
Abstract
The Indian pepper ‘Guijiangwang’ (Capsicum frutescens L.), one of the world’s hottest chili peppers, is rich in capsaicinoids. The accumulation of the alkaloid capsaicin and its analogs in the epidermal cells of the placenta contribute to the pungency of Capsicum fruits. To identify putative genes involved in capsaicin biosynthesis, RNA-Seq was used to analyze the pepper’s expression profiles over five developmental stages. Five cDNA libraries were constructed from the total RNA of placental tissue and sequenced using an Illumina HiSeq 2000. More than 19 million clean reads were obtained from each library, and greater than 50% of the reads were assignable to reference genes. Digital gene expression (DGE) profile analysis using Solexa sequencing was performed at five fruit developmental stages and resulted in the identification of 135 genes of known function; their expression patterns were compared to the capsaicin accumulation pattern. Ten genes of known function were identified as most likely to be involved in regulating capsaicin synthesis. Additionally, 20 new candidate genes were identified related to capsaicin synthesis. We use a combination of RNA-Seq and DGE analyses to contribute to the understanding of the biosynthetic regulatory mechanism(s) of secondary metabolites in a nonmodel plant and to identify candidate enzyme-encoding genes.
Similar content being viewed by others


Introduction
Capsaicinoids, which accumulate in the vesicles and vacuoles of epidermal cells of the placenta1, are responsible for the pungent taste of chili pepper (Capsicum sp.) fruits. Capsaicin and dihydrocapsaicin are the primary determinants capsaicinoid content of chili pepper fruits, accounting for 90% of the total content. As the source of their pungency, the concentration of capsaicinoids is the most important trait of peppers. Capsaicinoids are widely used in many scientific fields including the food, cosmetic, and pharmaceutical industries2. Thus, high capsaicinoid content is an important breeding goal for this crop plant. However, the limited material and dynamics of capsaicin synthesis complicate the genetics and gene discovery. Moreover, the molecular basis of capsaicin biosynthesis has not yet been explored. ‘Guijiaowang’ is a species of pepper that contains abundant capsaicinoids and, unlike other highly pungent pepper species, it displays obvious changes at different developmental stages. Therefore, we used this species to investigate capsaicin biosynthesis in pepper.
Capsaicinoids are synthesized via both the phenylpropanoid and the branched-fatty-acid pathways3. Numerous enzymes are involved in capsaicin biosynthesis, but many of these are not well characterized, and pathway regulation is not fully understood. Phenylalanine ammonia-lyase (PAL) is the first enzyme in the phenylpropanoid pathway, in which cinnamic acid is formed from phenylalanine4, followed by cinnamatc 4-hydroxylase (C4H), 4-Coumarate:Coenzyme A Ligase (4CL), hydroxycinnamoyl transferase (HCT), coumarate3-hydroxylase (C3H), caffeoyl-CoA 3-O-methyltransferase (CCoAOMT), and a putative aminotransferase (pAMT) to form vanillylamine. In the branched-fatty-acid synthesis pathway, the precursor 8-methyl-6-nonenoyl-CoA is produced via the fatty-acid synthesis cycle by a series of several enzymatic reactions5,6. In the end, vanillyamine and 8-methyl-6-nonenoyl-CoA, which are produced as two branched chains, are converted to capsaicin by acyl-transferase (AT)7. Capsaicinoid biosynthesis has long been of interest to researchers, and the pathway for the synthesis of all capsaicinoids has been proposed7. However, the regulation of the abundance of each analog in the fruit is not well understood. Over fifty gene families are involved in capsaicinoid biosynthesis8,9. Previous studies have indicated that the Pun1 locus, which is required for the presence of capsaicinoids in pepper, encodes a putative acyltransferase7,10,11. Quantitatively inherited traits that control developmental processes in pepper are influenced by many genes with small effects10. Thus, their direct participation in the capsaicinoid biosynthesis pathway has not been demonstrated12.
