
Abstract
While a plethora of studies support a therapeutic benefit of Nrf2 activation and ROS inhibition in diabetic nephropathy (dNP), the Nrf2 activator bardoxolone failed in clinical studies in type 2 diabetic patients due to cardiovascular side effects. Hence, alternative approaches to target Nrf2 are required. Intriguingly, the tetracycline antibiotic minocycline, which has been in clinical use for decades, has been shown to convey anti-inflammatory effects in diabetic patients and nephroprotection in rodent models of dNP. However, the mechanism underlying the nephroprotection remains unknown. Here we show that minocycline protects against dNP in mouse models of type 1 and type 2 diabetes, while caspase -3,-6,-7,-8 and -10 inhibition is insufficient, indicating a function of minocycline independent of apoptosis inhibition. Minocycline stabilizes endogenous Nrf2 in kidneys of db/db mice, thus dampening ROS-induced inflammasome activation in the kidney. Indeed, minocycline exerts antioxidant effects in vitro and in vivo, reducing glomerular markers of oxidative stress. Minocycline reduces ubiquitination of the redox-sensitive transcription factor Nrf2 and increases its protein levels. Accordingly, minocycline mediated Nlrp3 inflammasome inhibition and amelioration of dNP are abolished in diabetic Nrf2−/− mice. Taken together, we uncover a new function of minocycline, which stabilizes the redox-sensitive transcription factor Nrf2, thus protecting from dNP.
Similar content being viewed by others


Introduction
Diabetic nephropathy (dNP) is the leading cause of end-stage renal disease and the strongest predictor of mortality in diabetic patients worldwide1. Knowledge about the molecular mechanisms of diabetic nephropathy progressed over recent years2,3,4. Blockade of the renin-angiotensin-aldosterone system (RAAS) by ACE inhibitors (ACEI) or angiotensin II receptor blockers (ARB) is still the mainstay of therapy, but only delays the initiation and progression of dNP in type 1 and type 2 diabetic patients. Efficient therapeutic approaches halting or even reversing progression of dNP are lacking, necessitating the search for new treatment strategies. One potential therapeutic target in dNP is the transcription factor Nrf2, which induces antioxidant and anti-inflammatory effects5,6. Nrf2 is a cellular sensor of oxidative stress and a nuclear factor controlling and coordinating expression of genes protecting against cellular damage provoked by excess ROS and other cellular stressors. Nrf2 belongs to the basic leucine zipper (bZIP) transcription factors and is repressed by Keap1 (Kelch-like erythroid cell-derived protein with CNC homology (ECH)-associated protein 1, also known as INrf2, inhibitor of Nrf2) and the Cul3/Rbx1 E3 ubiquitin ligase complex under physiologic conditions7. Under resting conditions Nrf2 interacts with and is ubiquitinated by Keap1 and the Cul3/Rbs1 E3 complex, targeting it for proteasomal degradation. Following appropriate stimulation, e.g. oxidative stress, inhibitors of Nrf2 including Keap1, Cul3 and Rbx1 E3 are exported from the nucleus and themselves ubiquitinated, allowing unhindered nuclear import and activity of Nrf27. In parallel, Nrf2 is regulated by phosphorylation at specific serine or threonine residues7. To stimulate an antioxidant response Nrf2 dimerizes with members of the small Maf family or c-Jun and binds to antioxidant response elements (AREs) in the regulatory regions of cellular cytoprotective proteins, such as glutathione enzymes (glutathione reductase, microsomal glutathione S-transferase 2), heme metabolism enzymes (heme oxygenase 1, ferrochelatase) or antioxidant enzymes (manganese superoxide dismutase (MnSOD), peroxiredoxin 1, thioredoxin reductase 1)8,9. This allows Nrf2 to efficiently initiate a cytoprotective response and prevent increased oxidative stress and further cellular damage.
Pre-clinical studies using Nrf2 activators or Nrf2 deficient mice established that Nrf2 is a crucial endogenous modulator of ROS and protects from experimental dNP10,11. These pre-clinical data were supported by results form the BEAM trial, in which the Nrf2 activator bardoxolone improved kidney function in patients with chronic kidney disease and type 2 diabetes mellitus12. However, the subsequent and larger BEACON trial had to be prematurely terminated due to an increased rate of cardiovascular events, which may indicate off-target effects of bardoxolone13. This raises the question as to whether alternative approaches to restrict Nrf2 function are feasible, providing an effective yet safe therapeutic approach to dNP.
Minocycline, a tetracycline derivate with off-target effects such as modulation of mitochondrial function and apoptosis inhibition, has shown beneficial effects in patients with dNP and in pre-clinical kidney injury models14,15,16. A recent small study evaluating minocycline in patients with dNP revealed anti-inflammatory effects of minocycline in diabetic patients17. Furthermore, minocycline efficiently prevents dNP in type 1 diabetes models (streptozotocin) in mice and rats18,19. However, the mechanism underlying minocycline’s protective effect in dNP remains unknown. In renal ischemic-reperfusion injury models minocycline normalizes the Bax/Bcl-2 ratio, reduces cytochrome C release and inhibits apoptosis in the kidney, suggesting nephroprotective effects of minocycline through inhibition of apoptosis14,15. An increased apoptosis rates–largely based on results using the non-specific TUNEL assay–has been repeatedly demonstrated in dNP20. Accordingly, minocycline mediated cytoprotection in dNP has been associated with reduced apoptosis. However, we recently demonstrated that caspase-3 mediated apoptosis is not required for dNP development in mice21. Based on these data we speculated that minocycline must convey protection from dNP independent of apoptosis.
While homozygous caspase-3 deficiency fails to protect from dNP in mice, both homozygous and hemizygous caspase-1 deficiency is protective, consistent with a causal role of caspase-1 dependent inflammasome activation in dNP21,22. Furthermore, we previously demonstrated that increased reactive oxygen species (ROS) generation causes renal inflammasome activation and dNP22. Beyond inhibition of apoptosis minocycline inhibits ROS-formation, pro-inflammatory cytokines and IκBα degradation23,24–all pathways that are regulated by Nrf2 and of potential mechanistic relevance in dNP. Hence, we hypothesized that minocycline protects from dNP independent of apoptosis-inhibition by restricting excess ROS-generation potentially via a Nrf2-dependent mechanism, thus preventing inflammasome activation in dNP.
Results
Minocycline, but not a pan-caspase inhibitor, ameliorates dNP
To gain insight into the mechanism underlying the nephroprotective effect of minocycline in dNP we compared the efficacy of minocycline (5 mg/kg, intraperitoneal, i.p.) with that of the caspase-inhibitor CIX (inhibiting caspases -3, -6, -7, -8 and -10). Treatment was initiated in 8-week old db/db mice and mice were analyzed after 12-weeks of treatment at the age of 20 weeks. Both compounds efficiently reduced the frequency of TUNEL positive glomerular cells (as previously shown refs 18 and 21), yet only minocycline, but not the caspase-inhibitor CIX, ameliorated markers of dNP (albuminuria and extracellular matrix accumulation, as reflected by the fractional mesangial area, FMA; Fig. 1a,c,d), implying that minocycline protects from dNP independent of apoptosis inhibition. Blood glucose levels, body weight and liver enzymes did not differ among experimental mice (Fig. 1b, Supplementary Fig. S5a and data not shown). Minocycline likewise protected C57BL/6 mice with streptozotocin-induced hyperglycemia from developing hallmarks of dNP (Supplementary Fig. S1), corroborating previous data18. Again, minocycline had no effect on blood glucose levels, body weight, or liver enzymes (Supplementary Figs S1 and S5b). Taken together, minocycline provides nephroprotection in experimental dNP in both type 1 and type 2 diabetes models independent of apoptosis inhibition.

