/v) for 5 min at 20 °C. After centrifugation (4000 rpm, 5 min, 20 °C), the total soluble fraction was dried under a stream of N2 gas at 40 °C and the dry residue stored at −20 °C until required. The pellet (insoluble in methanol/water) consisted of a protein fraction.
For DNA purification, lyophilised cells (15 mg) were reconstituted with 1 mL water and extracted with an equal volume of phenol/chloroform in an ultrasound bath for 30 min. After centrifugation (4000 rpm, 5 min, 4 °C), the aqueous phase was filtered to remove proteins and the DNA was precipitated with an equal volume of isopropanol (1 mL). After gentle agitation, the DNA fragments were pelleted by centrifugation (4000 rpm, 5 min, 4 °C), washed twice with ethanol (70%) to remove impurities and dried under a stream of N2 gas at 40 °C.
13C and 15N abundance by elemental-analysis coupled to isotope ratio mass spectrometry (EA-IRMS)
Subcellular fractions (see above) were lyophilized for EA-IRMS analysis, in order to detect their 13C and 15N isotopes. From each sample ~0.7 mg was weighed with 10−5 g precision balance (Ohaus Discovery DV215CD, Pine Brook, New Jersey, USA) into each of three tin capsules (solids “light” 5 × 9 mm, Thermo Fisher Scientific, www.thermo.com) for isotope analysis. EA-IRMS analyses were performed as previously described7. The 13C and 15N abundance was first expressed as δ values (δ13C and δ15N, in ‰) as δ = R/Rst − 1 where R is the heavy-to-light isotope ratio and Rst stands for the isotope ratio in the international reference (V-PDB for 13C and air N2 for 15N).
The isotope abundance in sample fractions was expressed relative to that in total organic matter using the enrichment (in ‰) ε = δf − δtom where δf is the δ value in the fraction of interest (lipids, proteins, etc.) and δtom the (non-corrected) δ value in total organic matter. The enrichment ε is then positive if the sample fraction is enriched in the heavy isotope and negative if it is depleted. In this way, ε nearly equals the opposite of the isotope fractionation Δ (which is given by [δtom − δf]/[δf + 1] ≈ δtom − δf). For total organic matter in Figs 1 and 2 and Supplementary Figure S3, δ values were corrected to allow comparisons between patients (that may have had different nutritional preferences) and between cell lines (that had slightly different culture media), as follows:
In cultured cells, it was corrected for the δ value of the elemental C and N source using the classical relationships accounting for isotope fractionation: δcorr = (δav − Δ)/(Δ + 1) where Δ = (δsrce − δ)/(δ + 1), δsrce is the δ value of the elemental source and δav is δ value used as the reference (here, the average of δsrce values was used as the reference value; note that any change in this value would not change the pattern seen here, it would only change the absolute scale of δcorr values). δsrce was the δ15N and δ13C of source amino acids and sugars, respectively, in intact culture medium. When expressed as a fractionation (Fig. 2a), the isotope composition in total organic matter of cells is given as ε = δ − δsrce. That way, ε values are positive when it is enriched in the heavy isotope and negative when it is depleted, as for ε in individual cell fractions (see above).
For biopsies, δsrce could not, quite obviously, be known because food composition of patients was not controlled. We therefore had to use a correction that could be calculated independently of food composition. The major influence of nutrition on elemental composition and isotopes is the protein/fat ratio of ingested food39, with fat-rich and protein-poor food causing a general increase in %C (and decrease in %N) and a general decrease in both δ15N and δ13C in body tissues. Therefore, we renormalized the δ values in total organic matter by taking into account the %C and %N using δcorr = δ + (1 − f )τ where f is the elemental composition relative to the average and τ is the apparent isotope fractionation between the whole population δ value (average isotope composition of human tissues in European western (French) population (δa), −22‰ and 9‰ for 13C and 15N, respectively) and the average of our samples (δm): τ = δa − δm. For example, in this way, protein-rich samples (lean patients) that are naturally impoverished in elemental C and richer in 13C will have a negative correction for δ13C. Of course, such a correction was not ideal because the knowledge of the average isotope composition of ingested food would have been better. In practice, this correction was minimal (on average, 0.02‰ for 13C and 0.5‰ for 15N) and did not cause a substantial bias in our data.