A comparison of cDNA libraries prepared from the placenta of chili fruit indicated that PAL, Ca4H, CCoAOMT, Kas, and pAMT are differentially expressed. Moreover, their expression at the transcriptional level in the placenta is positively correlated with the degree of pungency in the chili pepper13. Biochemical analyses of placental tissues have shown that the independent silencing of CCoAOMT, Kas and pAMT can dramatically reduce capsaicinoid content in fruits12. Previous studies used suppression subtractive hybridization (SSH) to identify four genes that are involved in capsaicin metabolism. A BLAST search determined that these genes had high homology with pAMT, Kas, Acyltransferase (AT), and FAO13. They are specifically expressed in placental tissue and are accompanied by capsaicin accumulation. Liu et al. detected 52 candidate genes including PAT, TD and DHAD in the CapCyc model by transcriptome sequencing14. Qin and Kim detected 51 and 44 of these 52 genes, respectively, in chili genome-wide association studies. Partial genes have been used to study expression characteristics, polymorphisms, and phylogeny8,9.
Biological and morphological changes in resource utilization and allocation in tissues during development are accompanied by differences in gene expression patterns at various stages of fruit development14. In this paper, we illustrate a proposed pathway of capsaicin biosynthesis and analyze the expression of key genes involved in the biosynthetic pathway. The RNA-Seq platform15,16 was used to analyze the expression profiles of putative capsaicin biosynthetic genes in placental tissue, during five developmental stages of pepper fruit. The identified candidate genes could help to elucidate the molecular basis of pungency.
Methods
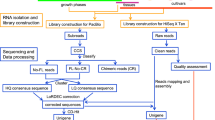
Plant material and cDNA library construction
The pepper seeds were obtained from “Hainan University, Haikou, China”. Fruits used in the experiments were grown in a growth room at 26 °C/20 °C (day/night temperatures) in 14 h light and 10 h dark per day. Individual flowers were tagged at anthesis. Fruits were randomly collected from different plants at 10, 20, 30, 40 and 50 days after pollination (DAP). Placental tissue (500 mg) from ten individual fruits for each time point were used for RNA extraction. The process was repeated in two independent biological replicates at each developmental stage.
Total RNA was isolated using the Total RNA Isolation System (Takara, Dalian, China) according to the manufacturer’s instructions. RNA quality was checked using an Agilent 2100 Bioanalyzer RNA Nano chip device (Agilent, Santa Clara, CA, USA), and total RNA concentrations were determined using a NanoDrop ND-1000 spectrophotometer (Nano-Drop, Wilmington, DE, USA). RNA with an OD260/280 between 1.8 and 2.2 and an OD260/230 ≥ 1.8 was used for the construction of cDNA libraries.
Determination of capsaicin content in fruit placenta
Placental tissues of fruits were harvested at 10, 20, 30, 40 and 50 DAP, dried at 50 °C for 6 h, then homogenized with neutralized glass powder (100 mg) using a mortar and pestle. The capsaicinoid extraction from the dried fruits followed the methods proposed by Alothman et al. (2012) with slight modifications. Dried pepper samples (0.2 g) were added to ethanol (70%, 2.0 ml) in a 10 mL glass bottle equipped with a Teflon-lined lid. Bottles were capped and placed in a water bath at 80 °C for 4 hours. The samples were sonicated for 50 minutes with a working frequency of 35 kHz. The extract was centrifuged at 12,000 × g for 15 min, and the supernatants were filtrated through a 0.45 μm syringe-mounted membrane filter. Samples were stored at −20 °C. Capsaicin analyses were performed by reversed-phase HPLC using a Kromasil Eternity-5-C18 column (4.6 mm × 250 mm). Samples were eluted using a water:methanol (30:70, v/v) mixture. The detection wavelength was set at 280 nm. The injection volume was 10 μL. All measurements were repeated in triplicate.
cDNA library construction and sequencing
The mRNA content was concentrated from total RNA using RNase-free DNase I (TaKaRa) and magnetic oligo (dT) beads. The mRNA was mixed with the fragmentation buffer and broken into short fragments (~200 bp long). Then, the first strand of cDNA was synthesized with a random hexamer primer. The second strand was synthesized using the SuperScript Double-Stranded cDNA Synthesis kit (Invitrogen, Camarillo, CA) and was purified via magnetic beads. The ends were repaired and tailed with a single 3′ adenosine. Subsequently, the cDNA fragments were ligated to sequencing adapters. Sequencing was accomplished using an Illumina HiSeq™ 2000 platform according to the manufacturer’s protocols.