Minocycline, but not the pan-caspase inhibitor CIX, protects from diabetic nephropathy in db/db mice.
Albuminuria (a) and extracellular matrix accumulation, as reflected by the fractional mesangial area (FMA, c,d) are decreased in minocycline (Mino) receiving db/db mice as compared to PBS-receiving db/db control mice (C, db/db). Application of CIX (+CIX, targeting caspases-3, -6, -7, -8 and -10) has no impact on these markers in db/db mice. Blood glucose levels are comparable among experimental groups (b). All treatment strategies were initiated at age 8 weeks and continued for 12 weeks. Mean values ± SEM (a,b,d). Representative PAS-stained glomeruli (c, size bar: 20 μm); *P < 0.05, ns: non-significant (a,b,d: ANOVA). Number of mice in each group is shown in parentheses in (a).
Inflammasome inhibition by minocycline in vivo
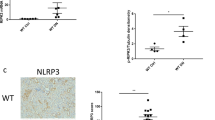
Recently, sterile inflammation rather than cell-death has been linked with dNP21,22. Of note, the polycaspase-inhibitor CIX does not target caspase-1, the interleukin-1 converting enzyme that is required for inflammasome activation. This raises the question as to whether minocycline ameliorates dNP by targeting the inflammasome. To evaluate this question we determined markers of inflammasome activation in db/db mice. In renal cortex extracts of db/db mice minocycline, but not CIX, reduced Nlrp3 expression and IL1β cleavage (Fig. 2a,b). Immunohistochemical assessment revealed a marked reduction of glomerular Nlrp3 and cleaved caspase-1 in minocycline but not in CIX receiving mice compared to control db/db mice (Fig. 2c,d). We observed colocalization of cleaved caspase-1 with podocytes and glomerular endothelial cells (GEnC) in histological sections of db/db mice (Fig. 2e,f and Supplementary Fig. S2a). Minocycline, but not CIX, markedly reduced glomerular (podocyte and GEnC) specific inflammasome activation (Fig. 2e,f and Supplementary Fig. S2b). These data establish that minocycline dampens diabetes induced renal and glomerular inflammasome activation.

Minocycline prevents glomerular inflammasome activation.
(a,b) In db/db mice treated with minocycline, but not in those treated with CIX, expression of Nlrp3 as well as the cleaved form of IL1β (cl-IL1β, b) are reduced in renal cortex extracts. (c,d) Immunohistochemical detection of Nlrp3 and cleaved caspase-1 demonstrates that minocycline, but not CIX, reduces markers of inflammasome activation within diseased glomeruli of db/db mice. (e,f) In glomeruli cleaved caspase-1 (cl-Casp1, red, e,f) co-localizes with the podocyte marker synaptopodin (Syn, green, e) and the endothelial marker CD34 (green, f). Minocycline, but not CIX application, reduces co-localization for both cell-types. Mean values ± SEM (a,b,d).; size bar (c,e): 20 μm; *P < 0.05 (a,b,d: ANOVA). Number of mice in each group is shown in parentheses in (a); (a,b) Representative cropped images without further modification are shown; exemplary uncropped images are provided in Supplementary Fig. 6.
Minocycline inhibits glucose-induced inflammasome activation in glomerular cells and normalizes glomerular filtration in vitro
To determine whether minocycline cell-autonomously inhibits inflammasome activation in podocytes and GEnC we incubated glucose challenged podocytes and GEnC with minocycline in vitro. Glucose induced Nlrp3 expression and caspase-1 activation in podocytes and GEnC, reflecting inflammasome activation22. Glucose-dependent inflammasome activation was prevented by minocycline both in podocytes and GEnCs (Fig. 3a,b). The effect of minocycline in regard to IL1β maturation was comparable to that of a caspase-1 inhibitor (Supplementary Fig. S3). Transfection with a constitutively active Nlrp3 mutant (Q705K) abolished minocycline’s suppressive effect on IL1β maturation in glucose stressed podocytes (Fig. 3c), demonstrating that minocycline mediated inhibition of IL1β-cleavage depends on Nlrp3.

Minocycline prevents inflammasome activation in glucose challenged podocytes and glomerular endothelial cells (GEnC) in vitro.
(a,b) High glucose (25 mM, 24 h), but not mannitol (25 mM, 24 h), induces the inflammasome markers Nlrp3 and cleaved IL1β in podocytes and glomerular endothelial cells in vitro. Minocycline (10 μM, 24 h) abrogates glucose-induced inflammasome activation. (c) Following transfection of human podocytes with a human Nlrp3 mutant (Q705K), resulting in constitutive active Nlrp3, minocycline (10 μM, 24 h) fails to suppress IL1β cleavage. (d,e) Minocycline prevents glucose-induced glomerular filtration barrier disruption in vitro; scheme reflecting the in vitro model used to mimic the glomerular filtration barrier (e) and bar graph summarizing results for albumin concentration in the lower chamber 3 hours after the addition of FITC-BSA to the upper chamber. Mean values ± SEM (a–c,e); *P < 0.05, ns: non-significant (a–c: Mann-Whitney-test; e: t-test); (a–c) representative cropped images without further modification are shown; exemplary uncropped images are provided in Supplementary Fig. 6.
To determine the effect of glucose-induced inflammasome activation and its inhibition by minocycline on the function of the glomerular filtration barrier we used a recently established in vitro model mimicking the glomerular filtration barrier (Fig. 3d and method section). Glucose, but not mannitol, markedly increased the transport of albumin across the in vitro filtration barrier consisting of podocytes and GEnC. The barrier disruptive effect of glucose was inhibited by minocycline (Fig. 3d,e), indicating that minocycline stabilizes the function of the glomerular filtration barrier through a direct effect on its cellular components.
Minocycline inhibits oxidative stress
Mitochondrial ROS can activate the inflammasome and minocycline has been shown to target mitochondria16,25. Hence we next evaluated whether minocycline inhibits diabetes-associated inflammasome activation by reducing mitochondrial ROS-generation. Indeed, in glucose stressed podocytes minocycline, but not the caspase-inhibitor CIX, prevented glucose-induced mitochondrial ROS generation, as reflected by staining with MitoSOX® and dihydrorhodamine (DHR) (Fig. 4a,b), two dyes detecting mitochondrial ROS-production. Similarly, treatment with minocycline, but not with CIX, markedly reduced glomerular 8-Oxo-dG (8-Oxo-2′-deoxyguanosine) staining and nitrotyrosine levels (Fig. 4c–e) while increasing levels of the mitochondrial antioxidant manganese superoxide dismutase (MnSOD) in renal cortex extracts of db/db mice (Fig. 4f). Thus minocycline conveys an antioxidant effect in glucose stressed podocytes and diabetic kidneys. This suggests that minocycline restricts glucose-induced inflammasome activation by inhibiting ROS generation. However, the mechanism through which minocycline restricts ROS generation and thus inflammasome activation remains unknown.

