
Abstract
G protein-coupled receptor kinase 5 (GRK5) is a regulator of cardiac performance and a potential therapeutic target in heart failure in the adult. Additionally, we have previously classified GRK5 as a determinant of left-right asymmetry and proper heart development using zebrafish. We thus aimed to identify GRK5 variants of functional significance by analysing 187 individuals with laterality defects (heterotaxy) that were associated with a congenital heart defect (CHD). Using Sanger sequencing we identified two moderately frequent variants in GRK5 with minor allele frequencies <10%, and seven very rare polymorphisms with minor allele frequencies <1%, two of which are novel variants. Given their evolutionarily conserved position in zebrafish, in-depth functional characterisation of four variants (p.Q41L, p.G298S, p.R304C and p.T425M) was performed. We tested the effects of these variants on normal subcellular localisation and the ability to desensitise receptor signalling as well as their ability to correct the left-right asymmetry defect upon Grk5l knockdown in zebrafish. While p.Q41L, p.R304C and p.T425M responded normally in the first two aspects, neither p.Q41L nor p.R304C were capable of rescuing the lateralisation phenotype. The fourth variant, p.G298S was identified as a complete loss-of-function variant in all assays and provides insight into the functions of GRK5.
Similar content being viewed by others

Introduction
Heterotaxy describes a very rare condition with an incidence of 1:10,0001. It is characterized by a deviation from the normal situs (situs solitus), a term which describes the normal asymmetric distribution of internal organs such as the heart, liver, spleen or pancreas. In contrast to individuals with situs inversus, which is a complete mirror image of organ placement, patients with heterotaxy appear to have a random distribution of their internal organs with for instance their heart apex being oriented rightward (dextrocardia), absence of the spleen (asplenia) or inverted gut looping (malrotation)2. As a consequence several complications can arise with congenital heart defects (CHD) being the most common and severe. Such CHDs with concomitant situs anomalies are clinically and genetically heterogeneous disorders. Oftentimes they consist of complex cardiovascular malformations such as transposition of the great arteries or double outlet right ventricle3.
G protein-coupled receptor kinase 5 (GRK5) is a versatile regulator of cellular signalling events, which also functions as a critical modulator of cardiac performance in the adult4. It further represents the only cardiac GRK for which clinically relevant genetic variations have been identified. The best-characterised variant so far is a single nucleotide polymorphism (SNP) rs17098707 resulting in an amino acid exchange in the N terminus. This p.Q41L variant has been found beneficial in a certain population of heart failure patients5 and was associated with fewer adverse cardiovascular events in hypertensive patients6. However, its full impact on signalling has not yet been clarified. Moreover, it has also been shown that p.Q41L may predispose to certain cardiac conditions such as Takotsubo cardiomyopathy7,8. Similarly, intronic SNPs which have been associated with atrial fibrillation after coronary bypass surgery9 will require further genetic and biochemical analyses to uncover their apparently clinically relevant but mechanistically unknown function.
In addition to its well-accepted function in the adult heart, we have recently shown that GRKs of the GRK4-6 family play a role during embryonic development10,11. Mice lacking both, GRK5 and GRK6 are embryonically lethal. Furthermore, depletion of either mouse GRK5 or its closest zebrafish homolog termed Grk5l results in elevated mTOR activity, which in turn impairs leftward gene expression in the lateral plate mesoderm, where the heart progenitors reside. As a consequence, a highly conserved pathway required for left-right patterning across species cannot be established properly, and abnormal internal asymmetry and cardiac development can be observed in zebrafish10.
These findings prompted us to screen GRK5 in DNA samples collected from 69 patients of German origin and in DNA samples obtained from 118 patients in the United States diagnosed with heterotaxy and concomitant CHD, in whom we previously excluded mutations in known heterotaxy genes. We detected functionally significant single nucleotide variants which suggest a potential contribution to the development of heterotaxy.
Results
Identification of GRK5 Sequence Variants
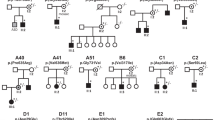
Sequencing of 69 individuals with CHD (Supplementary Table S1) and concomitant situs anomalies from the initial cohort identified 7 rare heterozygous variants (Table 1 and Supplementary Table S2). Their position in the gene and protein is shown in Fig. 1 and their minor allele frequencies are given in Table 2. Two variants, namely p.Q41L (rs17098707) and p.R304H (rs2230349), are moderately frequent with a minor allele frequency of <10%. Three additional variants, which are p.A119V (rs55980792), p.G298S (rs140946236) and p.D495D (rs149159651), are rare polymorphisms with minor allele frequencies of <1% (Table 2). In addition, we observed two novel coding variants, according to both ExAC Browser and dbSNP build 139, namely p.P464S and p.G549R. Notably, we detected two rare heterozygous variants in two affected individuals (Table 1). In individual 43 we detected both p.A119V and p.G298S, though further analysis revealed both to be paternally inherited. Individual 38 bears both p.R304H and p.G549R, however DNA samples from parents were not available for testing.

Variants of human GRK5 identified in heterotaxy patients.
(a) Cartoon displays the 16 exons and the detected genetic variations as well as their respective location within the coding region. The variants are shown at the nucleotide levels (e.g. c.122A < T) as well as the subsequent amino acid change in the protein (e.g. p.Q41L). Red bars mark variants more closely characterised in this study. The p.T425M variant of GRK5 identified in human patients corresponds to the p.S425M variant of zebrafish Grk5l used in this work. (b) Numbers and red spheres indicate location of relevant variants in the structure of bovine GRK5 (PDB entry 4WNK). Only position 298 is absolutely conserved as glycine in GRKs. Image provided by John J. G. Tesmer, University of Michigan, Ann Arbor.