Analysis and mapping of DGE reads
Image data generated by the Illumina HiSeq 2000 system was transformed from base calling data to sequence data. Raw reads were filtered to remove adaptor sequences, low quality tags (tags with unknown nucleotides N > 10%), and reads with more than 50% low quality (≤5) bases. The remaining reads were mapped to the reference genome (http://peppergenome.snu.ac.kr) using SOAPaligner/SOAP2. A maximum of two mismatches was determined to be acceptable for alignment purposes.
For gene expression analysis, expression levels were estimated based on the frequency of individual reads and then normalized to the number of reads per kb per million reads (RPKM). We used a probability ≥ 0.8 and a |log2Ratio| value ≥ 1 as thresholds to determine significant changes in gene expression. Finally, these differentially expressed tags were used for mapping and gene annotation.
Gene ontology (GO) and KEGG pathway enrichment analysis of differentially expressed genes (DEGs)
The Gene Ontology database (http://www.geneontology.org/) classifies genes into three general categories including “cellular component,” “molecular function” and “biological process”17. It can be used to predict the biological function of DEGs after mapping the sequencing reads to reference genes. After annotating expressed genes with GO terms, a GO functional classification was performed with WEGO.
All DEGs were mapped to terms in the KEGG database. Genes with a P-value ≤ 0.05 were considered to be differentially expressed. KEGG was used to graphically represent biological pathways and to display the up- or down-regulation of genes in a pathway.
qRT-PCR analysis for validating the DEGs
Total RNA was isolated from placental tissue at different developmental stages as described above. Genomic DNA was removed by treating with RNase-free Dnase I, and the first strand of cDNA was synthesized using 1 μg of total RNA with PrimeScript® Reverse Transcriptase (Takara) according to the manufacturer’s instructions.
Analysis of digital gene expression libraries
To investigate transcriptional changes in “Guijiaowang” at different stages of fruit development, the Illumina HiSeqTM 2000 platform was used to perform high throughput sequencing. Five libraries were constructed from samples that were collected at 10, 20, 30, 40 and 50 DAP. This was repeated twice.
Differentially expressed genes
At each stage of fruit development, mRNA was purified and cDNA libraries were created from two independent samples of whole fruits. These ten libraries were sequenced using the Illumina Hi SeqTM 2000, which produced a total of 16,870,295 raw reads. After filtering reads for quality, a total of 15,550,468 reads remained with a total length of 2,333 Mb.
Results
Changes in the placental capsaicin content during five stages of fruit development in ‘Gujiaowang’
Photographs of representative examples from the five stages of fruit development and capsaicin concentrations of chili pepper fruits at the different stages of development are presented in Fig. 1. The results showed limited (0.08 mg/g) capsaicin accumulation in 10 DAP fruit, 1.06 mg/g at 20 DAP, and 13.82 mg/g at 30 DAP. Capsaicin concentrations peaked at about 71.37 mg/g at 40 DAP and then decreased to 54.26 mg/g at 50 DAP. This is consistent with the pattern of capsaicin accumulation described above (Fig. 1).

‘Guijiaowang’ fruits and capsaicinoid content of fruit Placenta at different development stage.
Analysis of DGE libraries
Ten libraries were constructed from duplicate samples collected at 10, 20, 30, 40, and 50 days after pollination (DAP). Sequencing of these 10 libraries yielded a total of 225,880,313 clean reads after removing reads that contained an adaptor, Ns, or were of low quality. An overview of the sequencing and alignment statistics are shown in Table 1. The average read length was approximately 49 bases. The number of clean reads per library ranged from approximately 19 million to approximately 26 million. The proportion of clean tags all exceeded 99% for each library (Fig. S1).
Saturation analysis was performed to determine whether the sequencing depth was sufficient to detect genes for further analysis. The number of mapped genes increased proportionally with the number of reads (Fig. 2). However, the increase in the number of genes tapered off beyond two million clean reads, implying full saturation of the transcriptome. All of the raw sequence data produced are publicly available (accession number GSE55264).

Sequencing saturation analysis of ten sequenced samples.
Read mapping
The pepper reference genome (http://peppergenome.snu.ac.kr) was used to map the RNA-Seq reads. The proportion of clean reads mapped to the reference genes (Pepper.v.1.55.CDS.fa) in each library ranged from 53.78% and 56.38% (Table 1). Among all reads, 50.14–54.12% per library were uniquely mapped to the pepper reference genome, and 36.45–39.80% of reads were a perfect match to the reference gene.