Minocycline prevents glucose-induced ROS formation.
(a,b) Minocycline (10 μM, Mino, 24 h), but not CIX (5 μM, 24 h), prevents mitochondrial ROS formation (a: MitoSOX, b: Dihydrorhodamine, DHR, green) in glucose stressed murine podocytes in vitro. (c–f) Application of minocycline, but not CIX, prevents glomerular accumulation of the ROS marker 8-Oxo-2′-deoxyguanosine (8-Oxo-dG, c,d) and decreased nitrotyrosine (Nitro, e) levels while increasing mitochondrial manganese superoxide dismutase (MnSOD, f) in renal cortex extracts in 20 weeks old db/db mice as compared to PBS-receiving db/db controls (C). Representative 8-Oxo-2′-deoxyguanosine (8-Oxo-dG) stained images detected by HRP-DAB reaction, brown; hematoxylin counterstain, blue; size bar: 20 μm (c). Mean values ± SEM (a–f). Size bars (b,c): 20 μm; *P < 0.05 (a,b,d–f: ANOVA). Number of mice in each group is shown in parentheses in (d–f) representative cropped images without further modification are shown; exemplary uncropped images are provided in Supplementary Fig. 6.
Stabilization of Nrf2 by minocycline
The transcription factor Nrf2, which initiates antioxidant defense mechanisms, ameliorates dNP in mice10,11. As increased ROS generation causes inflammasome activation and aggravates dNP we hypothesized that minocycline restricts glucose-induced inflammasome activation through a Nrf2-dependent mechanism22. First, to determine whether Nrf2 modulates glucose induced inflammasome activation we reduced Nrf2 expression via knock down in podocytes. Loss of endogenous Nrf2 expression markedly enhanced the glucose induced inflammasome activation, as reflected by the increase of cleaved IL1β, as compared to glucose challenged control (non-specific siRNA) podocytes (Supplementary Fig. S4), establishing that Nrf2 restricts glucose-induced inflammasome activation in podocytes.
To determine whether minocycline modulates Nrf2 expression we analyzed glucose stressed podocytes. Exposure to high glucose concentration slightly (although not significantly) induced Nrf2 expression, which was further induced by minocycline, but not by the caspase-inhibitor CIX (Fig. 5a). In line with these in vitro results minocycline, but not the caspase-inhibitor CIX, induced Nrf2 expression in the renal cortex (Fig. 5b) and in glomeruli (Fig. 5c,d) of db/db compared to untreated db/db and db/m control mice. These data demonstrate that minocycline increases endogenous Nrf2 expression in dNP.

Minocycline increases Nrf2 expression in glucose challenged podocytes in vitro and in diabetic mice in vivo.
(a) In glucose stressed murine podocytes minocycline (10 μM, 24 h), but not CIX (5 μM, 24 h), increases Nrf2 protein levels in vitro. (b–d) In 20 weeks old db/db mice minocycline, but not CIX, induces Nrf2 protein levels in renal cortex extracts and in glomeruli (c,d) as compared to db/db control (C) mice. Representative Nrf2 stained images detected by HRP-DAB reaction, brown; hematoxylin counterstain, blue; size bar: 20 μm (c). Mean values ± SEM (a–d). *P < 0.05 (a: Mann-Whitney-test; b,d: ANOVA). Number of mice in each group is shown in parentheses in (b); (a,b) representative cropped images without further modification are shown; exemplary uncropped images are provided in Supplementary Fig. 6.
The above immunoblotting and immunohistochemical analyses demonstrate increased Nrf2 protein levels following minocycline treatment in vitro and in vivo. To further elucidate the underlying mechanism we determined Nrf2 mRNA expression in minocycline treated podocytes (Fig. 6a). Minocycline did not affect Nrf2 mRNA expression (Fig. 6a), indicating that Nrf2 is regulated posttranslationally by minocycline. Posttranslational regulation of Nrf2 stability via ubiquitination and protein degradation is well established26,27. To determine Nrf2 protein stability we blocked its resynthesis using cycloheximide (CHX, Fig. 6b). In the presence of cycloheximide Nrf2 protein levels declined, but Nrf2 protein levels remained stable if minocycline was added in addition to cycloheximide (Fig. 6b). This suggests that minocycline regulates Nrf2 via the ubiquitin-proteasomal system. Indeed, minocycline reduced overall glucose-induced ubiquitination (Fig. 6c) and–importantly–it specifically prevented glucose induced Nrf2 ubiquitination in podocytes (Fig. 6d). These results suggest that minocycline maintains Nrf2 protein levels in glucose stressed podocytes by limiting ubiquitination and proteasomal degradation of Nrf2.

Minocycline increases Nrf2 protein stability and suppresses Nrf2 ubiquitination.
(a) Minocycline (+Mino) has no impact on Nrf2 mRNA expression in murine podocytes compared to untreated controls (Ctrl). (b) After incubation of podocytes with the protein synthesis inhibitor cycloheximide (CHX, 10 mg/ml for 24 h) Nrf2 protein levels decline. Application of minocycline stabilizes Nrf2 levels. (c) After glucose challenge (25 mM for 24 h) protein ubiquitination (IB: Ubiquitin) is increased compared to unstressed or mannitol incubated (25 mM for 24 h) controls. Minocycline reduces glucose-induced protein ubiquitination; representative immunoblot. (d) Minocycline reduces glucose-induced Nrf2 ubiquitination. Immunoprecipitation (IP) of Nrf2 from whole cell lysates of control (5 mM glucose), glucose challenged (25 mM for 24 h), or glucose plus minocycline (10 μM, 24 h) exposed podocytes. Mean values ± SEM (a,b,d); *P < 0.05, ns: non-significant (a,b,d: Mann-Whitney-test). (a–d) Representative cropped images without further modification are shown; exemplary uncropped images are provided in Supplementary Fig. 6.
Nrf2 is required for minocycline dependent nephroprotection
To ascertain that the nephro- and cytoprotective effect of minocycline depends on Nrf2 in vivo we determined the effect of minocycline on dNP, inflammasome activation, ROS-generation and glomerular cell death in Nrf2 deficient (Nrf2−/−) diabetic mice. In Nrf2−/− diabetic mice minocycline failed to reduce Nlrp3 expression, IL1β cleavage or glomerular 8-Oxo-2′-deoxyguanosine (8-Oxo-dG) (Fig. 7a–c). Furthermore, minocycline failed to protect Nrf2−/− mice from dNP, as indicated by a failure to reduce albuminuria and extracellular matrix accumulation (Fig. 7d,e). Again, minocycline had no impact on blood glucose levels in Nrf2−/− diabetic mice (Fig. 7f). Taken together, the cyto- and nephroprotective effect of minocycline depends on Nrf2, which is stabilized by minocycline posttranslationally and reduces ROS-generation and inflammasome activation in renal cells.

