
Abstract
The central rift of the Red Sea has 25 brine pools with different physical and geochemical characteristics. Atlantis II (ATIID), Discovery Deeps (DD) and Chain Deep (CD) are characterized by high salinity, temperature and metal content. Several studies reported microbial communities in these brine pools, but few studies addressed the brine pool sediments. Therefore, sediment cores were collected from ATIID, DD, CD brine pools and an adjacent brine-influenced site. Sixteen different lithologic sediment sections were subjected to shotgun DNA pyrosequencing to generate 1.47 billion base pairs (1.47 × 109 bp). We generated sediment-specific reads and attempted to annotate all reads. We report the phylogenetic and biochemical uniqueness of the deepest ATIID sulfur-rich brine pool sediments. In contrary to all other sediment sections, bacteria dominate the deepest ATIID sulfur-rich brine pool sediments. This decrease in virus-to-bacteria ratio in selected sections and depth coincided with an overrepresentation of mobile genetic elements. Skewing in the composition of viruses-to-mobile genetic elements may uniquely contribute to the distinct microbial consortium in sediments in proximity to hydrothermally active vents of the Red Sea and possibly in their surroundings, through differential horizontal gene transfer.
Similar content being viewed by others

Introduction
Tectonically induced deep, hypersaline, anoxic marine brine pools are found in the Red Sea, Mediterranean Sea and the Gulf of Mexico. The unique geochemical and physical characteristics of these topographic depressions were more studied than studies of biological life1.
The Red Sea rift contains 25 brine pools2,3,4. The rift is formed by the slow spreading motion of the African and Arabian plate, extending from north (Dead Sea rift) to south (Afar triple junction of the Aden ridge and East African rift)3,5. The origin of the Red Sea deeps and their brine pools is evident from the general structure of strata of the Red Sea bed. Evaporite strata, rich in minerals, were formed by tectonic and climatic changes that caused the desiccation of basins and seas6. Fissuring around the deep causes minerals to be leached by the invading deep water through the evaporite layer and hydrothermal activity may further influence brine formation3,7. Despite shared properties of Red Sea brine pools they are characterized by differential high heavy metal content and metalliferous sediments1. The Atlantis II area includes the largest brine pool Atlantis II (ATIID), Discovery Deep (DD) and Chain Deep (CD)8. Oxygen levels are low in ATIID and DD, the subjects of this study, measuring <1 ml/l below 2000 m depth, but Oxygen is fully absent at higher temperatures-in the ATIID lower brine layer3,9,10. Sources of light are scarce and limited to bioluminescence as the deeps are well beyond the photic zone11. Consequently, this deep oligotrophic extreme environment forms a niche of extremophilic organisms that are well adapted to such harsh conditions.
Both ATIID and DD brine pools have different geochemical and physical characteristics3. A slow rise in temperature over the last 50 years in ATIID was reported while DD remains fairly unchanged3,12. This high heat flux contributed to the ATIID brine pool richer mineral content, higher salinity and brine stratification13. The ATIID brine pool is the largest of all brines, covering an area of 55 km2. Its maximum depth is 2,170 m, and the accumulated metalliferous sediment varies in thickness from 10 to 30 m3,4,14. Its brine reaches up to 270 psu13, and it’s latest recorded temperature was 68 °C9,10. The DD brine pool is a smaller pool, covering 11.5 km2 with a maximum depth of 2,237 m3,14. The deep lies on the periphery of ATIID. The brine temperature was relatively low (48 °C)9,10. Metalliferous sediments in the deep brines have varying thickness and composition depending on the richness and precipitation of minerals from the overlying brine9. Chain Deep lacks hydrothermal activity and its temperature reaches up to 50.3 °C8.