15N-abundance and amino acid content using gas chromatography - combustion - isotope ratio mass spectrometry (GC-C-IRMS)
When cells were confluent, the culture medium was kept (10 mL from ~108 cells) and cells (~108) were washed with PBS and then lysed in 36 mL of 80% methanol. After homogenization, methanolic extracts were dried under a stream of N2 gas at 40 °C. Amino acids were prepared as their N-pivaloyl, O-isopropyl esters for GC-C-IRMS analysis and used for both quantification (μmol) and 15N values (‰) as previously described40.
13C-abundance in metabolites using liquid chromatography coupled to chemical oxidation and isotope ratio mass spectrometry (LC-co-IRMS)
For LC-co-IRMS analysis, ~107 cells were used in 12 mL of 80% methanol. LC-co-IRMS analysis was performed as previously described, using water as a mobile phase and a RS Pak KC-811 column (Shodex)41. δ13C values were corrected using pure standard solutions at different concentrations. In practice, such corrections due to non-linearity were very small, about 0.2‰.
GC-MS metabolomics
Human biopsies
If material remained after lyophilization and lipid extraction, an additional ~10–15 mg of tissue was ground and dissolved in a standard solution of DL-2-Aminobutyric acid (0.2 mM, 950 μL) and methanol/water (950 μL, 80:20 v/v) was added to extract the soluble metabolites. After centrifugation (14000 rpm, 15 min, 4 °C), the soluble fraction was mixed with standard solution of ribitol (2 mM, 95 μL). The solution was centrifuged again and three aliquots of 200 μL were dried using a speed-vac before derivatization and GC-MS analysis as described previously41,42.
Cultured cells and extracellular medium
About 5 × 107 cells were dissolved in a solution of methanol/water (7 mL, 60:40 v/v) containing 500 μL of a standard solution D,L-2-aminobutyric acid (0.2 mM). After centrifugation (14000 rpm, 15 min, 4 °C), the soluble fraction was mixed with another standard solution of ribitol (2 mM, 50 μL). The solution was centrifuged again and three aliquots of 1.4 mL, i.e. 20% of ~5 × 107 cells, were dried using a speed-vac before derivatization and GC-MS analysis. For extracellular medium, the same standard solutions [D,L-2-aminobutyric acid (0.2 mM, 500 μL) and ribitol (2 mM, 50 μL) and methanol/water solution (500 μL, 60:40 v/v)] were added to 500 μL of media collected from the cultured cells. After a centrifugation, three aliquots of 300 μL were prepared for GC-MS analysis. All metabolomics data were normalized to the specific number of cells so that the exo- and endo-metabolome could be directly compared.
RT-qPCR
RNA was isolated from cell lines and 500 ng RNA was reverse transcribed as previously described43. Quantitative PCR was done using the Maxima SYBR Green/ROX qPCR Master Mix (Life Technologies) and the MX4000 instrument (Stratagene, Basel, Switzerland), according to the manufacturer’s instructions. The endogenous housekeeping genes RPLPO, HPRT1 and ACTB1 were used for normalization. Relative quantification was carried out by using the ΔΔCt method. Primer sequences are listed in Supplementary Table S5.
Statistics
Pair-wise comparisons were carried out using Student-Welsh tests, with a p-threshold value of 0.01 unless otherwise stated in text. Metabolomics were examined using unsupervised analysis with principal component analysis (PCA) or supervised analysis with orthogonal partial least square discriminant analysis (OPLS-DA), both carried out with Simca (Umetrics). Heat maps and hierarchical clustering (HCL) were done using the open source software MeV44. The HCL was carried out using the cosine correlation as a measure of proximity.