Nrf2 is required for minocycline-mediated protection from diabetic nephropathy.
(a–c) Nrf2 deficiency (Nrf2−/−) abolishes minocycline’s inhibitory effect on Nlrp3 (a), cl-IL1β (b) and glomerular 8-Oxo-2′-deoxyguanosine (8-Oxo-dG) (c) induction in diabetic C57BL/6 mice (DM). (d,e) Minocycline fails to reduce albuminuria (d) and fractional mesangial area (FMA, representative PAS-stained histological images, e) in Nrf2−/− mice. (f) Glucose levels are comparable in diabetic Nrf2−/− mice without (DM) or with (DM + Mino) minocycline application. Representative immunoblots (a) and bar graphs summarizing results (b–d,f). Representative 8-Oxo-2′-deoxyguanosine (8-Oxo-dG) stained images detected by HRP-DAB reaction, brown; hematoxylin counterstain, blue; size bar: 20 μm (c,e). Ctrl indicates non-diabetic Nrf2−/− control mice. Mean value ± SEM (b–f); *P < 0.05, ns: non-significant (b–e: t-test). Number of mice in each group is shown in parentheses in (a); (a) representative cropped image without further modification is shown; exemplary uncropped images are provided in Supplementary Fig. 6.
Discussion
Within the current study we provide new insights into the nephroprotective mechanism of minocycline in dNP. Minocycline reduces glomerular ROS-generation and inflammasome activation via the transcription factor Nrf2. Functionally, minocycline protects the endothelial-podocyte filtrations barrier in vitro and ameliorates diabetic kidney disease in vivo. Mechanistically minocycline maintains renal protein levels of Nrf2, possibly by reducing its ubiquitination and degradation. The inhibition of inflammasome activation via Nrf2 identifies a new minocycline-dependent cytoprotective mechanism that is of potential translational relevance in dNP, but may also have implications beyond dNP28,29. Furthermore, the protection from dNP through inflammasome inhibition and the failure of the poly-caspase inhibitor CIX (targeting caspases-3,-6,-7,-8,-10, but not caspase-1) strengthens the recently described pathogenetic role of inflammasome activation and sterile inflammation in dNP while supporting the notion that caspase-3-dependent apoptosis is negligible21,22.
Sterile inflammation is an established hallmark of dNP and is increasingly recognized as a disease aggravating mechanism in dNP. Inflammasome activation has been mechanistically linked with excess ROS generation30. Hence, a crucial pathophysiological role of inflammasome mediated sterile inflammation is entirely compatible with excess ROS generation, which is considered to be a unifying pathomechanism in dNP31,32. However, while inhibition of excess ROS-generation is beneficial in experimental dNP, clinical studies evaluating antioxidants failed to demonstrate a benefit33. The failure of clinical studies using non-selective ROS scavengers has been attributed to the inadvertent inhibition of physiological ROS functions34. Hence, alternative approaches aiming to restrict excess ROS formation are needed. Possible approaches include the inhibition of specific ROS sources, e.g. mitochondrial ROS35, or the amplification of endogenous ROS-scavenging pathways, e.g. by inducing the redox-sensitive transcription factor Nrf210. Alternatively, targeting ROS-dependent pathomechanisms, such as inflammasome activation, may allow nephroprotection without interfering with the physiological functions of ROS. Inflammasome inhibition provides nephroprotection in several renal injury models and amelioration of dNP in human diabetic patients has been reported in few cases21,22,36,37.
Based on the current results minocycline may be an interesting therapeutic approach to restrict ROS-mediated inflammasome activation in dNP. We establish that minocycline reduces ubiquitination of Nrf2 and increases Nrf2 protein stability. Nrf2 co-ordinately induces the expression of antioxidant defence-enzymes, such as NQO1, NQO2, HO-1 and MnSOD8,38. The regulation of Nrf2 in murine models of dNP, as observed in the current studies, raises the question as to whether minocycline may simultaneously target other pathways involved in dNP. Thus, Nrf2 interacts with Notch1 and NF-kB signaling and hence with pathways thought to be relevant in dNP39,40. Intriguingly, just like Nrf2 both Notch1 and NF-kB are regulated by ubiquitination. Further studies are required to evaluate whether minocycline targets multiple pathways linked with dNP, potentially via ubiquitination2,3,4. The versatility of minocycline allowing it to target multiple pro-inflammatory pathways may be an important factor for its well-established cyto-protective effects. Furthermore, recent evidence suggests that multiple cellular stress pathways, such as mitochondrial, inflammatory and oxidative stress, need to be simultaneously therapeutically targeted to yield a favourable outcome in dNP41. Therefore targeting Nrf2 and interacting pathways with a well-tolerated drug such as minocycline could prove to be a promising approach to dNP.
Pre-clinical studies using Nrf2 activators or Nrf2 deficient mice and in vitro work established a crucial function of Nrf2 in modulating glucose induced ROS generation and suggest that Nrf2 activators may be a suitable pharmaceutical approach to limit glucose-induced excess ROS generation42,43,44,45 and to convey nephroprotection in diabetes10,11. However, after initial promising results in clinical studies12,46 a phase III study evaluating the Nrf2 activator bardoxolone in type 2 diabetic patients with stage IV CKD (chronic kidney disease) was prematurely stopped because of increased of heart failure rates in the treatment group, which may reflect off-target effects6,13. Minocycline may present an interesting alternative in this regard. Minocycline has been in clinical use for decades, e.g. as a long-term treatment for acne in adolescence or for gingivitis in diabetic patients and is generally well-tolerated47,48,49. Thus, clinical experience and safety data on minocycline are on hand. Correspondingly, minocycline was well tolerated in the current rodent study, as mice did not show any obvious signs of toxicity and liver enzymes (ALT, AST) remained stable (Supplementary Fig. S5). This contrasts a previous study using the bardoxolone methyl analogue RTA 405 in ZDF rats, in which liver toxicity and an impaired physical status of RTA 405 treated animals was observed50. In addition, minocycline is inexpensive and readily available and, based on the current results, targets crucial pathomechanisms in dNP (excess ROS formation and sterile inflammation). Interestingly, small pilot studies demonstrated that oral application of minocycline is not only a safe, but also an effective treatment in diabetic patients with macular edema and neuropathy28,29. Very recently, the anti-proteinuric effect of minocycline was prospectively evaluated in 14 patients with dNP17. After 24 weeks of minocycline treatment a non-significant reduction of urine protein/creatinine and microalbumin/creatinine ratios was apparent in the minocycline, but not in the placebo treated group17. Interestingly, inflammatory markers (urine IL6 and osteoprotegerin) were significantly reduced in the minocycline group, which is in agreement with the anti-inflammatory effect of minocycline identified within this study. Based on the current and this recent clinical pilot study sufficiently powered studies, possibly for longer period, evaluating the efficacy of minocycline in patients with dNP are now warranted17,18,19.