Several culture-dependent and independent studies addressed microbial communities in the Red Sea brine pools, but only two studies addressed the selectively metal rich brine pool sediments15,16. The general microbial community in the ATIID sediment was similar to the overlying water column, as revealed by 16S rRNA and 454 shotgun sequencing16. A comparative 16S rRNA pyrotag analysis of sections from the ATIID and DD brine sediments identified unique microbial consortia in the deepest sulfur-rich ATIID sediment section and the deepest nitrogen-rich DD sediment section15. However, the microbial evolution in relation to differences in environmental conditions of the pool sediments remains to be explored. We hypothesize that lithologically different sections and the geochemical gradient will possess a complementary, genetically resistant microbial community to overcome the diversity of these extreme environments. Additionally, different horizontal genetic transfer mechanisms may have contributed to the evolution of the genetic makeup of microbes in this unique ecosystem. The lithologically different sediment sections, sampled from the 3.5m sediment core, had different depth. Seven Atlantis II sediment samples were studied (ATII-1;-6), the sample closest to the sea bed were samples ATII-1a and the deepest, ATII-6, was 3.4m from the sea-bed. Similarly Seven DD sediment samples were studied (DD-1;-7); DD-1 being the closest to the sea bed and the deepest, DD-7, was 3.4m from the sea-bed15. One sample from CD and another BI site were analyzed, both were a few centimeters from the seabed (Supplementary table 1).
To unveil such microbial communities we performed shotgun metagenomic sequencing and comprehensive analysis of 14 sediment sections of ATIID and DD, and two samples from CD and a Brine Influenced site (BI). A map illustrating the sampling sites in the Red Sea Atlantis II area is presented (Fig. 1). We aimed to identify sediment-specific reads for each sediment section. Through a custom in-silico approach and the classical homology-based alignment to reference sequences approach, we report the phylogenetic and metabolic potential uniqueness of the deepest ATIID brine pool sediments. Our results suggest a distinct microbial consortium, in proximity to hydrothermally active vents, which may have evolved through alternative horizontal genetic transfer mechanisms.

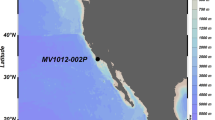
Sampling sites of Red Sea Atlantis II area.
Samples were collected during KAUST Red Sea Expedition on April 2010. Sampling locations of the 1-Atlantis II (ATII), 2-Discovery Deep (DD), 3-Chain Deep (CD) and 4-the brine-influenced (BI) sites are presented. The figure was generated using Google Earth v 7.1.4.1529 © 2015 Google and data provided to the software by Basarsoft, US Dept. of Sate Geographer, Image Landsat, Data SIO, NOAA, U.S. Navy, NGA GEBCO. The overlaid bathymetric contours © 2014, GRID-Arendal.
Results
Metagenomes from Red Sea ATIID, DD, CD and BI sediments
Out of ≈3.7 × 106 reads (1.47 × 109 bp) reads generated by shotgun sequencing of DNA from all sampled brine pools and brine-influenced sediment sites, ≈3.2 × 106 reads (1.32 Gbp) were retained after quality control (QC) processing (Table 1 and Supplementary table 2a). None of the samples showed an appreciable change in GC content following QC (Supplementary table 2b).
Subtraction of ATIID water column and brine seawater reads from the sediment reads was performed to generate sediment specific reads. Atlantis II water column reads were used to subtract common reads from ATIID, DD, BI and CD sediment reads. This filtration step resulted in the utilization of 6.5 × 105, 1 × 106, 6.9 × 104 and 8.1 × 104 reads for ATIID, DD, BI and CD, respectively. The sediment specific reads, are presented for each sample/section (Supplementary table 3). Alpha rarefaction analysis of identified taxa in the sediment datasets, before or after subtraction of water column reads (Supplementary Figure 1), reached an asymptote in the diversity of the sediment-specific datasets compared to unfiltered datasets.