These data prompted us to analyse those exons of GRK5 in which we detected rare variants in an independent cohort of 118 affected individuals. In addition to identifying individuals bearing p.Q41L and p.R304H, we found two further variants, p.R304C (rs145397190) and p.T425M (rs77323445) with minor allele frequencies of <1% (Table 2). In summary, we found two moderately frequent and seven rare variants in our cohorts of CHD patients with concomitant situs anomalies. All patients except for one carrying p.Q41L were heterozygous carriers of the GRK5 variants. To determine the possible effect of these single nucleotide variants on GRK function, analysis was performed in heterologous cell systems and zebrafish. We concentrated our analysis on four amino acid changes. Substitutions p.G298S and p.R304C were chosen because both mutated residues were conserved in zebrafish (see protein alignment in Supplementary Figure S1). In addition, we included p.T425M. In zebrafish the threonine at position 425 is a serine, which is also an uncharged amino acid that can be phosphorylated by serine-threonine kinases. Hereafter, this variant will be named p.S425M. These three variants were further chosen as they were predicted to be possibly damaging for GRK5’s function (Table 2). For such predictions we applied SIFT and Polyphen-2 (Polymorphism Phenotyping v2) algorithms, which calculate how harmful a genetic variant is likely to be based on the structure, function and degree of amino acid conservation12,13. The lower a SIFT score, the higher the likelihood that a variant is detrimental. We also included the previously reported variant p.Q41L which had been proposed to be beneficial to cardiac function under some conditions, but deleterious to the heart under other5,6,7,8. We refrained from analysing the two novel variants as the closest zebrafish homolog of human GRK5 already contains a serine corresponding to position 464 in humans and arginine at position relative to position 549 in humans (Supplemental Figure S1). The position of the detected variants with respect to the exon sequence of GRK5 is depicted in Fig. 1, which also highlights the position of the four variants analysed in more detail in the crystal structure of GRK5.
The Impact of GRK5 Variants on GPCR Desensitisation
In the classical paradigm of G protein-coupled receptor (GPCR) signalling, agonist binding induces a conformational change in receptors that in turn activates signalling of the heterotrimeric G protein14. GRKs serve as the terminating enzymes of active signalling. To address this, we examined their effect on signalling of murine angiotensin II receptor type1a (AT1a) (Fig. 2a) and the human chemokine receptors CCR2b (Fig. 2b), CCR3 (Fig. 2c) and CXCR4 (Fig. 2d). Both, AT1a and CXCR4 receptors have previously been shown to be regulated by GRKs, including GRK515,16. For all four receptors we made use of a luciferase-based system, in which the activation of the transcriptional regulatory element SRE.L by a Gαq coupled receptor is measured and which we had established for the measurement of G protein-mediated chemokine receptor signalling17,18. Since CCR3 and CXCR4 predominantly couple to Gαi, measurement of signalling was facilitated by co-expression with a chimeric Gαqi5, in which the last five amino acids of Gαq are exchanged for those of Gαi. By this change, signalling of Gαi coupled receptors can be redirected to Gαq proteins19,20. In this setting, stimulation of either receptor activates Rho GTPases, which cause cytoskeletal rearrangements and transcriptional activity of SRF, for which SRE.L functions as readout21,22,23.

Mutations in Grk5l alter its ability for receptor desensitisation.
We tested the ability of Grk5l variants to terminate signalling of four different receptors in a luciferase reporter assay that is based on the Gq-Rho-SRE signal transduction axis. Individual receptors were transiently expressed in HEK293 cells together with wild-type Grk5l or different variants thereof. As positive control for receptor desensitisation, bovine GRK5 was co-expressed with the receptor. All panels show representative blots. One-way ANOVA was applied and all variants were compared to wild-type Grk5l; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant. Bar graphs display mean ± SEM of three independent experiments done in triplicates. (a) Murine AT1a receptor stimulated with 30 μM angiotensin II: All variants except for p.G298S display the same efficiency in the desensitisation of AT1a receptors. (b) Human CCR2b receptor stimulated with 50 nM CCL2: The p.G289S shows significantly reduced ability to terminate signalling compared to wild-type Grk5l, while p.S425M is even more potent. (c) Human CCR3 receptor stimulated with 50 nM CCL11: As CCR3 is not Gq-coupled a Gαqi5 construct was co-transfected, which enables Gi-receptors to stimulate Gq signalling cascades. Grk5l inhibits signalling to a similar extent as bovine GRK5. The p.G298S variant lacks the ability to desensitise CCR3 receptors, while the lower inhibition seen by p.R304C may be accounted to lower expression levels. (d) Human CXCR4 stimulated with 50 nM CXCL12: As CXCR4 is not Gq-coupled a Gαqi5 construct was co-transfected, which enables Gi-receptors to stimulate Gq signalling cascades. On its own, Gαqi5 does not alter receptor signalling. p.G298S and also p.R304C are less effective in inhibiting receptor signalling, whereas p.Q41L and p.S425M appeared to be more potent than wild-type Grk5l.
For all tested receptors agonist stimulation robustly induced activation of luciferase indicating that active signalling is conferred. Co-expression of bovine GRK5 abrogated signalling, which could be similarly observed for co-expression of Grk5l. p.Q41L which has been shown to be more potent in desensitising β2 adrenergic receptors24, desensitised the receptors to a similar if not almost greater extent than wild-type Grk5l. p.G298S, on the other hand, failed to terminate G protein signalling under all experimental conditions. p.R304C as well as p.T425M both retained the ability for receptor desensitisation although to different extends. Most likely the reduced capability of p.R304C to block CCR3 (Fig. 2c) and CXCR4 (Fig. 2d) signalling was due to the lower expression levels, which could not be elevated by increasing the amount of transfected plasmid or by different plasmid preparations. We also tested the effect of Gαqi5 alone, which produced only marginal, if any activation of SRE.L. Taken together, we confirmed that p.Q41L is as effective as wild-type GRK in receptor desensitisation and found that the glycine at position 298 is crucial for receptor desensitisation, indicating a functional effect of this variant in GPCR signalling.