The mouse models used in this study for diabetic nephropathy are known to develop functional and morphological renal abnormalities51,52. However, in comparison to the human disease only features of early diabetic nephropathy are replicated. The typical nodular glomerulosclerosis of late stage dNP in humans is not apparent in STZ induced diabetic C57BL/6 or db/db mice51. Therefore the potential of minocycline to target advanced pathological features of human dNP cannot be assessed. Future studies using mouse models of more advanced dNP, such as eNOS-deficient mice53,54, may be better suitable to evaluate the therapeutic benefit of minocycline in advanced dNP. Alternatively, observational clinical studies may provide insights into the role of minocycline in advanced dNP, e.g. in patients with established dNP and receiving minocycline for a different indication (e.g. acne or infections). Furthermore, the question whether minocycline may provide an added value on top of established therapies such as ACE-inhibition or reverses established dNP remains to be addressed in future studies.
The cytoprotective effects of minocycline, which was originally identified as a tetracycline-derived antibiotic, are well documented and have been linked with modulation of mitochondrial function, normalization of the Bax/Bcl-2 ratio, reduced cytochrome C release and apoptosis inhibition14,15. However, the exact mechanism of minocycline mediated cytoprotection remains unknown. By linking minocycline’s protective effect with sustained Nrf2 protein levels and reduced Nrf2 ubiquitination the current data provides new insights into the cytoprotective action of minocycline. The importance of the ubiquitin-proteasomal system for the regulation of Nrf2 is well established7 and hence it appears plausible that the minocycline mediated reduction of Nrf2 ubiquitination is functionally relevant. We acknowledge that the exact mechanism through which minocycline inhibits Nrf2 ubiquitination and the relevance of this for Nrf2 protein stabilization or sub-cellular localization remain to be evaluated55. Of note, the current results are in line with recent reports demonstrating that proteasomal inhibitors induce anti-oxidant effects56. Furthermore, the proteasomal inhibitor MG132 conveys nephroprotective effects in diabetic rats, which have been linked with increased Nrf2, superoxide dismutase-1 (SOD1), catalase (CAT) and glutathione peroxidase (GPx) expression and reduced nitrotyrosine levels in the kidney of diabetic rat57. However, while in the latter study the causality between increased Nrf2 expression and nephroprotection by MG132 remained unanswered, we demonstrate a mechanistic relevance of sustained Nrf2 expression for minocycline mediated nephroprotection using genetically Nrf2 deficient mice. Based on the current results minocycline posttranslationally upregulates Nrf2 in glucose stressed podocytes in vitro by inhibiting its glucose-induced ubiquitination. Further studies are needed to identify the mechanism through which minocycline limits glucose induced Nrf2 ubiquitination, e.g. the potential involvement of deubiquitinating enzymes. Such mechanistic details will then allow further studies, e.g. podocyte specific inhibition of the corresponding deubiquitinating enzyme, to provide in vivo evidence for the proposed mechanism.
The association of dNP and sterile inflammation is now established21,22. Based on the current and previous work we propose that glucose induced ROS and inflammasome activation drive the disease processes in dNP21,22,57. Therapeutic approaches directly targeting the inflammasome or controlling the inflammasome indirectly by restricting unchecked ROS generation via Nrf2 and/or limiting proteasomal degradation of Nrf2, as shown here for minocycline, may constitute promising new approaches to combat onset and progression of dNP.
Material and Methods
Mice
Nrf2−/− (Nfe2l2), db/db (Lepr db/db) and non-diabetic db/m control mice (C57BL/6J background) were obtained from Jackson Laboratories, Bar Harbor, ME, USA. In the current study we used littermates which have been backcrossed for at least 10 generations on a C57BL/6J background. All animal experiments were conducted in accordance with the relevant guidelines by the local Animal Care and Use Committee and approved by the local Animal Care and Use Committee (Landesverwaltungsamt Halle, Germany; Licences: 2-1085, 2-1179 and 2-1104).
Diabetic nephropathy models
We used two different mouse models of dNP within the current study. First, we use the db/db mice, in which treatment was initiated at age 8-weeks and mice were sacrificed after 12 weeks of treatment at the age of 20 weeks. In non-treated db/db mice indices of dNP, e.g. albuminuria and glomerular extracellular matrix accumulation (FMA, fractional mesangial area), are markedly increased in 20 weeks old db/db mice58.
In addition we used the streptozotocin (STZ) plus unilateral nephrectomy model of dNP22,59,60. In this model diabetes was induced by injections of STZ (i.p., 40 mg/kg body weight, freshly dissolved in 0.05 M sterile sodium citrate, pH 4.5) for five consecutive days two weeks after unilateral nephrectomy. Age-matched control mice received 100 μL PBS i.p. for five consecutive days. Mice were considered diabetic if blood glucose levels were above 300 mg/dL 16 d after the last STZ injection. Blood and tissue samples were obtained after 10 weeks of persistent hyperglycemia in uni-nephrectomised diabetic mice. Age-matched littermates served as controls.
In vivo intervention studies
A subset of mice was treated with the poly-caspase inhibitor CIX (peptide sequence: Ac-DEVD-CMK) (20 mg/kg body weight daily; intraperitoneally), known not to affect IL1β processing (Merck, Millipore) or minocycline (5 mg/kg body weight daily, intraperitoneally)18,61. Control mice received an equal volume of PBS. Interventions with minocycline or CIX or were initiated in 8 weeks old db/db mice to determine its preventive effect. In uninephrectomized, STZ treated mice treatment with minocycline was initiated 2 weeks after establishment of stable hyperglycemia and continued for 10 weeks. Control mice were injected with PBS.
Histology and immuohistochemical analysis
Histology and immunohistochemical analysis was performed as previously described18,22,59,62. Freshly sacrificed mice were first perfused with ice cold PBS (10 ml) and then with 4% buffered paraformaldehyde (5 ml). Tissues were furthered fixed with 4% buffered paraformaldehyde for 2 days at 4 °C, then embedded in paraffin and processed for sectioning. Extracellular matrix deposition in glomeruli was assessed by Periodic acid–Schiff staining. Histological evaluations were performed in accordance with the Diabetes Complications Consortium guidelines (DCC) (www.diacomp.org). The fractional mesangial area (FMA) was calculated following the current DCC protocol. Briefly, 4–5 μm thick sections were stained with Periodic acid-Schiff reagent. At least 30 different superficial glomeruli/mouse were randomly chosen for analysis. For every investigated glomerulus, total glomerular area and glomerular tuft area were determined by tracing the outline of the Bowman’s capsule and the tuft, respectively, using ImageJ. The FMA was calculated as the percentage of the glomerular area relative to the tuft area22,59. Immunohistochemical detection of anti-8-oxoguanine, Nrf2, Nlrp3, synaptopodin, cleaved caspase 1, or nitrotyrosine was performed using corresponding specific primary and secondary antibodies. Control images, obtained following incubation with non-specific primary antibodies, were used for background correction. Specificity of caspase-1 and Nlrp3 antibodies was confirmed using sections of corresponding knock-out mice as negative controls. The Image Pro Plus software (version 6.0) and Image J software were used for image analysis. All histological analyses were performed by two independent blinded investigators. Immunohistochemistry and immunofluorescence images were captured with an Olympus Bx43-Microscope (Olympus, Hamburg, Gemany). Confocal images were obtained at 40x magnification using a Zeiss LSM 4 Pascal microscope (Carl Zeiss, Jena, Germany).