Identification of sediment specific taxa in brine sediments
Domain level taxonomical classification (Fig. 2) showed bacterial related reads prevalence in the ATIID-1a and ATIID-1b datasets, contrary to all other ATIID, DD, BI or CD sections, which were dominated by viral related reads (Table 2). ATIID-1a and ATIID-1b had 32.7 and 20.09% bacterial reads compared to the remaining sediment samples that had 9.37% ± 0.29. Principal Component Analysis (PCA) of sediment-specific reads, based on species-level taxonomical classification, revealed the first three principal components accounted for 97.7% of the data variability, and the first principal component alone accounted for 91.5% of the variability. PCA showed that ATIID-1a and ATIID-1b sections were distant from all other sections, confirming their unique composition. All sections had a large proportion of no-match reads (68.86 ± 0.63% and 67.16 ± 0.94% in total un-subtracted and sediment specific datasets, respectively) and only 30% of the reads were annotated.

Domain distribution of metagenomic reads in ATIID, DD, CD and BI sediment sections.
Classification of the metagenomic reads at the domain level, following elimination of water column reads. The bar graph shows the prevalence reads of bacterial, viral and archaeal origin. Unknown reads for each sample (not illustrated) represent the rest of the bar up to 100%. The classified bacterial community in ATIID-1a and ATIID-1b sections is high compared to the remaining samples. Reads of viral origin are higher in the remaining ATIID, DD, CD and BI samples. The figure was generated by MEGAN software v. 4.70.4 with modifications.
A distinct cluster of taxa, mostly bacteria, was identified in the deepest ATIID sections (Supplementary Figure 3). Out of 2,553 taxa identified, 684 sediment specific taxa showed a significant difference among sediment sections. Consequently, two-group comparison between ATIID-1a & 1b groups vs. all other sections, identified a distinct ATIID-1a & 1b cluster (Supplementary table 4). On the other hand, all sections differed from ATIID-1a & 1b mainly in the viral community.
The distinct ATIID-1a & 1b cluster includes: 1) Archaea: Candidatus Parvarchaeum (Euryarchaeota), Nanoarchaeum equitans (Nanoarchaeota), and Candidatus Nitrosoarchaeum koreensis (Thaumarchaeota), 2) Viruses: only ssDNA virus Nanoviridae and 3) diverse bacterial community taxa. These include a distinct consortium of Actinobacteria, Aquifica, Chloroflexi, Synechococcus, Acaryochloris, Firmicutes as well as α, β and γ- proteobacteria (Supplementary table 3).
The combination of PhAnToMe and GAAS analysis confirmed a dramatic decrease in abundance and diversity of known viruses in ATIID-1a&b sections. These deepest two layers of the Red Sea (ATIID-1) showed a low relative abundance of known viral sequences (1.19% and 2.35%, respectively) (Table 2). The ATIID subsections also demonstrated the lowest viral alpha-diversity, and their viral fraction was largely dominated by gokushoviruses (30.5% and 54.5% of total virome in ATIID-1a and -1b, respectively) followed by uncultured Mediterranean phages (52.3% and 24.8% of total virome in ATIID-1a and -1b, respectively). On the other hand, different Mediterranean phage types and Cyanophages were the most abundant in the remaining sediment sections (85.4% ± 0.3 and 4.8% ± 0.1 for each phage, respectively). Additionally, the virome analysis revealed the dominance of several Mediterranean phage-like viruses in the Red Sea sediment metagenome. The uppermost Discovery Deep section, closest to the seabed (DD-7) was the richest (1,662 different species) and most virally diverse section (Shannon diversity = 5.44).