Genes detected per subset
The number of genes expressed in the five stages of fruit development ranged from 23,564 genes at 40 DAP to 25,137 genes at 10 DAP (Table 2). These values represent the sum of the number of genes derived from two biological replicates at each developmental stage. The total number of genes expressed in at least one of the samples was 28,434. A Venn diagram (Fig. 3) illustrates the intersections between the expressed genes detected at the five developmental stages (10, 20, 30, 40, and 50 DAP). A total of 20,181 genes were expressed in all five stages of development, whereas 2,093 genes were expressed in only one stage. This indicated that while many genes were involved in the overall process of fruit development, far fewer genes were functionally unique to the individual developmental stages of ‘Guijiaowang’ fruit.

Each set represent a stage of development. Numbers in each intersection represent the number of genes detected with at least one read (gene tag) in these disjoint sets (intersections).
Changes in gene expression profiles during different developmental stages
The distribution of a gene’s coverage in each sample was analyzed to evaluate the quality of the RNA-Seq dataset. The term “gene coverage” reflects the proportion of the full gene sequence represented in the RNA-Seq reads (Fig. 4). More than 64.8% of genes had greater than 50% coverage. DGE profile analysis was used to analyze gene expression in the five stages of ‘Guijiaowang’ fruit placental development. The expression level of each gene was calculated using normalized RPKM (Reads Per Kb per Million reads) for each developmental stage18.

The number of up-regulated and down-regulated genes between 10 DAP and 20 DAP; 20 DAP and 30DAP; 30 DAP and 40 DAP; 40DAP AND 50DAP are summarized.
DEGs between samples were identified using an algorithm developed by Audic et al.19. The changes in gene expression between consecutive time points was analyzed (Fig. 5). In the early stage of fruit development, 925 genes were differentially expressed between 10 and 20 days. Among these genes, 251 were up-regulated and 674 were down-regulated in 20-day old placental tissue compared with those in 10-day old tissue. Between 20 and 30 DAP, 770 DEGs were observed, with 372 up-regulated and 398 down-regulated. A total of 634 DEGs were detected between 30 and 40 DAP, with 198 up-regulated and 436 down-regulated. Most DEGs were observed between 40 and 50 DAP, with 924 up-regulated and 425 down-regulated relative to the 40 DAP time point. These data are presented in a Venn diagram in Fig. 6. Eighty-one significantly differentially expressed genes were found at different times during placental development. Seventy-five genes were differentially expressed at both 20–30 and 30–40 DAP.

Distribution of genes coverage from ten sequenced samples.

Veen diagram show the number of DGEs identified between consecutive time points.
Functional classification of DEGs in the fruit placenta during different developmental stages
To annotate differentially expressed gene sequences, we performed a BLAST search against the NCBI nonreductant (NR) database (-p blastx -e 1e-5 -m 7), and the results were annotated using Blast2GO and the GO database. For the 10 vs. 20 DAP comparison, of 925 DEGs, 679 genes could be assigned to at least one GO annotation category. The corresponding proportion for the 20 vs. 30 DAP comparison was 580 out of 770; for the 30 vs. 40 DAP comparison, it was 450 of 634; and for the 40 vs. 50 DAP comparison, it was 901 of 1349. Furthermore, for the 10 vs. 20 DAP comparison, 430 genes were categorized as “cellular component,” 534 genes had a molecular function, and 511 genes were involved in a biological process. The respective distributions in the 20 vs. 30 DAP, 30 vs. 40 DAP, and 40 vs. 50 DAP comparisons were 364, 460 and 432, 259, 377, and 342, 506, 734, and 653. Most of these genes were associated with cells, cell parts, extracellular regions, macromolecular complexes, membranes, membrane parts, organelles, and organelle parts. The Gene Ontology significant enrichment analysis of DEGs (10 vs. 20 DAP, 20 vs. 30 DAP, 30 vs. 40 DAP, and 40 vs. 50 DAP) with a P-value cut-off of ≤0.05 is shown in an additional file. In the “cellular component” ontology category, there were 13 and 14 enriched terms in the 10 vs. 20 and 20 vs. 30, comparisons, respectively. No enriched “cellular component” ontology terms were found in the 30 vs. 40 and 40 vs. 50 DAP comparisons (Fig. 7).