Determination of albuminuria and liver enzymes
Mouse urine albumin and creatinine were measured as previously described22,59. Briefly, the day before sacrificing animal at the end of experimental time, mice were placed individually in metabolic cages and 12 hour urine samples were collected. Urine albumin was determined using an ELISA for mouse albumin according to the manufacturer’s instructions and urine creatinine was determined using a commercially available assay of a modified version of the Jaffe method (X-Pand automated platform, Siemens). Serum activity of the liver enzymes ALT (alanin aminotransferase) or AST (aspartat aminotransferase) was determined according to current protocols by the IFCC (International Federation of Clinical Chemistry) with a pyridoxalphosphate-dependent UV-reaction at 37 °C.
Cell culture, Nrf2 knockdown and Nlrp3 gain of function
Conditionally immortalized human and mouse wild-type podocytes were cultured as described previously18,62. In brief, podocytes were grown on 10 cm2 cell culture plates coated with 0.2% collagen type 1 at 33 °C in the presence of interferon-γ (10 U/ml) to enhance expression of the thermosensitive T antigen. Under these conditions, cells proliferate and remain undifferentiated. To induce differentiation, podocytes were grown at 37 °C in the absence of interferon-γ for 14 days. Experiments were performed after 14 days of differentiation. Differentiation was confirmed by determining expression of synaptopodin and Wilms tumor-1 protein. 293T cells (American Type Culture Collection, Rockville, MD) were cultured in DMEM with 10% FBS and penicillin/streptomycin. Mouse glomerular endothelial cells (GEnC) were obtained from J. Anders (Munich, Germany). GEnC were maintained in RPMI medium with 10% FCS and 1% antibiotics at 37 °C in a humidified 5% CO2 incubator.
Mouse Nrf2 gene expression was knocked down using siRNA against Nrf2 gene transcripts. Briefly, 0.25–1 × 106 cells were seeded in 6 well culture plate. Upon reaching 60–80% confluence, siRNA (final conc. 30p mol) was added to diluted lipofectamine®RNAiMAX reagent in 1:1 ratio. Cells were then incubated for 5 minutes at room temperature. siRNA lipid complex (final conc. 25 pmol) was added to cells and Incubated for 3 days at 37 °C. The transfection efficiency was ascertained by Nrf2 protein expression, as determined by immunoblotting.
A Nlrp3 mutant resulting in expression of an constitutive active IL1β was used to generate cells with a gain-of-function of Nrlp3, as previously described22.
Glomerular filtration barrier in vitro assay
To determine glomerular filtration barrier we used a recently established in vitro model mimicking central aspects of the glomerular filtration barrier with some modifications63,64. This model consists of a polyethyleneterephthalate (PET) membrane (Millipore membrane, Millicell Hanging Cell Culture Insert) with 1 μm pores that is coated on both sides with collagen type IV. First inserts were placed upside-down in a larger well and podocytes were seeded on the membrane-side facing upwards. Once attached (~6 h), the insert was turned around, placed in a well of appropriate size and podocytes were allowed to differentiate for 10 days. Then GEnC were placed on the side of the membrane now facing upwards. Cells were cultured under resting conditions for additional four days using podocyte and endothelial cell specific medium in the corresponding chamber. This in vitro model enables functional permeability assays by determining the transmembrane passage of fluorescently labeled proteins with a defined molecular weight (such as FITC-labelled albumin). To mimic hyperglycemic conditions corresponding medium containing 25 mM of glucose was added to both chambers, while control cells were kept at low glucose concentrations (5 mM). After 48 hours medium in both chambers was changed to serum-free medium (SFM, RPMI 1640). After 1 h FITC-labeled BSA (Sigma) was added to the upper chamber at a final concentration of 0.5 mg/ml. After further 3 h the concentration of FITC-labeled BSA in the lower chamber was measured. The fluorescence was measured using fluorometric spectrophotometer at 495 nm and the concentration of FITC-BSA was calculated by reference to a set of standard dilutions. The amount of FITC-labeled BSA accumulating in the lower chamber reflects the barrier permeability.
Immunoblotting and Immunoprecipitation
Immunoblotting and Immunoprecipitation were performed as previously described18,22,59,62. In brief, for immunoblotting whole cell lysates were prepared in RIPA buffer (50 mM Tris at pH 7.4, 1% Nonidet P-40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA and 1 mM Na3VO4, supplemented with protease inhibitor cocktail). Lysates were centrifuged (10,000 × g for 20 min at 4 °C) and supernatant was kept, while the pellet containing debris was discarded. The protein concentration in supernatants was quantified using BCA reagent. Equal amounts of protein were electrophoretically separated on 10% (vol/vol) or 12.5% (vol/vol) SDS polyacrylamide gels, transferred to PVDF membranes and probed with the desired primary antibodies overnight at 4 °C. Membranes were then washed with PBST and incubated with anti-mouse (1:2,000), anti-goat IgG (1:2000), anti-rat IgG (1:2000) or anti-rabbit IgG (1:2000) horseradish peroxidase-conjugated antibodies, as indicated. Blots were developed with the immobilon western chemiluminiscent HRP substrate. To compare and quantify levels of proteins, the density of each band was measured by using ImageJ software. Equal protein loading was confirmed by immunoblotting with β-actin or GAPDH antibody.
Immunoprecipitation of podocyte total cellular proteins was done as described elsewhere62. Total cellular proteins were extracted with RIPA containing complete protease inhibitor cocktail. Samples were sonicated (set at 10% of maximum power) for 10 sec followed by 1 min incubation on ice. This process was repeated five times. Lysates were combined with 5 μg of specific antibody and incubated overnight at 4 °C on rotating shaker. Immunoprecipitates were purified with protein A/G agarose beads and washed with PBS containing protease inhibitor cocktail. Immunoprecipitates were fractionated by SDS-PAGE (10%), transferred to membranes and subjected to immunoblotting with appropriate primary and secondary antibodies as described above.
Mitochondrial ROS production
Mitochondrial specific ROS were measured as described elsewhere22 using MitoSOX™ following manufacturer’s instructions. Briefly, mouse podocyte were grown in 96-wells cell culture plate. Upon reaching 90% confluence, cells were washed once with PBS and then incubated with 5 μM MitoSOX™ for 10 min at 37 °C in the dark. Cells were washed 3x with wash buffer (Hank’s balanced salt solution, HBSS) and mitochondrial specific ROS were measured using a fluorescent plate reader (excitation/emission of 510/580 nm).
RT-PCR
RNA was isolated using Trizol reagent according to manufacturer’s instructions. cDNA was generated using 1 μg total RNA following treatment with DNAse (5U/5 μg RNA) followed by reverse transcription using RevertAid™ H Minus First Strand cDNA Synthesis kit (Fermentas, Heidelberg, Germany). PCR primers used for expression of Nrf2 analyses are as follows: forward 5′-CGCTGGAAAAAGAAGTGGGC-3′, reverse 5′-AGTGACTGACTGATGGCAGC-3′. PCR conditions were optimized (95 °C for 2 min, then 35 cycles of 94 °C, for 20 sec; 60 °C for 20 sec; 72 °C for 30 sec; final extension at 72 °C for 12 min) to detect the logarithmic increase of the amplimer, which was separated on a 1.8% agarose gel and visualized by ethidium bromide staining. Expression was normalized to β-actin. Reactions lacking reverse transcriptase served as negative controls.