Selected sediment specific metabolic potential
PCA of functional assignments using SEED classification17 showed that the 16 sediment sections generated three principal components that account for 79.3% of the variability in the data, with the first PCA component describing 51.2% of the variance. Similar to the phylogenetic classification, ATIID-1a & 1b were quite distant from other sediment samples (Supplementary Figure 4a). Repeating the analysis with KEGG orthologous group classification18 generated similar results (Supplementary Figure 4b). As expected, diverse SEED subsystems involved in several metabolic pathways were detected. Most relevant were Sulfur, nucleotide metabolism and DNA replication and repair, which seem to be responsible for distinguishing ATIID-1a & 1b from the rest of the sediment sections (Fig. 3). Sulfur metabolism pathway in these ATIID-1a & 1b sections represented 0.38 ± 0.01% of the total metagenomic reads, a fraction that is significantly higher, compared to other sections (Fig. 3a).

Sulfur metabolism, nucleotides metabolism, DNA replication and repair in all sediment metagenomic reads.
Visualization of (a) Sulfur metabolism utilizing the SEED classification and (b) Differentially represented KEGG orthologous groups in nucleotides metabolism, DNA replication and repair categories. Frequencies are normalized to total identified reads per sample. The figure was generated by MEGAN software v. 4.70.4 with modifications.
A two-group comparison between the sediment specific and total unfiltered datasets, based on SEED subsystems and KEGG functional classifications, allowed the identification of functions likely to be unique to the sediment. Arylsulfatase, aryl-sulfate sulfohydrolase, involved in organic sulfur assimilation and internalin-like proteins involved in virulence were unique to the sediment specific samples. The same comparison using KEGG classification identified homologues to the K05747 orthologous group (Wiskott-Aldrich syndrome protein) involved in cell motility (Supplementary Figure 5).
Abundance and diversity of mobile genetic elements
We analyzed three different mobile genetic elements: plasmids, integrons and insertion sequences (IS). Interestingly, ATIID-1a&b showed the highest abundance and diversity of the three mobile elements in almost all cases (Fig. 4a,b). In general, IS were more abundant than integrons and plasmids. The lowest IS abundance was detected in sections 1&7 of DD. Integrons could not be detected in 10 out of 16 (62.5%) different lithologic sections (ATIID-3,4,5&7; DD-1,2,3&6; CD and BI). The lowest abundance of plasmids was found in BI. All detected IS were classified into their corresponding families, and IS family abundance was computed for each dataset (Fig. 4c). Based on the level and types of IS families, ATIID-1a and b showed a unique cluster. The correlation coefficient (r) and P-value illustrate a positive correlation between bacteria and MGEs abundance and a negative correlation between viruses and MGEs abundance (Supplementary table 6).

Abundance and diversity of mobile genetic elements in the Red Sea metagenomes.
(a) Abundance of mobile genetic elements. (b) Diversity of mobile genetic elements. (c) Heat map based on the abundance of insertion sequence families. ATIID, Atlantis II Deep; DD, Discovery Deep; CD; Chain Deep, BI, Brine Influenced; IS, Insertion sequences. Charts (a,b) were generated by GraphPad Prism software, while heatmap (c) was generated by heatmap package in R environment.
Discussion
A Red Sea brine pool sediment metagenomic study
This study was launched to enrich our knowledge about the biological diversity of the Red Sea brine pool sediments, which are understudied compared to the water column. A challenge to any marine metagenomic study is ruling out contamination with overlying water column, and to address this we computationally eliminated reads in common with the ATIID water column and overlying brine and generated sediment-specific reads (Supplementary table 3). This filtration step was justified by a previous study demonstrating no apparent differences in community structure at the same depth in water from the ATIID and the DD sites of the Red Sea19. These eliminated reads are not necessarily contamination of overlying water column, but may represent shared communities with the sediment, e.g. due to a frequent exchange. This study exclusively focused on sediment specific microbes and metabolic potential.