Y-axis (right) represents the number of the genes in each category; Y-axis (left) represents the percentage of a specific category of the genes within that main category.
In the “molecular function” category, the 10 vs. 20 and 20 vs. 30 DAP comparisons had the most enriched terms (four and six terms, respectively, Fig. 7). Only 39 “protein binding” genes were enriched in the 30 vs. 40 DAP comparison, and 42 “nucleic-acid binding transcription factor activity” genes were enriched in the 40 vs. 50 DAP comparison. In the “biological process” category, “chromosome organization,” “chromatin organization,” “macromolecular complex subunit organization,” “protein-DNA complex subunit organization,” and “nucleosome organization” were significantly enriched in the 10 vs. 20 and 20 vs. 30 DAP comparisons. Only “response to inorganic substance” was significantly enriched in the 30 vs. 40 DAP comparison, and 7 terms were enriched in the 40 vs. 50 DAP comparison (Fig. S2).
We used pathway enrichment analysis on the DEGs in the four sample comparisons (10 vs. 20, 20 vs. 30, 30 vs. 40 and 40 vs. 50 DAP). Only DEGs with a P-value of ≤0.005 were used for the analysis. In the 10 vs. 20 DAP comparison, 35 significantly enriched pathways were detected, including those involved in phenylpropanoid, flavonoid, fatty-acid, and unsaturated fatty-acid biosynthesis; phenylalanine metabolism; and valine, leucine, and isoleucine synthesis and degradation. A total of 28 significantly enriched pathways were detected in the 20 vs. 30 DAP comparison. These included pathways involved in secondary metabolite biosynthesis; alanine, aspartic-acid, glutamic-acid, and pyruvic-acid metabolism; and fatty-acid elongation. A total of 14 significantly enriched pathways existed in the 30 vs. 40 DAP comparison, including secondary metabolite biosynthesis; valine, leucine, and isoleucine synthesis and degradation; and pyruvic-acid and phenylalanine metabolism. A total of 28 significantly enriched pathways existed in the 40 vs. 50 DAP comparison, including secondary-metabolite, phenylpropanoid, and fatty-acid biosynthesis; and pyruvic-acid and phenylalanine metabolism (additional excel file s1).
Identification of potential capsaicinoid biosynthetic pathway genes
The capsaicinoid biosynthetic pathway proposed by Mazourek20 contains 52 candidate enzymes that are related to capsaicinoid biosynthesis (Fig. 8). Using a BLAST search against the NCBI NR database, 131 genes were annotated as genes involved in the capsaicinoid pathway, except for ferredoxin-dependent glutamate synthase and 3-isopropylmalate dehydrogenase, which were undetected (additional excel file s2). The results showed that many genes encoded the same enzymes. These genes showed different expression patterns, indicating that they may play different regulatory roles in capsaicinoid synthesis.

Putative modles of capsaicin biosynthetic pathway.
Capsaicin typically accounts for about 70% of the total capsaicinoids. Figure 8 summarizes the main pathway of capsaicin synthesis according to Mazourek et al.20. It includes phenylpropanoid and benzenoid metabolism, as well as medium-length, branched-chain fatty acid biosynthesis and L-valine metabolism. A total of 79 genes are predicted to encode enzymes|involved in capsaicin biosynthesis. To understand the expression patterns of these genes, the expression patterns were clustered (Fig. 9). We found two groups of genes that clustered together and whose expression patterns were correlated with capsaicin accumulation. These included: PAL, C4H, Acyl-CoA synthetase (ACS), NADH-GOGAT, BCKDH, and AT. These genes may play important roles in capsaicin accumulation. This finding was consistent with that of Kim et al.8. Other genes showed different expression patterns; for example, gene expression in group 3 was higher than gene expression in the aforementioned groups at the other time points.

Gray bar indicated that the gene expression is too low to detect.