Statistical Analysis
The data are summarized as means ± SEM (standard error of the mean). Statistical analyses were performed with Student’s t-test, ANOVA, Mann-Whitney-Test, as appropriate and post-hoc comparison with the method of Tukey. The Kolmogorov-Smirnov (KS) test or D’Agostino-Pearson-Normality-test were used to determine whether the data are consistent with a Gaussian distribution. StatistiXL (www.statistixl.com) and Prism 5 (www.graphpad.com) software were used for statistical analyses. All data presented involving cell culture is representative of at least three independent repeat experiments. Statistical significance was accepted at values of p < 0.05.
Additional Information
How to cite this article: Shahzad, K. et al. Stabilization of endogenous Nrf2 by minocycline protects against Nlrp3-inflammasome induced diabetic nephropathy. Sci. Rep. 6, 34228; doi: 10.1038/srep34228 (2016).
References
Martinez-Castelao, A., Navarro-Gonzalez, J. F., Gorriz, J. L. & de Alvaro, F. The Concept and the Epidemiology of Diabetic Nephropathy Have Changed in Recent Years. J Clin Med 4, 1207–1216 (2015).
Reidy, K., Kang, H. M., Hostetter, T. & Susztak, K. Molecular mechanisms of diabetic kidney disease. J Clin Invest 124, 2333–2340 (2014).
Zoja, C., Zanchi, C. & Benigni, A. Key pathways in renal disease progression of experimental diabetes. Nephrol Dial Transplant 30 Suppl 4, iv54–iv59 (2015).
Forbes, J. M. & Cooper, M. E. Mechanisms of diabetic complications. Physiol Rev 93, 137–188 (2013).
Choi, B. H., Kang, K. S. & Kwak, M. K. Effect of redox modulating NRF2 activators on chronic kidney disease. Molecules 19, 12727–12759 (2014).
Ruiz, S., Pergola, P. E., Zager, R. A. & Vaziri, N. D. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Int 83, 1029–1041 (2013).
Niture, S. K., Khatri, R. & Jaiswal, A. K. Regulation of Nrf2-an update. Free Radic Biol Med 66, 36–44 (2014).
Huang, X. S. et al. Nrf2-dependent upregulation of antioxidative enzymes: a novel pathway for hypoxic preconditioning-mediated delayed cardioprotection. Mol Cell Biochem 385, 33–41 (2014).
Suzuki, T., Motohashi, H. & Yamamoto, M. Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol Sci 34, 340–346 (2013).
Zheng, H. et al. Therapeutic potential of Nrf2 activators in streptozotocin-induced diabetic nephropathy. Diabetes 60, 3055–3066 (2011).
Jiang, T. et al. The protective role of Nrf2 in streptozotocin-induced diabetic nephropathy. Diabetes 59, 850–860 (2010).
Pergola, P. E. et al. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N Engl J Med 365, 327–336 (2011).
de Zeeuw, D. et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N Engl J Med 369, 2492–2503 (2013).
Kelly, K. J. et al. Minocycline inhibits apoptosis and inflammation in a rat model of ischemic renal injury. Am J Physiol Renal Physiol 287, F760–F766 (2004).
Sutton, T. A. et al. Minocycline reduces renal microvascular leakage in a rat model of ischemic renal injury. Am J Physiol Renal Physiol 288, F91–F97 (2005).
Wang, J. et al. Minocycline up-regulates Bcl-2 and protects against cell death in mitochondria. J Biol Chem 279, 19948–19954 (2004).
Shah, A. P. et al. Effects of Minocycline on Urine Albumin, Interleukin-6 and Osteoprotegerin in Patients with Diabetic Nephropathy: A Randomized Controlled Pilot Trial. PLoS One 11, e0152357 (2016).
Isermann, B. et al. Activated protein C protects against diabetic nephropathy by inhibiting endothelial and podocyte apoptosis. Nat Med 13, 1349–1358 (2007).
Kumar Bhatt, L. & Addepalli, V. Minocycline with aspirin: an approach to attenuate diabetic nephropathy in rats. Ren Fail 33, 72–78 (2011).
Susztak, K., Raff, A. C., Schiffer, M. & Bottinger, E. P. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes 55, 225–233 (2006).
Shahzad, K. et al. Caspase-1, but Not Caspase-3, Promotes Diabetic Nephropathy. J Am Soc Nephrol (2016).
Shahzad, K. et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int 87, 74–84 (2015).
Alano, C. C., Kauppinen, T. M., Valls, A. V. & Swanson, R. A. Minocycline inhibits poly(ADP-ribose) polymerase-1 at nanomolar concentrations. Proc Natl Acad Sci USA 103, 9685–9690 (2006).
Ryan, M. E., Usman, A., Ramamurthy, N. S., Golub, L. M. & Greenwald, R. A. Excessive matrix metalloproteinase activity in diabetes: inhibition by tetracycline analogues with zinc reactivity. Curr Med Chem 8, 305–316 (2001).
Zhou, R., Yazdi, A. S., Menu, P. & Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225 (2011).
Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 53, 401–426 (2013).
Gao, C., Huang, W., Kanasaki, K. & Xu, Y. The role of ubiquitination and sumoylation in diabetic nephropathy. Biomed Res Int 2014, 160692 (2014).
Cukras, C. A., Petrou, P., Chew, E. Y., Meyerle, C. B. & Wong, W. T. Oral minocycline for the treatment of diabetic macular edema (DME): results of a phase I/II clinical study. Invest Ophthalmol Vis Sci 53, 3865–3874 (2012).
Syngle, A., Verma, I., Krishan, P., Garg, N. & Syngle, V. Minocycline improves peripheral and autonomic neuropathy in type 2 diabetes: MIND study. Neurol Sci 35, 1067–1073 (2014).
Zhou, R., Tardivel, A., Thorens, B., Choi, I. & Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol 11, 136–140 (2010).
Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 414, 813–820 (2001).
Forbes, J. M., Coughlan, M. T. & Cooper, M. E. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes 57, 1446–1454 (2008).
Gao, L. & Mann, G. E. Vascular NAD(P)H oxidase activation in diabetes: a double-edged sword in redox signalling. Cardiovasc Res 82, 9–20 (2009).
Miki, H. & Funato, Y. Regulation of intracellular signalling through cysteine oxidation by reactive oxygen species. J Biochem 151, 255–261 (2012).
Perez-Matute, P., Zulet, M. A. & Martinez, J. A. Reactive species and diabetes: counteracting oxidative stress to improve health. Curr Opin Pharmacol 9, 771–779 (2009).
Anders, H. J. & Muruve, D. A. The inflammasomes in kidney disease. J Am Soc Nephrol 22, 1007–1018 (2011).
Balasubramaniam, G., Almond, M. & Dasgupta, B. Improved renal function in diabetic patients with acute gout treated with anakinra. Kidney Int 88, 195–196 (2015).
Jaiswal, A. K. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic Biol Med 36, 1199–1207 (2004).