Subcellular Localisation of GRK5 Depends on Glycine 298
One characteristic of GRK5 is the predominant association with the cytosolic side of the plasma membrane through N- and C-terminally located basic or hydrophobic amino acids25,26,27. Considering that this membrane localisation is a crucial requirement for receptor desensitisation by a GRK we assessed the subcellular localisation of the newly identified GRK5 variants in a heterologous cell system and in zebrafish. Transient transfection of wild-type Grk5l revealed the expected membrane association with little or no expression in the cytosol. Similarly, p.Q41L strongly localised to the plasma membrane with some protein remaining in the cytoplasm. Mutation of glycine 298 to serine, however, completely abrogated membrane association of Grk5l and resulted in cytosolic retention with a small fraction locating to the nucleus. When arginine 304 was exchanged for cysteine the kinase appeared to be evenly distributed between the cytosol and the plasma membrane. In addition, weak fluorescence could be seen in the nucleus. Finally, Grk5l S425M, which had been indistinguishable to wild-type Grk5l in the desensitisation assay, nicely associated with the plasma membrane (Fig. 3 upper panels).

Subcellular distribution of Grk5l variants.
Upper panel: HEK293 cells were transiently transfected with GFP fusion constructs of wild-type or mutated Grk5l (cloned into pEGFP-N3) and analysed using confocal microscopy. Representative images of three transfections shown. Lower panel: Expression of Grk5l variants in zebrafish produced subcellular distribution of the kinases similar to the results in HEK293 cells. Images show expression in the epidermis. At least five different embryos each from two different injection days were assessed.
We next wondered whether the same subcellular pattern occurred in vivo and injected capped RNA encoding either wild-type Grk5l or one of the four variants fused to GFP into fertilised zebrafish eggs. As readout we analysed GFP fluorescence in the epidermis at 24 hours post fertilisation (hpf). Similar to expression in a heterologous cell system, Grk5l wild-type, p.Q41L and p.S425M localised to the membrane in zebrafish embryos. Expression of p.G298S yielded only cytosolic localisation, while p.R304C was again evenly distributed between cytosol and membrane (Fig. 3 lower panels). These results confirm and extend our desensitisation studies. Moreover, they indicate that membrane association may depend on additional domains within GRK5 in addition to the amphipathic helix.
Grk5l variants localise to cilia
Left-right asymmetry and subsequent heart development depend on cilia2. Previously, we have found that Grk5l regulates cilia function and that it localizes to cilia10. We have thus investigated whether mutation of Grk5l changes ciliary localization. Interestingly and in contrast to the differences in membrane localization, we could detect all four variants in cilia of cultured fibroblasts (Fig. 4) suggesting that any contribution to the development of heterotaxy-driven CHD is likely caused by an inability to regulate signal transduction.

Variants of Grk5l localise to cilia.
Human forearm fibroblast cells immortalised by hTERT were transfected with pEGFP-N3 plasmids carrying HA-tagged wild-type or mutated Grk5l cDNA, followed by serum starvation. Cells were then probed for acetylated-tubulin and the HA tag, respectively, to detect primary cilia and expressed Grk5l variants. Assessment by confocal microscopy revealed ciliary localisation of all variants. Acetylated-tubulin is shown in red in panels to the left. Localisation of Grk5l variants are represented in green in middle panels, while panels to the right show an overlay.
Impact of GRK5 Variants on Left-Right Asymmetry Development
Finally, we tested whether any of the GRK5 variants had an impact on the development of left-right asymmetry. Bilateral asymmetry determines the oriented morphogenesis of internal organs with respect to each other, but also within the organs itself, particularly the heart2,3. Thus, the development of a CHD may be propagated if one of the variants would induce aberrant left-right asymmetry development. First, we monitored the side-specific expression of the lateralisation gene southpaw (spaw). Spaw is expressed exclusively in the left lateral plate mesoderm during somitogenesis28. When symmetry breaking fails such as in the case of MO-mediated Grk5l depletion10, spaw can be found in a bilateral expression or may be solely present on the right site of the body (Fig. 5a). We have shown in our initial study that the asymmetry defect in Grk5l knockdown embryos can be rescued by reconstitution with Grk5l10. We have thus tested whether the four variants retained this ability to recue asymmetry. Compared to rescue with wild-type Grk5l, none of the variants could significantly rescue correct spaw expression in the lateral plate mesoderm. Nevertheless, p.S425M showed a tendency towards a partial rescue (Fig. 5b). To further investigate whether any of the variants affected situs development, we scored abdominal organ placement by examining the localization of the endocrine pancreas (Fig. 5c,d). Again, co-injection of RNA encoding p.Q41L, p.G298S and p.R304C did not correct pancreas positioning with respect to the midline (Fig. 5d). p.S425M, interestingly and concordant with the tendency of spaw rescue in the left lateral plate mesoderm, rescued pancreas placement (Fig. 5d).

Functional analysis of GRK5 variations in zebrafish.
(a) Whole mount in situ hybridisation of 20–22 somite stage zebrafish embryos for the leftward gene southpaw (spaw). Embryos were either left uninjected (NI) or were injected with a 5 basepair mismatch control MO (CMO) or a translation blocking MO against Grk5l, respectively. To analyse the functionality of the different Grk5l variants, fertilised eggs were simultaneously injected with capped RNAs for wild-type Grk5l or one of the four variants. Control embryos predominantly express spaw in the left lateral plate mesoderm (left panel), while. Grk5l depleted embryos show aberrant spaw expression bilaterally or right of the midline (middle and right panel). (b) Summary of 4 to 17 independent experiments with 114–432 embryos: embryos were scored for spaw expression left (L) or right (R) from the midline, for bilateral (B) or absent expression (A) See supplement for injections of RNA only. (c) WMISH of 48 hours post fertilisation (hpf) zebrafish embryos for the insulin gene to detect placement of the pancreas upon rescue attempts with Grk5l variant RNAs. In controls, pancreas placement is predominantly on the right side of the midline (indicated by dashed line), while Grk5l depletion renders embryos with left-sided pancreas. (d) Summary of 3 to 6 independent experiments with 42–133 embryos: pancreas position is indicated by L (left) or R (right). See supplement for injections of RNA only. (e) WMISH of 48 hpf zebrafish embryos for cardiac myosin light chain 2 (cmlc2) to monitor heart looping. Left panel shows a correctly looped heart (D), the middle displays an unlooped heart (N) and the right panel shows an inversely looped heart (L). (d) Summary of 3 to 6 independent experiments with 42–133 embryos scored for heart looping after co-injection of Grk5l MO and different rescue RNAs. See supplement for injections of RNA only. In all panels anterior to the top. Fisher’s exact test was applied to compare efficiency of rescue of variant to wt Grk5l and p values are indicated to the right.