Additional Information
How to cite this article: Tea, I. et al.13C and 15N natural isotope abundance reflects breast cancer cell metabolism. Sci. Rep. 6, 34251; doi: 10.1038/srep34251 (2016).
References
Graham, D. Y. et al. Campylobacter pylori detected noninvasively by the 13C-urea breath test. The Lancet 1, 1174–1177 (1987).
Schmidt, H.-L., Robins, R. J. & Werner, R. A. Multi-factorial in vivo stable isotope fractionation: causes, correlations, consequences and applications. Isotopes Environ Health Stud 51, 155–199 (2015).
Bartstow, T. J., Cooper, D. M., Epstein, S. & Wasserman, K. Changes in 13CO2/12CO2 consequent to exercise and hypoxia. J Appl Physiol 66, 936–942 (1989).
Perkins, S. E. & Speakman, J. R. Measuring natural abundance of 13C in respired CO2: variability and implications for non-invasive dietary analysis. Funct Ecol 15, 791–797 (2001).
Balter, V. et al. Natural variations of copper and sulfur stable isotopes in blood of hepatocellular carcinoma patients. Proc Natl Acad Sci USA 4, 982–985 (2015).
De Berardinis, R. J., Sayed, N., Ditworth, D. & Thompson, C. B. Brick by brick: metabolism and tumor cell growth. Curr Op Gen Dev 18, 54–61 (2008).
Tea, I., Martineau, E., Giraudeau, P., Akoka, S. & Barillé-Nion, S. Method for characterising the origin and/or condition of diseased or healthy cells and uses thereof in biology. Patent WO 2012123886-A1/US2014-0051116 (2012).
Colonese, A. C. et al. Archaeological bone lipids as palaeodietary markers. Rapid Commun Mass Spectrom 29, 611–618 (2015).
Arneson, L. S. & MacAvoy, S. E. Carbon, nitrogen and sulphur diet-tissue discrimination in mouse tissues. Revue Can J Zool 83, 989–995 (2005).
Franck, C. J., McCreery, R. L. & Redd, D. C. B. Raman spectroscopy of normal and diseased human breast tissues. Anal Chem 67, 777–783 (1995).
Lazebnik, M. et al. A large-scale study of the ultrawideband microwave dielectric properties of normal, benign and malignant breast tissues obtained from cancer surgeries. Phys Med Biol 52, 6093–6115 (2007).
Petzke, K. J., Boeing, H., Klaus, S. & Metges, C. C. Carbon and Nitrogen Stable Isotopic Composition of Hair Protein and Amino Acids Can Be Used as Biomarkers for Animal-Derived Dietary Protein Intake in Humans. J Nutr 135, 1515–1520 (2005).
DeNiro, M. J. & Epstein, S. Influence of diet on the distribution of nitrogen isotopes in animals. Geochim Cosmochim Acta 45, 341–351 (1981).
DeNiro, M. J. & Epstein, S. Influence of diet on the distribution of carbon isotopes in animals. Geochim Cosmochim Acta 42, 495–506 (1978).
O’Connell, T. C., Kneale, C. J., Tasevska, N. & Kuhnle, G. G. C. The Diet-Body Offset in Human Nitrogen Isotopic Values: A Controlled Dietary Study. Am J Phys Anthropol 149, 426–434 (2012).
Tang, X. et al. A joint analysis of metabolomics and genetics of breast cancer. Breast Cancer Res 16, 415 (2014).
Budczies, J. et al. Remodeling of central metabolism in invasive breast cancer compared to normal breast tissue – a GC-TOFMS based metabolomics study. BMC Genomics 13, 334 (2012).
Todor, I. N., Lukyanova, N. Y. & Chekhun, V. F. The lipid content of cisplatin- and doxorubicin-resistant MCF-7 human breast cancer cells. Exp Oncology 34, 97–100 (2012).