Unique microbial structure and metabolic potential in Red Sea sediments
Comparison of phylogeny data generated from the current study to the previous 16S rRNA taxa assignment15 revealed 10-folds increase in the microbial richness (Supplementary table 5). This could be due to the amplification bias generated from 16S V6V4 rRNA pyrotags20. Approximately 30% of the sediment-specific reads were well annotated reads and the remaining 70% of the reads had no match to the current database (Fig. 2). Many of these annotated sediment-specific reads were homologous to genes of dsDNA phages: myoviruses, siphoviruses and podoviruses, all of which were homologous to phages infecting cyanobacteria of the Synechococcus and Prochlorococcus genera, which inhabit the photic zone of surface water21,22. The reason that these reads were retained even after the filtrations step may be due to the modular nature of these viruses and their broad host range21,22, therefore escaping elimination. Alternatively if such reads were generated from free phages, and not prophage, then these viruses might have originally stemmed from the overlying water column and sunk down but evolved, over time, in these extreme environments.
To identify sediment-specific metabolic potential, we compared functional classifications of sediment-specific and total unfiltered reads. This included selected sulfur assimilation (Arylsulfatase), cell motility (“Wiskott-Aldrich syndrome protein”) and virulence processes (internalin-like proteins) (Supplementary Figure 5). The identification of internalin-like proteins to be unique to the sediment specific samples is in agreement with the role of viral infection and suggests that these deep marine sediments are hotspots of viral abundance and potentially of viral infection. Reads assigned to DNA repair and nucleotide metabolism, were enriched in sections other than ATIID-1a and b, this could be due to the observed prevalence of viruses, and the new requirement imposed on the infected hosts (Fig. 3).
Marked skew in viruses-to-prokaryote-to-Mobile Genetic Elements ratio in sulfur-rich deep marine sediment sections
Our previous study revealed a relatively high total sulfur content in ATIID-1a & 1b, apparent from the CHNS profiling15. Comparison of the S-rich ATII-1 sections with other sections revealed several abundant taxa and proteins known to be involved in sulfur metabolism (Fig. 3, Supplementary table 5). This finding further suggests that sulfur metabolism shapes the microbial community structure15,23, despite the fact that this pathway was less prevalent than other microbial metabolic potential identified in the metagenome.
The prevalence of viruses in all sediment sections, with the exception of the S-rich ATIID-1a & 1b sections, has been previously reported in the deep sea and is suggested to recycle biomass and maintain diversity of the microbial community22,24,25. We suggest that the higher virus-to-bacteria ratio suggests a higher virus-to-host ratio and a higher rates of lysogenic vs. lytic infection in these habitats26, however more data need to be generated on the host abundance and diversity in these sediments.
Comparative metagenomic analysis showed the deeper ATIID-1a & 1b sediment layers to be distinct from the remaining sediment sections. ATIID-1a & 1b sediment microbial communities were comprised of mainly bacterial taxa. Mobile genetic elements (MGE) were overrepresented in the ATIID-1a & 1b metagenomes including plasmids, insertion sequences and integrons (Fig. 4a,b). Under selected environmental stresses a competitive role may be implied between MGEs and viruses, because of the inverse correlation observed and that the environmental stress in ATIID-1a & 1b sections favors the role of MGEs in horizontal gene transfer and microbial evolution.
This study is an environmental genomics study of three Red Sea brine pool sediments and an adjacent brine-influenced site. The different sampling locations and the lithologically dissected sediment cores downstream analysis revealed an accurate precision of microbial stratification derived from a similarly accurate precision in evolutionary mechanism. The skew of the virus-to-bacteria ratio in only the deeper, sulfur-rich samples, not common in other deep-sea marine systems including hydrothermal vents, suggests an alternative horizontal gene transfer mechanism that contribute largely to the evolution of microbes in the specified narrow section of deep sea sediments. The skew in the viruses-to-mobile genetic elements ratio in only these deeper and sulfur-rich samples suggests that mobile genetic elements contribute largely to the evolution of microbes at these sites. Further studies should examine the role of viruses-to-mobile genetic elements ratio in these selected deep-sea habitats. Additionally, habitat preferences for the prevalence of such alternative horizontal transfer mechanism need to be explored.