The most interesting parameter in the light of this study is the analysis of cardiac looping, which becomes randomized in zebrafish with impaired left-right asymmetry establishment. Under healthy control conditions the zebrafish heart undergoes looping from 36 hours post fertilization (hpf) on. At 48 hpf, the heart is shaped like an S, where the ventricle lies left and above of the atrium (Fig. 5e). When asymmetry is disturbed, inversely looped hearts or hearts, which completely fail to loop can be observed (Fig. 5e). MO-mediated knockdown of Grk5l results in randomization of heart looping with roughly half of the embryos still developing normally looped hearts. Complementation with wild-type Grk5l or p.S425M through co-injection of capped RNA reproducibly corrected heart looping. Co-injection of any of the other variants, however did not rescue indicating that the detected variants may contribute to the development of heterotaxy-driven CHD (Fig. 5f).
Last, but not least, to rule out a potential dominant negative effect of p.Q41L, p.G298S and p.R304C and since the detected patient variants were mostly heterozygous, we injected the respective RNAs into wild-type embryos without Grk5l MO. Embryos injected with either of the three RNAs were indistinguishable from control injected embryos for spaw localisation, pancreas placement or heart looping indicating that the rescue experiment results are not explained by variant inhibition of endogenous Grk5l. Interestingly, injection with RNA encoding p.S425M interestingly affected spaw expression, although it did not impair asymmetry developmentof developing organs at later stages (Supplementary Figures S2–4). Further studies would be required to better understand the unexpected effect on spaw.
Discussion
The term CHD describes the developmental malformation of the heart and its connected structures. It affects a large number of newborns and differs greatly regarding the type and complexity of malformation and thus severity29. Genetically, single gene causes of CHDs such as TBX5 mutations in Holt-Oram-Syndrome or mutations in the cardiac differentiation factor NKX2.530,31 explain a relatively small proportion of all known cases. While the ability to provide a cytogenetic or molecular diagnosis for causes of syndromic or isolated CHD are increasing, up to 80% of cases are still without an etiologic explanation32. Furthermore, mutations in single genes cannot explain the variability in the severity of different CHDs. Therefore it is the generally assumed that the majority of CHDs are multifactorial, involving combinatorial genetic and environmental interactions. Heterotaxy spectrum CHD has the highest relative risk amongst CHD subtypes, indicating a strong genetic basis33,34,35. Those defects arise when internal bilateral asymmetry cannot be established. In our previous study, we have established GRK5 as a novel determinant of left-right asymmetry and heart development in mice and zebrafish10. Herein, we aimed to identify functional variants of GRK5 using patients diagnosed with a situs anomaly with CHD.
Several genetic analyses, including GWAS studies, have investigated an impact of GRK5 variations on the cardiovascular system5,6,7,8,9. Those studies have concentrated on adult individuals and omitted patients with CHD. Here, we fill this gap and provide data on additional GRK5 variants which have not been previously described. Since we were interested only in the coding region of GRK5, we performed Sanger sequencing of exons. Using this approach we found 8 rare missense variants plus the previously reported p.Q41L allele5. We chose to further investigate three rare and highly conserved variants with a low SIFT score and a Polyphen-2 prediction indicating an impairment in function. In addition, we included the known p.Q41L variant as it has previously been shown to be more potent in receptor desensitisation than wild-type GRK55,24.
To evaluate the functionality of these variants, and therefore the possibility they are risk alleles, we tested four characteristics of GRK5: the ability to desensitise GPCRs, predominant localisation to the plasma membrane or the cilium and finally whether the variants would be able to rescue the lateralisation defect upon Grk5l knockdown in zebrafish embryos. Consistent with previous data we found that p.Q41L was functional, if not even more potent, in terminating G protein-mediated signalling of GPCRs24. This could be explained by the position of the variant, which is in the membrane interface (Fig. 1b). Since leucine likely binds better to the membrane than glutamine, there may be more GRK5 readily available for phosphorylation of activated receptors. Interestingly, however, the p.Q41L variant failed to rescue Grk5l knockdown embryos, despite robust expression in zebrafish. This controversy mirrors other discrepancies regarding p.Q41L, which appears to prevent negative outcomes in certain heart failure patients5 while predisposing at the same time to other cardiovascular conditions such as atrial fibrillation7,9. It also suggests that GRK5 variants may confer different outcomes depending on the cell type, disease state, interaction partner or the signalling cascade. Further studies will be needed to better understand those context-dependent actions of p.Q41L.
What about the other three rare variants? Recently, the crystal structure of GRK5 has been published26,27. According to that, p.G298S is likely to destabilize the kinase domain as the side chain of the serine would introduce steric clashes. Consistent with that p.G298S acted as a non-functional mutant in all assays tested. p.R304C and p.S425M, however, reside in areas of GRK5 that are conserved among GRKs and are solvent exposed. Thus their potential roles in folding or function are unknown. In light of this, some of the results we obtained are unexpected. Both, p.R304C and p.S425M retained the canonical GRK ability to desensitise receptors. During left-right asymmetry development however, which is disturbed in heterotaxy patients, p.R304C appears to be non-functional. Thus, depending on the conformational flexibility of GRK5 any of the three variants may reside in a crucial domain. Interestingly, we also observed that p.R304C cannot be expressed to high levels in cells, which explains the reduced activity in receptor desensitization. In contrast to this, it is nicely expressed in zebrafish upon RNA injection. This suggests an additional regulation at the transcriptional or post-transcriptional level in the presence of this SNP. Having said that, even when using capped RNA, we did not obtain a rescue of asymmetry development by p.R304C implying some functional deficiency in addition to an impairment of plasmid-driven expression. These data are somewhat in accordance with the prediction of Polyphen-2, which classified this mutation as probably damaging.
p.S425M, on the other hand, could be expressed at high levels in cells and fish. It functioned very similar to wild-type GRK5 in the cellular assays. When co-injected with Grk5l MO it resulted in a rescue in left-right asymmetry development with respect to abdominal organ placement and heart looping. This is in stark contrast to the prediction of PolyPhen-2, which classified this mutation as possibly damaging.