Schmidt, H. L. Fundamentals and systematics of the non-statistical distribution of isotopes in natural compounds. Naturwissenschaften 90, 537–552 (2003).
Wise, D. R. & Thompson, C. B. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci 35, 427–433 (2010).
Rishavy, M. A., Cleland, W. W. & Lusty, C. J. 15N isotope effects in glutamine hydrolysis by carbamyl phosphate synthetase: evidence for a tetrahedral intermediate in the mechanism. Biochemistry 39, 7309–7315 (2000).
Kim, S. C. & Raushel, F. M. Isotopic probes of the argininosuccinate lyase reaction. Biochemistry 25, 4744–4749 (1986).
Schmidt, H. L. & Medina, R. Possibilities and scope of the double isotope effect method in the elucidation of the mechanisms of enzyme catalyzed reactions. Isotopes Environ Health Studies 27, 1–4 (1991).
Marlier, J. F. & O’Leary, M. H. Carbon kinetic isotope effects on the hydration of carbon dioxide and the dehydration of bicarbonate ion. J Am Chem Soc 106, 5054–5057 (1984).
Tcherkez, G., Mahé, A. & Hodges, M. 12C/13C fractionations in plant primary metabolism. Trends Plant Sci 16, 499–506 (2011).
Marlier, J. F. & O’Leary, M. H. Carbon kinetic isotope effects on the hydration of carbon dioxide and the dehydration of bicarbonate ion. J Am Chem Soc 106, 5054–5057 (1984).
Lyon, R. C., Cohen, J. S., Faustino, P. J., Megnin, F. & Myers, C. E. Glucose metabolism in drug-senstitive and drug-resistant human breast cancer cells monitored by magnetic resonance spectroscopy. Cancer Res 48, 870–877 (1988).
Perkins, S. E. & Speakman, J. R. Measuring natural abundance of 13C in respired CO2: variability and implications for non-invasive dietary analysis. Funct Ecol 15, 791–797 (2001).
Hatch, K. A., Pinshow, B. & Speakman, J. R. The analysis of the 13C/12C ratio in exhaled CO2: its advantages and potential application to field research to infer diet, changes in diet over time and substrate metabolism in birds. Int Comp Biol 42, 21–33 (2002).
Cerussi, A. et al. In vivo absorption, scattering and physiologic properties of 58 malignant breast tumors determined by broadband diffuse optical spectroscopy. J Biomed Optics 11, 044005 (2006).
Pavlova, N. N. & Thompson, C. B. The emerging hallmarks of cancer metabolism. Cell Metabolism 23, 27–47 (2016).
Delage, B. et al. Arginine deprivation and argininosuccinate synthetase expression in the treatment of cancer. Int. J. Cancer 126, 2762–2772 (2010).
Gerner, E. W. & Meyskens, F. L. Polyamines and cancer: old molecules, new understanding. Nat Rev 4, 781–792 (2004).
Slupsky, C. M. et al. Urine metabolite analysis offers potential early diagnosis of ovarian and breast cancer. Clin Cancer Res 16, 5835–5841 (2010).
Budczies, J. et al. Glutamate enrichment as new diagnostic opportunity in breast cancer. Int J Cancer 136, 1619–1628 (2015).
Singh, R., Pervin, S., Karimi, A., Cederbaum, S. & Chaudhuri, G. Arginase activity in human breast cancer cell lines: Nω-hydroxy-L-arginine selectively inhibits cell proliferation and induces apoptosis in MDA-MB-468 cells. Cancer Res 60, 3305–3312 (2000).
Straus, B., Cepelak, I. & Festa, G. Arginase, a new marker of mammary carcinoma. Clin Chim Acta 210, 5–12 (1992).
Lloyd, S. M., Arnold, J. & Sreekumar, A. Metabolomic profiling of hormone-dependant cancers: a bird’s eye view. Trends in Endocrinology and Metabolism 26, 477–485 (2015).