Wakabayashi, N., Slocum, S. L., Skoko, J. J., Shin, S. & Kensler, T. W. When NRF2 talks, who’s listening? Antioxid Redox Signal 13, 1649–1663 (2010).
Sweetwyne, M. T. et al. Notch1 and Notch2 in Podocytes Play Differential Roles During Diabetic Nephropathy Development. Diabetes 64, 4099–4111 (2015).
Mooradian, A. D. Targeting Select Cellular Stress Pathways to Prevent Hyperglycemia-Related Complications: Shifting the Paradigm. Drugs 76, 1081–1091 (2016).
Tan, Y. et al. Diabetic downregulation of Nrf2 activity via ERK contributes to oxidative stress-induced insulin resistance in cardiac cells in vitro and in vivo. Diabetes 60, 625–633 (2011).
Velmurugan, G. V., Sundaresan, N. R., Gupta, M. P. & White, C. Defective Nrf2-dependent redox signalling contributes to microvascular dysfunction in type 2 diabetes. Cardiovasc Res 100, 143–150 (2013).
Foresti, R. et al. Nrf2 activators modulate oxidative stress responses and bioenergetic profiles of human retinal epithelial cells cultured in normal or high glucose conditions. Pharmacol Res 99, 296–307 (2015).
Chapple, S. J., Siow, R. C. & Mann, G. E. Crosstalk between Nrf2 and the proteasome: therapeutic potential of Nrf2 inducers in vascular disease and aging. Int J Biochem Cell Biol 44, 1315–1320 (2012).
Pergola, P. E. et al. Effect of bardoxolone methyl on kidney function in patients with T2D and Stage 3b-4 CKD. Am J Nephrol 33, 469–476 (2011).
Garner, S. E. et al. Minocycline for acne vulgaris: efficacy and safety. Cochrane Database Syst Rev 8, CD002086 (2012).
Garrido-Mesa, N., Zarzuelo, A. & Galvez, J. Minocycline: far beyond an antibiotic. Br J Pharmacol 169, 337–352 (2013).
Lessem, J. & Hanlon, A. A post-marketing study of 2805 patients treated for periodontal disease with Arestin. J Int Acad Periodontol 6, 150–153 (2004).
Zoja, C. et al. Analogs of bardoxolone methyl worsen diabetic nephropathy in rats with additional adverse effects. Am J Physiol Renal Physiol 304, F808–F819 (2013).
Alpers, C. E. & Hudkins, K. L. Mouse models of diabetic nephropathy. Curr Opin Nephrol Hypertens 20, 278–284 (2011).
Brosius, F. C. 3rd et al. Mouse models of diabetic nephropathy. J Am Soc Nephrol 20, 2503–2512 (2009).
Kosugi, T., Heinig, M., Nakayama, T., Matsuo, S. & Nakagawa, T. eNOS knockout mice with advanced diabetic nephropathy have less benefit from renin-angiotensin blockade than from aldosterone receptor antagonists. Am J Pathol 176, 619–629 (2010).
Nakagawa, T. et al. Diabetic endothelial nitric oxide synthase knockout mice develop advanced diabetic nephropathy. J Am Soc Nephrol 18, 539–550 (2007).
Niture, S. K., Jain, A. K., Shelton, P. M. & Jaiswal, A. K. Src subfamily kinases regulate nuclear export and degradation of transcription factor Nrf2 to switch off Nrf2-mediated antioxidant activation of cytoprotective gene expression. The Journal of biological chemistry 286, 28821–28832 (2011).
Meiners, S. et al. Nontoxic proteasome inhibition activates a protective antioxidant defense response in endothelial cells. Free Radic Biol Med 40, 2232–2241 (2006).
Luo, Z. F. et al. Prevention of diabetic nephropathy in rats through enhanced renal antioxidative capacity by inhibition of the proteasome. Life Sci 88, 512–520 (2011).
Sharma, K., McCue, P. & Dunn, S. R. Diabetic kidney disease in the db/db mouse. American journal of physiology. Renal physiology 284, F1138–F1144 (2003).
Bock, F. et al. Activated protein C ameliorates diabetic nephropathy by epigenetically inhibiting the redox enzyme p66Shc. Proc Natl Acad Sci USA 110, 648–653 (2013).
Gil-Bernabe, P. et al. Exogenous activated protein C inhibits the progression of diabetic nephropathy. J Thromb Haemost 10, 337–346 (2012).
Shahzad, K. et al. Minocycline reduces plaque size in diet induced atherosclerosis via p27(Kip1). Atherosclerosis 219, 74–83 (2011).
Madhusudhan, T. et al. Defective podocyte insulin signalling through p85-XBP1 promotes ATF6-dependent maladaptive ER-stress response in diabetic nephropathy. Nat Commun 6, 6496 (2015).
Li, M. et al. Three-dimensional podocyte-endothelial cell co-cultures: Assembly, validation and application to drug testing and intercellular signaling studies. Eur J Pharm Sci 86, 1–12 (2016).
Satchell, S. C., Anderson, K. L. & Mathieson, P. W. Angiopoietin 1 and vascular endothelial growth factor modulate human glomerular endothelial cell barrier properties. J Am Soc Nephrol 15, 566–574 (2004).
Acknowledgements
This work was supported by grants from the Deutsche Forschungsgemeinschaft (SH 849/1-2 to KS; IS 67/4-3, IS 67/5-3 and SFB 854/B26N to BI; Me 1365/8-1 and SFB 854/A1 to PRM; WA 3663/2-1; SFB1118 to PPN), the EFSD (European Foundation for the Study of Diabetes, BI), Hopp Stiftung and German Diabetes Center (PPN) and a DAAD scholarship to MMA. We thank Kathrin Deneser, Julia Judin, Juliane Friedrich, Rene Rudat and Rumiya Makarova (all affiliated with the Institute of Clinical Chemistry and Pathobiochemistry, Otto-von-Guericke-University, 39120 Magdeburg, Germany) for excellent technical support.
Author information
Authors and Affiliations
Contributions
K.S., F.B. and M.M.A. designed, performed and interpreted experiments; I.G., S.N. H.W., Sh.K. and S.R., conducted mouse experiments and ex vivo analyses; P.P.N. and P.R.M. interpreted data and B.I. designed and interpreted the experimental work and prepared the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Shahzad, K., Bock, F., Al-Dabet, M. et al. Stabilization of endogenous Nrf2 by minocycline protects against Nlrp3-inflammasome induced diabetic nephropathy. Sci Rep 6, 34228 (2016). https://doi.org/10.1038/srep34228
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep34228
This article is cited by
-
Regulation of synaptic connectivity in schizophrenia spectrum by mutual neuron-microglia interaction
Communications Biology (2023)
-
Tetracycline ameliorates silica-induced pulmonary inflammation and fibrosis via inhibition of caspase-1
Respiratory Research (2022)
-
Novel insights into NOD-like receptors in renal diseases
Acta Pharmacologica Sinica (2022)
-
Selenium Attenuates Doxorubicin-Induced Cardiotoxicity Through Nrf2-NLRP3 Pathway
Biological Trace Element Research (2022)
-
An arylthiazyne derivative is a potent inhibitor of lipid peroxidation and ferroptosis providing neuroprotection in vitro and in vivo
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