The most intriguing variant is p.G298S. This alteration lies within the kinase domain of GRK5, but not in close proximity to the ATP pocket26,27. Nevertheless, p.G298S was classified as probably damaging by Polyphen-2 and had the lowest score using the SIFT algorithm, which suggested that it would show the most pronounced loss of function in our experiments. This was indeed the case: exchange of glycine for serine at position 298 renders cytoplasmic distribution of the kinase, although the domains relevant for association with the membrane’s phospholipids are in the outer most termini of GRK525,26,27. One explanation for this could be that p.G298S is not folded correctly and thus retained in the cytoplasm. Secondly, p.G298S is unable to inhibit GPCR signalling, suggesting that unlike GRK2 it remains in the cytosol even upon receptor activation. Last, but not least, p.G298S is incapable of correcting aberrant spaw expression in Grk5l knockdown embryos and unable to rescue normally lateralized organogenesis. Together, these data suggest that p.G298S is a loss-of-function variant of GRK5 and potentially confers moderate-risk to the development of CHD.
CHDs associated with heterotaxy have only been survivable with the advances of surgical approaches over the last several decades. From an evolutionary genetic standpoint, this indicates that genetic causes of heterotaxy would be biased toward de novo mutations in affected individuals or combinatorial interactions of rare susceptibility alleles. Heterozygous rare variants identified in GRK5 are good candidates to support the latter mechanism. We have investigated deletions as well as gene duplications in heterotaxy36 and reviewed the data relating to CHD in general37. The majority of genetic causes identified so far show reduced penetrance and variable expressivity, which implies high genetic complexity38. It is very likely that these specific types of CHD result in some cases from the inheritance of multiple susceptibility alleles that disrupt molecular signalling in a synergistic fashion which in turn can be further worsened by additional environmental factors such as exposure to teratogens during pregnancy39. CHDs are typically thought to be inherited as a complex trait, and Mendelian inheritance patterns are rare. Thus, identifying potential susceptibility alleles for CHDs is an important goal for delineation of genetic contribution and risk assessment40. Furthermore, variants which are rare in the population are often predicted to be potentially damaging by bioinformatics prediction programs. The results presented on several genetic variants of GRK5, a gene previously identified as important for left-right patterning and normal heart development, indicate the complexity of dissecting allelic function in vitro and in vivo. Based on our data in zebrafish heart looping experiments we speculate that the p.G298S variant in GRK5 confers moderate susceptibility to CHD and situs abnormalities and may propagate the disease in combination and interaction with other susceptibility alleles and environmental factors. Overall, our results show that multiple functional assays may be required to determine allelic effects not only GRK5, but most likely also of other candidate genes and indicate zebrafish as a robust model for dissecting the complex human variations and combinatorial interactions underlying protein function and potentially heterotaxy.
Methods
Materials
Cell culture media and supplements were purchased from Life Technologies Invitrogen and Sigma. JetPrime® was from Polyplus. CXCL12 and CCL11 were purchased from Peprotech.
Patient Cohorts and Sequencing
Two heterotaxy cohorts with concomitant CHD were analysed: DNA samples of the first cohort (n = 69) were provided by the National Register for Congenital Heart Defects (Berlin, Germany) (collection and distribution were approved by the local ethics committee in Berlin). Here, according to German laws no ethnic data was collected. Medical records regarding the heart phenotype and situs were provided with the samples. These samples were screened for potential genetic variations in all 16 exons of GRK5 as well as for known mutations in genes associated with situs defects, namely ZIC3, ACVR2B, LEFTY2, CFC1, NODAL and FOXH1. A second cohort (n = 118) with phenotype information including imaging and diagnostic studies, clinical genetic testing results, and pathology results were collected at Cincinnati Children’s Hospital Medical Center (CCHMC) under a protocol approved by the CCHMC Institutional Review Board. In these probands, exons 2, 5, 9, 13, 14 and 15 of GRK5 were screened. This study was approved by the local ethics committee at Ulm University and by the CCHMC Institutional Review Board. The study was performed in accordance with the Declaration of Helsinki protocols. Prior to inclusion into the study patients or their respective parents gave their informed consent.
DNA samples from whole blood were isolated by standard procedures. For gene analysis, we designed intronic primers to PCR amplify (ReadyMix Taq PCR Mix, Sigma Aldrich or FastStart PCR Master, Roche Applied Sciences) all coding exons and the exon-intron boundaries of GRK5, ZIC3, ACVR2B, LEFTY2, CFC1, NODAL and FOXH1. Primer pairs for the amplification of coding exons, and the approximately 50 base pairs (bp) of flanking UTR or intronic sequences, are available upon request. The purified PCR products were sequenced using BigDye terminator version 3.1 chemistry on an ABI3700 Genetic Analyzer (Applied Biosystems). Sequence traces were assembled, aligned, and analysed using Sequencher (Gene Codes) or Mutation Surveyer (SoftGenetics) software.
Cloning
For experiments in zebrafish N-terminally HA-tagged Grk5l was cloned into pCS2+ and pCS2+ GFP via EcoRI. Site-directed mutagenesis was performed with a PCR based approach and capped RNA was prepared using Ambion’s mMessage mMachine Kit from linearised plasmids. For subcellular localization studies in HEK293 cells and hTert immortalised fibroblasts N-terminally tagged wild-type and variant Grk5l was cloned into pEGFP-N3 via BamHI and HindIII. cDNAs of human CXCR4 (accession number NM_001008540) and human CCR3 (accession number XM_006712960) were amplified from spleen and monocyte cDNA and cloned into pcDNA3.1 + with a N-terminal HA-epitope. To generate chimeric Gαqi5 in pcDNA3.1+, the last five amino acids of Gαq in EE-epitope-tagged human Gαq (GNA0Q0EI00, UMR cDNA Resource Center) were exchanged for the sequence encoding Gαi2 (according to Conklin et al.41). The reporter plasmids pSRE.L and pRL-TK were obtained from Dr. Dianqing Wu (New Haven, CT) and Promega, respectively.