Phylogenetic analysis of AT genes and expression variation
Pungent gene 1 (Pun1) proteins encoded by AT3 are believed to catalyze the last steps in the capsaicin biosynthetic pathway7. A total of 21 genes were annotated as putative acyltransferases in our study, including AT3. The expression patterns of these differed between the developmental stages. To examine the relationships among them, a phylogenetic tree was constructed using the amino acid sequences of the 21 genes (Fig. 10). All genes were classified into three groups. Group A contained pun1; group C had the fewest genes, and three of them had expression patterns that paralleled capsaicin content.

Molecular phylogenetic tree of the amino acid sequences of 21 acyltransferase genes.
Clustering analysis of DEGs during placental development
To identify new genes that could contribute to capsaicin accumulation, we examined differences in DEG expression patterns by comparing various combinations of samples (10 vs. 20, 20 vs. 30, 30 vs. 40, 40 vs. 50, 10 vs. 30 and 20 vs. 40 DAP). Based on capsaicin accumulation patterns, we identified 20 new candidate genes expressed in the placenta of ‘Guijiaowang’ fruit. All of these genes were up-regulated in the 20–40 day interval, and then expression significantly declined from 40–50 days (Table 3). This included genes involved in the metabolic pathways of phenylpropanoid biosynthesis, phenylacetic acid degradation, phenylalanine degradation, and glutathione metabolism. This is the first time that these genes have been implicated in capsaicin synthesis and accumulation.
Quantitative real-time PCR (qPCR) validation
To confirm the gene expression levels detected by RNA-Seq, nine randomly selected candidate genes were evaluated using qPCR. The actin gene was used as an internal control (Table 4). The results of the qPCR analyses supported the RNA-Seq analysis (Fig. 11).

Real-time quantitative validation of digital genes expression data.
Discussion
Previous studies have indicated that the Pun1 locus, which is required for the presence of capsaicinoids in pepper, encodes a putative acyltransferase7,11. Quantitative traits that control developmental processes in pepper are influenced by many genes with small effects. The present study detected 135 known capsaicinoid synthetase-coding genes, but not ferredoxin-dependent glutamate synthase and 3-isopropylmalate dehydrogenase. We identified 20 new candidate genes and reported their involvement in capsaicin synthesis and accumulation for the first time.
AT catalyzes the final step in the capsaicin-biosynthetic pathway, which is one of the most important steps in the pathway21. Among the 21 detected acyltransferase-coding genes, CA01g32960 and CA02g19270 had expression patterns that paralleled capsaicin accumulation. Their levels of expression at 20–40 DAP were significantly different from those at 10 and 20 DAP. They may have a regulatory effect on the synthesis and content of capsaicin. The CA01g32970 gene was specifically expressed from 20–40 days and may play an important role in the regulation of capsaicin synthesis.
The production of cinnamic acid from phenylalanine is catalyzed by PAL. PAL formation and accumulation are involved in capsaicin biosynthesis22. Five PAL-coding genes were detected, and the expression of CA00g95510 and CA05g20790 were found to be positively correlated with capsaicin accumulation, suggesting that these genes play a significant role in the capsaicin synthesis pathway and contribute to capsaicinoid accumulation.
Ferulic acid as a phenylpropanoid intermediate that is produced from caffeoyl-CoA by the action of the enzyme caffeoyl-CoA 3-O-methyltransferase (CCoAOMT)12. The increased expression of CCoAOMT also increased capsaicinoid accumulation. This study detected five CCoAOMT-coding genes and showed that the expression of CA02g14470 was positively correlated with capsaicin accumulation.
pAMT is the enzyme that catalyzes the final step in the phenylpropanoid pathway, in which vanillylamine is formed from vanillin23. Decreasing or silencing pAMT gene expression leads to a significant reduction in capsaicin content in placental tissue10. However, no definitive correlation was observed between changes in capsaicin content and the expression of two pAMT genes. Therefore, additional experiments will be required to fully understand the functions of the pAMT genes.
Numerous enzymes are involved in the branched fatty-acid synthesis pathway, including ketoacyl-ACP synthase I (KAS), malonyl CoA-ACP transacylase (MCAT), acyl carrier protein (ACL), ketoacyl-ACP reductase (KR), hydroxyacyl-ACP dehydratase (DH), enoyl-ACP reductase (ENR), and thioesterase (TE)24. Studies have shown that the fatty-acid synthase genes Acl1, FatA, and Kas are differentially expressed in Capsicum fruits and that the transcript levels of the three genes are positively correlated with the degree of pungency. We detected 11 Kas-coding genes. The expression of two (CA07g11150 and CA03g35350) of these was correlated with capsaicin accumulation, suggesting that these genes may be involved in regulating capsaicin content.