Voigt, C. C., Rex, K., Michener, R. H. & Speakman, J. R. Nutrient routing in omnivorous animals tracked by stable carbon isotopes in tissue and exhaled breath. Oecologia. 157, 31–40 (2008).
Tea, I. et al. Simultaneous determination of natural-abundance δ15N values and quantity of individual amino acids in proteins from milk of lactating women and from infant hair using an isotope ratio mass spectrometry. Rapid Commun Mass Spectrom 27, 1345–1353 (2013).
Dellero, Y., Lamothe-Sibold, M., Jossier, M. & Hodges, M. Arabidopsis thaliana ggt1 photorespiratory mutants maintain leaf carbon:nitrogen balance by reducing RuBisCO content and plant growth. Plant J 83, 1005–1018 (2015).
Bathellier, C. et al. On the resilience of nitrogen assimilation by intact roots under starvation, as revealed by isotopic and metabolomic techniques. Rapid Commun Mass Spectrom 23, 2847–2856 (2009).
Bah, N. et al. Bcl-xL controls a switch between cell death modes during mitotic arrest. Cell Death Dis 5, e1291 (2014).
Saeed, A. I. et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34, 374–378 (2003).
Melzer, E. & Schmidt, H. L. Carbon isotope effects on the pyruvate dehydrogenase reaction and their importance for relative carbon-13 depletion in lipids. J Biol Chem 262, 8159–8164 (1987).
Gilbert, A., Silvestre, V., Robins, R. J. & Remaud, G. S. Accurate quantitative isotopic 13C NMR spectroscopy for the determination of the intramolecular distribution of 13C in glucose at natural abundance. Anal Chem 81, 8978–8985 (2009).
Acknowledgements
The authors thanks Dr. P. Giraudeau, Pr. S. Akoka and Dr. J.M. Chao de la Barca and Pr. P. Reynier for preliminary tests for metabolome analysis by NMR and discussion about the manuscript, respectively. The authors are grateful to M. Grand for 13C lipid analysis by EA-IRMS analyses. The authors thank the support of the Agence Nationale de la Recherche through a research grant Jeunes Chercheurs (under contract 12-000101), the metabolomics platform CORSAIRE from the Biogenouest network, the funding from SATT (Nantes University Technology Transfer Services), from PLAISIR Pays de la Loire Association for International Structure on “Isotopomic Research” French Regional International Strategy Grant and the Australian Research Council through a Future Fellowship (under contract FT14 0100645). The authors thank the company Elementar France for kindly providing equipment to carry out LC-co-IRMS analyses.
Author information
Authors and Affiliations
Contributions
I.T. and G.T. designed and supervised the project. E.M., I.A. and C.M. collected and generated the isotopic data and performed isotopic analyses; J.L. performed qPCR analyses; Metabolomics data were performed by F.G. and S.B.-N. provided tissue and cell samples. S.B.-N. and A.C.B. advised on breast cancer metabolism context. G.T. and I.T. performed statistical analyses, prepared the figures and tables and wrote the manuscript; S.B.-N. and A.C.B. revised the manuscript; all authors viewed, had the opportunity to comment on and approved the final draft.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Tea, I., Martineau, E., Antheaume, I. et al. 13C and 15N natural isotope abundance reflects breast cancer cell metabolism. Sci Rep 6, 34251 (2016). https://doi.org/10.1038/srep34251
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep34251
This article is cited by
-
The CAT-SIR is out of the bag: tumors prefer host rather than dietary nutrients
BMC Biology (2021)
-
Distinct nitrogen isotopic compositions of healthy and cancerous tissue in mice brain and head&neck micro-biopsies
BMC Cancer (2021)
-
Natural abundance isotope ratios to differentiate sources of carbon used during tumor growth in vivo
BMC Biology (2021)
-
IntLIM: integration using linear models of metabolomics and gene expression data
BMC Bioinformatics (2018)
-
Nitrogen isotopes provide clues to amino acid metabolism in human colorectal cancer cells
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