Cell Culture
HEK293 cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2 in Dulbecco´s modified Eagle´s medium (DMEM, Sigma Aldrich) supplemented with 10% (v/v) fetal calf serum (FCS), 100 units/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine, 25 mM HEPES, and 1 mM sodium pyruvate. MEMα (Life technologies) supplemented with 10% FCS, 100 units/ml penicillin and 100 μg/ml streptomycin was used to culture hTert immortalised fibroblasts. Cilia formation in fibroblasts was induced by a 48 hours culture in MEMα containing 0.1% FCS.
Desensitization Assay
Luciferase assays were performed using the Dual-Luciferase® Reporter Assay System (Promega) as described previously42. HEK293 cells were seeded into 24- or 48-well plates at a density of 1,6 × 105 and 8 × 104 cells per well, respectively, and were grown for 24 h in 1 ml or 0.5 ml of the same medium per well prior to transfection. For transfection with JetPrime® (Polyplus-transfection Inc.), plasmid DNA was mixed with 2 μl transfection reagent per μg of DNA in transfection buffer. In each transfection, the amounts of DNA were kept constant between the samples by adding the corresponding empty vector. Plasmid amounts transfected were: 30 ng per well of pSRE.L and pRL-TK, 300 ng per well of either empty vector pcDNA3.1 + and vector pcDNA3.1 + encoding Gαqi5 (Control-Mock, 50 ng/well), 200 ng per well murine AT1a43, 200 ng per well HA-epitope-tagged CCR2b, 200 ng per well HA-epitope-tagged pCXCR4, 200 ng per well HA-epitope-tagged pCCR3, and 100 ng per well of the individual GRK5 plasmids. The plasmid encoding mouse At1a was a kind gift of Lutz Hein. Cells were transfected with the reporter gene plasmid pSRE.L that carries the firefly luciferase gene under the control of a modified transcriptional regulatory element, referred to as SRE.L. To correct for variations in transfection efficiency, cells were co-transfected with a reporter plasmid pRL-TK, carrying a Renilla reniformis luciferase gene under the control of the Herpes simplex virus thymidine kinase (HSV-TK) promoter, which provides low-level, constitutive expression of the Renilla luciferase. Twenty-four hours after transfection, cells were stimulated with the indicated chemokine ligands and incubated for another 7 hours. After a single wash with 0.5 ml buffer A (10 mM Na2HPO4, 1.8 mM KH2PO4, 140 mM NaCl, 2.7 mM KCl, pH 7.4), the cells were lysed with 80 μl (24-well plates) or 40 μl (48-well plates) Passive Lysis Buffer (Promega) under gentle agitation on a rocking plate for 15 min at room temperature. The lysate was centrifuged for 10 min at 12,000 × g and the supernatants were collected for luciferase assays. For measurement of reporter gene activity, 10 μl of cleared lysate was combined with 25 μl of Luciferase Assay Reagent II to record the firefly luciferase activity. Twenty-five μl of Stop & Glo® Reagent was added to allow determination of Renilla luciferase activity.
Data from representative experiments are shown as means ± standard deviation of triplicate determinations performed on independently transfected cells.
Western Blotting
HEK293 cells transfected as for the signalling assay were lysed in a SDS based lysis buffer44 and treated with Benzonase (Pierce Thermo Fisher). Proteins were separated on Bolt® 4–12% Bis-Tris Plus Gels (Life technologies) and blotted onto nitrocellulose. Primary antibodies to detect GRK5 (ARP54750_P050, 1:1000) and GAPDH (clone 6C5, 1:500) were purchased from Aviva Antibodies and Acris antibodies, respectively. Signals were obtained using near infrared-labelled secondary antibodies (Li-COR) and a Li-COR Odyssey SA system.
Subcellular localization studies
HEK293 cells and fibroblasts were transfected with Attractene Transfection reagent (Qiagen) according to the manufacturer’s instructions (0.5 μg plasmid per well of a 6-well plate). Fertilized eggs were injected using capped RNA encoding GFP fusion constructs of wild-type Grk5l and Grk5l variants (see also section below). Cilia in fibroblasts were immunostained using a mouse-anti-acetylated antibody (1:500, clone 6-11B-1, Sigma) and an Alexa568-labelled secondary antibody. Cells as well as injected embryos were mounted in Vectashield mounting medium (Vectorlabs).
Zebrafish Husbandry, Manipulation and Analysis
All zebrafish procedures in this study followed the protocols approved by local authorities in Germany (registry no. 0140) and complied with the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes. Zebrafish were maintained in a water-recycling tank system (Tecniplast) and exposed to a 14 hours light and 10 hours dark cycle. Embryos generated from natural wild-type matings (EK and AB strains) were injected at the 1–2 cell stage using an Eppendorf Femtojet and pulled glass capillaries. For both, rescue experiments as well as subcellular localization studies, 500 ng capped RNA was injected into the yolk. Rescue RNAs encoded HA-tagged Grk5l or variants thereof, that was in vitro transcribed from pCS2+ plasmids. GFP-tagged RNAs were transcribed from pCS2+ GFP vectors (a vector map is available on request). The MO to knock down Grk5l has been extensively validated and described previously10,45. After injection embryos were allowed to develop at 28.5 °C to the desired stages, before they were fixed for subsequent analysis. Whole mount in situ hybridisation was performed according to standard protocols using a DIG-labelled RNA probe against southpaw (spaw)10,46.
Microscopy
Transfected cells were analysed with a Leica TCS SP5II confocal microscopy. Single plane pictures were taken. Zebrafish embryos were either imaged using a Leica MZ 125 equipped with an IC80 HD or by confocal microscopy.
Statistical Analysis
Due to the restriction that no ethnic data may be recorded in Germany, no statistical analysis was done for the prevalence of GRK5 variants. Statistical analysis for functional assessment of the variants was performed in Prism4 or Prism6 (GraphPad Software Inc.). The precise statistical test as well as number of experiments and/or animals is indicated in the respective figure legends. Bar graphs are presented as means ± SEM.
Additional Information
How to cite this article: Lessel, D. et al. The analysis of heterotaxy patients reveals new loss-of-function variants of GRK5. Sci. Rep. 6, 33231; doi: 10.1038/srep33231 (2016).