ACS is the enzyme that catalyzes the final step in the branched-chain fatty-acid pathway, in which 8-methyl-6-nonenoyl-CoA is formed from 8-methylpentanoic acid25. A total of 11 ACS-coding genes were detected. Three genes (CA01g01120, CA04g10340, and CA07g08100) had expression patterns that were related to capsaicin accumulation. These genes may play crucial roles in the regulation of capsaicin synthesis.
Among the 20 new candidate genes, five genes (CA06g15270, CA00g86910, CA10g13930, CA04g13110, and CA02g04610) are involved in the known capsaicin biosynthetic pathway. These genes can be used to further explore the regulation of capsaicin synthesis.
Conclusions
In this study, expressed unigenes involved in secondary metabolism in the placenta of ‘Guijiaowang’ pepper fruit were identified using DGE. Candidate enzyme-encoding genes were identified and subsequently analyzed for expression patterns and phylogenetic relationships. The identified genes confirmed the complexity of the regulatory network thought to be involved in capsaicinoid biosynthesis. The genes identified in this study provide potential targets for controlling the production of capsaicinoid in Capsicum breeding programs.
Additional Information
How to cite this article: Zhang, Z.-X. et al. Discovery of putative capsaicin biosynthetic genes by RNA-Seq and digital gene expression analysis of pepper. Sci. Rep. 6, 34121; doi: 10.1038/srep34121 (2016).
References
Bosland, P., Coon, D. & Cooke, P. Novel formation of ectopic (nonplacental) capsaicinoid secreting vesicles on fruit walls explains the morphological mechanism for super-hot chile peppers. J. Amer. Soc. Hort 140, 253 (2015).
Prasad, B. C. N. et al. Characterization of capsaicin synthase and identification of its gene (csy1) for pungency factor capsaicin in pepper (Capsicum sp.). PNAS 103, 13315 (2006).
Aza-González, C., Núñez-Palenius, H. G. & Ochoa-Alejo, N. Molecular biology of capsaicinoid biosynthesis in chili pepper (Capsicum spp.). Plant Cell Reports 30, 695–706 (2011).
Perucka, I. & Materska, M. Phenylalanine ammonia-lyase and antioxidant activities of lipophilic fraction of fresh pepper fruits Capsicum annum L. Innovative Food Science Emerging Technologies 2, 189–192 (2001).
Suzuki, T., Kawada, T. & Lwai, K. Biosynthesis of acyl moieties of capsaicin and its analogues from valine and leucine in Capsicum fruits. Plant Cell Physiol 22, 23–32 (1981).
Sukrasno, N. & Yeoman, M. M. Phenylpropanoid metabolism during growth and development of Capsicum frutescens fruits. Phytochemistry 32, 839–844 (1993).
Stewart, C. Jr. et al. The Pun1 gene for pungency in pepper encodes a putative acyltransferase. Plant J 42, 675–688 (2005).
Kim, S. et al. Genome sequence of the hot pepper provides insights into the evolution of pungency in Capsicum species. Nat Genet 46, 270–278 (2014).
Qin, C. et al. Whole-genome sequencing of cultivated and wild peppers provides insights into Capsicum domestication and specialization. Proc Natl Acad Sci USA 111, 5135–5140 (2014).
Chaim, A. B. et al. QTL mapping of fruit-related traits in pepper (Capsicum annuum). Theoretical and Applied Genetics 102, 1016–1028 (2001).
Ogawa, K. et al. Evidence of capsaicin synthase activity of the Pun1-encoded protein and its role as a determinant of capsaicinoid accumulation in pepper. BMC Plant Biology 15, 93 (2015).
Abraham-Juárez, M. d. R. et al. Virus-induced silencing of Comt, pAmt and Kas genes results in a reduction of capsaicinoid accumulation in chili pepper fruits. Planta 227, 681–695 (2008).
Curry, J. et al. Transcripts for possible capsaicinoid biosynthetic genes are differentially accumulated in pungent and non-pungent Capsicum spp. Plant Science 148, 47–57 (1999).