References
Lin, A. E., Ticho, B. S., Houde, K., Westgate, M. N. & Holmes, L. B. Heterotaxy: associated conditions and hospital-based prevalence in newborns. Genet Med 2, 157–172, doi: 10.109700125817-200005000-00002 (2000).
Casar Tena, T., Burkhalter, M. D. & Philipp, M. Left-right asymmetry in the light of TOR: An update on what we know so far. Biol Cell 107, 306–318, doi: 10.1111/boc.201400094 (2015).
Ramsdell, A. F. Left-right asymmetry and congenital cardiac defects: getting to the heart of the matter in vertebrate left-right axis determination. Dev Biol 288, 1–20, doi: 10.1016/j.ydbio.2005.07.038 (2005).
Huang, Z. M., Gold, J. I. & Koch, W. J. G protein-coupled receptor kinases in normal and failing myocardium. Front Biosci (Landmark Ed) 16, 3047–3060 (2011).
Liggett, S. B. et al. A GRK5 polymorphism that inhibits beta-adrenergic receptor signaling is protective in heart failure. Nat Med 14, 510–517, doi: 10.1038/nm1750 (2008).
Lobmeyer, M. T. et al. Polymorphisms in genes coding for GRK2 and GRK5 and response differences in antihypertensive-treated patients. Pharmacogenet Genomics 21, 42–49, doi: 10.1097/FPC.0b013e328341e911 (2011).
Novo, G. et al. G-protein-coupled receptor kinase 5 polymorphism and Takotsubo cardiomyopathy. J Cardiovasc Med (Hagerstown) 16, 639–643, doi: 10.2459/JCM.0000000000000120 (2014).
Spinelli, L. et al. L41Q polymorphism of the G protein coupled receptor kinase 5 is associated with left ventricular apical ballooning syndrome. Eur J Heart Fail 12, 13–16, doi: 10.1093/eurjhf/hfp173 (2010).
Kertai, M. D. et al. G protein-coupled receptor kinase 5 gene polymorphisms are associated with postoperative atrial fibrillation after coronary artery bypass grafting in patients receiving beta-blockers. Circ Cardiovasc Genet 7, 625–633, doi: 10.1161/CIRCGENETICS.113.000451 (2014).
Burkhalter, M. D., Fralish, G. B., Premont, R. T., Caron, M. G. & Philipp, M. Grk5l Controls Heart Development by Limiting mTOR Signaling during Symmetry Breaking. Cell Rep 4, 625–632, doi: 10.1016/j.celrep.2013.07.036 (2013).
Philipp, M., Berger, I. M., Just, S. & Caron, M. G. Overlapping and opposing functions of G protein-coupled receptor kinase 2 (GRK2) and GRK5 during heart development. J Biol Chem 289, 26119–26130, doi: 10.1074/jbc.M114.551952 (2014).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat Methods 7, 248–249, doi: 10.1038/nmeth0410-248 (2010).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4, 1073–1081, doi: 10.1038/nprot.2009.86 (2009).
Freedman, N. J. & Lefkowitz, R. J. Desensitization of G protein-coupled receptors. Recent Prog Horm Res 51, 319–351; discussion 352–313 (1996).
Buss, M. C. et al. The WIP1 oncogene promotes progression and invasion of aggressive medulloblastoma variants. Oncogene 34, 1126–1140, doi: 10.1038/onc.2014.37 (2015).
Heitzler, D. et al. Competing G protein-coupled receptor kinases balance G protein and beta-arrestin signaling. Mol Syst Biol 8, 590, doi: 10.1038/msb.2012.22 (2012).
Palmesino, E., Moepps, B., Gierschik, P. & Thelen, M. Differences in CXCR4-mediated signaling in B cells. Immunobiology 211, 377–389, doi: 10.1016/j.imbio.2005.12.003 (2006).
Vatter, P., Schuhholz, J., Koenig, C., Pfreimer, M. & Moepps, B. Ligand-dependent serum response factor activation by the human CC chemokine receptors CCR2a and CCR2b is mediated by G proteins of the Gq family. J Leukoc Biol 99, 979–991, doi: 10.1189/jlb.2MA0815-386R (2016).
Kostenis, E., Waelbroeck, M. & Milligan, G. Techniques: promiscuous Galpha proteins in basic research and drug discovery. Trends Pharmacol Sci 26, 595–602, doi: 10.1016/j.tips.2005.09.007 (2005).
Milligan, G. & Rees, S. Chimaeric G alpha proteins: their potential use in drug discovery. Trends Pharmacol Sci 20, 118–124 (1999).
Chikumi, H., Vazquez-Prado, J., Servitja, J. M., Miyazaki, H. & Gutkind, J. S. Potent activation of RhoA by Galpha q and Gq-coupled receptors. J Biol Chem 277, 27130–27134, doi: 10.1074/jbc.M204715200 (2002).
Hill, C. S., Wynne, J. & Treisman, R. The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell 81, 1159–1170 (1995).
Mao, J., Yuan, H., Xie, W., Simon, M. I. & Wu, D. Specific involvement of G proteins in regulation of serum response factor-mediated gene transcription by different receptors. J Biol Chem 273, 27118–27123 (1998).
Wang, W. C., Mihlbachler, K. A., Bleecker, E. R., Weiss, S. T. & Liggett, S. B. A polymorphism of G-protein coupled receptor kinase5 alters agonist-promoted desensitization of beta2-adrenergic receptors. Pharmacogenet Genomics 18, 729–732, doi: 10.1097/FPC.0b013e32830967e9 (2008).
Thiyagarajan, M. M. et al. A predicted amphipathic helix mediates plasma membrane localization of GRK5. J Biol Chem 279, 17989–17995, doi: 10.1074/jbc.M310738200 (2004).
Homan, K. T. et al. Crystal Structure of G Protein-Coupled Receptor Kinase 5 in Complex with a Rationally Designed Inhibitor. J Biol Chem 290, 20649–20659, doi: 10.1074/jbc.M115.647370 (2015).
Komolov, K. E., Bhardwaj, A. & Benovic, J. L. Atomic Structure of G Protein-Coupled Receptor Kinase 5 (GRK5) Reveals Distinct Structural Features Novel for GRKs. J Biol Chem 290, 20629–20647, doi: 10.1074/jbc.M115.647297 (2015).
Long, S., Ahmad, N. & Rebagliati, M. The zebrafish nodal-related gene southpaw is required for visceral and diencephalic left-right asymmetry. Development 130, 2303–2316 (2003).
Gilboa, S. M., Salemi, J. L., Nembhard, W. N., Fixler, D. E. & Correa, A. Mortality resulting from congenital heart disease among children and adults in the United States, 1999 to 2006. Circulation 122, 2254–2263, doi: 10.1161/CIRCULATIONAHA.110.947002 (2010).
Basson, C. T. et al. Mutations in human TBX5 [corrected] cause limb and cardiac malformation in Holt-Oram syndrome. Nat Genet 15, 30–35, doi: 10.1038/ng0197-30 (1997).
Schott, J. J. et al. Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science 281, 108–111 (1998).
Cowan, J. R. & Ware, S. M. Genetics and genetic testing in congenital heart disease. Clin Perinatol 42, 373–393, ix, doi: 10.1016/j.clp.2015.02.009 (2015).
Richards, A. A. & Garg, V. Genetics of congenital heart disease. Curr Cardiol Rev 6, 91–97, doi: 10.2174/157340310791162703 (2010).
Shiraishi, I. & Ichikawa, H. Human heterotaxy syndrome - from molecular genetics to clinical features, management, and prognosis. Circ J 76, 2066–2075 (2012).
Oyen, N. et al. Recurrence of congenital heart defects in families. Circulation 120, 295–301, doi: 10.1161/CIRCULATIONAHA.109.857987 (2009).
Fakhro, K. A. et al. Rare copy number variations in congenital heart disease patients identify unique genes in left-right patterning. Proc Natl Acad Sci USA 108, 2915–2920, doi: 10.1073/pnas.1019645108 (2011).
Lander, J. a. & Ware, S. M. Copy number variation in congenital heart defects. Curr Genet Med Rep 2, 168–178 (2014).
Roessler, E. et al. Reduced NODAL signaling strength via mutation of several pathway members including FOXH1 is linked to human heart defects and holoprosencephaly. Am J Hum Genet 83, 18–29, doi: 10.1016/j.ajhg.2008.05.012 (2008).
Kuehl, K. S. & Loffredo, C. Risk factors for heart disease associated with abnormal sidedness. Teratology 66, 242–248, doi: 10.1002/tera.10099 (2002).
McBride, K. L. & Ware, S. M. Modifying Mendel: approaches for identification of susceptibility alleles for human cardiovascular malformations. Circ Cardiovasc Genet 5, 274–276, doi: 10.1161/CIRCGENETICS.112.963579 (2012).
Conklin, B. R., Farfel, Z., Lustig, K. D., Julius, D. & Bourne, H. R. Substitution of three amino acids switches receptor specificity of Gq alpha to that of Gi alpha. Nature 363, 274–276, doi: 10.1038/363274a0 (1993).
Moepps, B. et al. Constitutive serum response factor activation by the viral chemokine receptor homologue pUS28 is differentially regulated by Galpha(q/11) and Galpha(16). Cell Signal 20, 1528–1537, doi: 10.1016/j.cellsig.2008.04.010 (2008).
Hein, L., Meinel, L., Pratt, R. E., Dzau, V. J. & Kobilka, B. K. Intracellular trafficking of angiotensin II and its AT1 and AT2 receptors: evidence for selective sorting of receptor and ligand. Mol Endocrinol 11, 1266–1277, doi: 10.1210/mend.11.9.9975 (1997).
Burczyk, M. et al. Phenotypic regulation of the sphingosine 1-phosphate receptor miles apart by g protein-coupled receptor kinase 2. Biochemistry 54, 765–775, doi: 10.1021/bi501061h (2015).
Soderblom, E. J., Philipp, M., Thompson, J. W., Caron, M. G. & Moseley, M. A. Quantitative label-free phosphoproteomics strategy for multifaceted experimental designs. Anal Chem 83, 3758–3764, doi: 10.1021/ac200213b (2011).
Thisse, C. & Thisse, B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat Protoc 3, 59–69, doi: 10.1038/nprot.2007.514 (2008).
Acknowledgements
We thank John Tesmer for help with the crystal structure of GRK5 and the interpretation of the results, Lutz Hein for the AT1a plasmid and Sabrina Matysik and Sandra Burczyk for excellent fish care. We are also grateful to Martina Burczyk for critical reading of the manuscript. This work was supported by a grant from the Deutsche Stiftung für Herzforschung [MP] and by a Burroughs Wellcome Fund Clinical Scientist Award in Translational Research #1008496 and American Heart Association Established Investigator Award AHA 13EIA13460001 [SMW]. The lab of MP is further supported by grant from the Deutsche Forschungsgemeinschaft (PH144/4-1). TCT is a scholar of the International Graduate School in Molecular Medicine of Ulm University (funded by the German Excellence Initiative of the DFG). The Competence Network for Congenital Heart Defects/National Register for Congenital Heart Defects (Germany) is funded by the Federal Ministry of Education and Research (BMBF), Support Code FKZ 01GI0601 and the DZHK (German Centre for Cardiovascular Research).
Author information
Authors and Affiliations
Contributions
D.L., T.M., T.C.T., B.M., M.D.B. and M.P. performed experiments; D.L., T.M., T.C.T., B.M., U.M.M.B. and M.P. analysed data; D.L., C.K., S.M.W. and M.P. conceived the study; M.-P.H., O.T., A.R., A.S., S.S., U.M.M.B. and S.M.W. were involved in patient recruitment, phenotyping and provided patient material; D.L., M.D.B., S.M.W. and M.P. wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Lessel, D., Muhammad, T., Casar Tena, T. et al. The analysis of heterotaxy patients reveals new loss-of-function variants of GRK5. Sci Rep 6, 33231 (2016). https://doi.org/10.1038/srep33231
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep33231
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