Liu, S., Li, W., Wu, Y., Chen, C. & Lei, J. De novo transcriptome assembly in chili pepper (Capsicum frutescens) to identify genes involved in the biosynthesis of capsaicinoids. PLoS One 8, e48156 (2013).
Ma, H., Lu, Z., Liu, B., Qiu, Q. & Liu, J. Transcriptome analyses of a Chinese hazelnut species Corylus mandshurica. BMC Plant Biol 13, 152 (2013).
Hornett, E. A. & Wheat, C. W. Quantitative RNA-Seq analysis in non-model species: assessing transcriptome assemblies as a scaffold and the utility of evolutionary divergent genomic reference species. BMC Genomics 13, 361 (2012).
Gene Ontology Consortium. Creating the Gene Ontology Resource: Design and Implementation. Genome Research 11, 1425–1433 (2001).
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5, 621–628 (2008).
Audic, S. & Claverie, J. M. The significance of digital gene expression profiles. Genome Res 7, 986–995 (1997).
Mazourek, M. et al. A dynamic interface for capsaicinoid systems biology. Plant Physiol 150, 1806–1821 (2009).
Díaz, J., Pomar, F., Bernal, A. & Merino, F. Peroxidases and the metabolism of capsaicin in Capsicum annuum L. Phytochemistry Reviews 3, 141–157 (2004).
Ochoa-Alejo Judith, N. & Gómez-Peralta, E. Activity of Enzymes Involved in Capsaicin Biosynthesis in Callus Tissue and Fruits of Chili Pepper (Capsicum annuum L.). Journal of Plant Physiology 141, 147–152 (1993).
Yaqin Lang. et al. Functional loss of pAMT results in biosynthesis of capsinoids, capsaicinoid analogs, in Capsicum annuum cv. CH-19 Sweet. The plant journal 59, 953–961 (2009).
Nishida, I. Plastid metabolic pathways for fatty acid metabolism. Molecular Biology and Biotechnology of Plant Organelles 543–564 (2004).
Thiele, R., Mueller-Seitz, E. & Petz, M. Chili Pepper Fruits: Presumed Precursors of Fatty Acids Characteristic for Capsaicinoids. J. Agric. Food Chem 56, 4219–4224 (2008).
Acknowledgements
This research was supported by; the Youth Scientific Foundation from Hainan University (qnjj1218), the Fundamental Scientific Research Funds for CATAS-TCGRI (1630032014045), the Cultivation Project of Excellent Master Thesis for Hainan University (201273), and Scientific research project of Department of Education in Hainan Province(hnky2016HD-2).
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: S.-H.C. and Z.-X.Z. Performed the experiments: Z.-X.Z., Z.-M.H., S.-N.Z. and Z.-M.C. Analyzed data: Z.-X.Z. and G.-F.L. Contributed materials: S.-S.L. Wrote the paper: Z.-X.Z.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhang, ZX., Zhao, SN., Liu, GF. et al. Discovery of putative capsaicin biosynthetic genes by RNA-Seq and digital gene expression analysis of pepper. Sci Rep 6, 34121 (2016). https://doi.org/10.1038/srep34121
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep34121
This article is cited by
-
Surviving mutations: how an Indonesian Capsicum frutescens L. cultivar maintains capsaicin biosynthesis despite disruptive mutations
Genetic Resources and Crop Evolution (2023)
-
Transcriptome profiling of Capsicum annuum using Illumina- and PacBio SMRT-based RNA-Seq for in-depth understanding of genes involved in trichome formation
Scientific Reports (2021)
-
Efficient enhancement of capsaicinoids biosynthesis in cell suspension cultures of Capsicum chinense Jacq. cv. ‘Umorok’ by elicitors and differential gene expression analysis of elicited cultures
Plant Cell, Tissue and Organ Culture (PCTOC) (2020)
-
Phosphoinositide-specific phospholipase C signalling mediates expression of two phenylalanine ammonia lyase genes induced by salicylic acid in Capsicum chinense cells
Journal of Plant Biochemistry and Biotechnology (2020)
-
Gene expression related to the capsaicinoids biosynthesis in the Capsicum genus: Molecular and transcriptomic studies
Brazilian Journal of Botany (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