Methods
Sampling and generation of metagenomic datasets
Sampling was performed in spring 2010, on board the Hellenic Center for Marine Research (HCMR) research vessel Aegaeo15. Locations of the sampling sites are presented in Fig. 1. The 3.5 m sediment core, was used to sample lithologically distinct seven Atlantis II and seven Discovery Deep samples. DD-1 and ATII-1a, were the deepest samples, ATII-1b was a few centimeters away, but was lithologically distinct. The sediment samples, closest to the seabed, were ATII-6 and DD-7, at 3.4 m deep from the sea-bed. Only one sample was isolated from CD and BI sites, and both were a few centimeters away from the seabed15 (Supplementary table 1). Sampling equipment, physical and CHNS profile of the four sites, the two brine pools ATIID, DD, CD and BI sites are presented in supplementary table 2 and were previously reported15. DNA was extracted from each sample. 0.5–1 g with PowerSoil® DNA isolation kit (MO-BIO, Calsbad, CA) at the American University in Cairo (AUC)27. 1 μg of DNA, from each sample, was subjected to direct metagenomic DNA 454 shotgun pyrosequencing library was constructed as recommended by GS FLX Roche Titanium guide, and the Double SPRI-TE Method (Beckman Coulter) was used for nucleic acid extraction via magnetic bead DNA fragment size selection. The libraries were sequenced with 454 GS FLX Titanium technology (454 Life Sciences) at the AUC laboratory.
Bioinformatic analyses
Raw metagenomic datasets quality control (QC) processing
Metagenomic datasets in raw sequence flow format from 454 GS FLX/FLX+ Data Processing Software were exported and pre-processed using the PRINSEQ software28,29. Read ends with base quality value <15 (corresponding to about ≈97% base calling accuracy) and sequences of length below 60 bp were trimmed. The low complexity filter was used to exclude sequences having entropy <50. An overall quality filter was applied, allowing for a mean quality score of ≥15 along the whole trimmed sequence length. Sequences with ambiguity characters, >5% of their entire length, were removed. Finally exact and near identical (down to 98% similarity) 454 artificial replicates “ghost reads sequencing artifact”30 were removed using CD-HIT-454 software31 default criteria.
Generation of sediment-specific reads
QC reads from the 16 sediment datasets were pooled, followed by mpiBLASTn32,33 alignment with reads from datasets of overlying water column and brine34. ATIID brine/water samples were subtracted from all sediment reads, additionally ATIID and DD brine reads were subtracted from ATIID and DD sediment section datasets, respectively. Default criteria with soft masking enabled, were used in alignment, and reads from each section were filtered out if they had a length of ≥40 bp and a minimum identity of 90% to the reads from any of its target water or brine datasets were filtered out.
Further virome analyses utilizing both GAAS35 and PhAnToMe (PHage ANnotation TOols and MEthods) - http://www.phantome.org/Downloads/36 as performed to estimate the taxonomic composition and average genome size of phage-like elements in the metagenomic samples. With GAAS, the filtering criteria used an E-value threshold of (1 × 10e−6) and a minimum alignment similarity of 75%. The PhAnToMe database used was downloaded on September 1, 2015.
Protein-based phylogeny and function profiling of metagenomic datasets
The sediment reads were annotated by mpiBLASTx32,33 alignment to NCBI nr protein database (Jan. 2013). Default search criteria and soft filtering were enabled and a maximum of 100 hits per query read were used by the MEGAN37 software lowest common ancestor classifier algorithm. Additionally, built-in RefSeq mapping of NCBI nr to SEED17 and KEGG databases18 was used to generate function profiles. The generated profiles for the individual 16 sections were combined and used to build frequency read counts for downstream comparisons. Initially, we filtered frequency data to features with at least one statistically significant difference between any two datasets, using multiple pairwise two-tailed Fisher’s Exact Tests (FET)38,39 and a sequential Bonferroni adjusted p-value threshold of 0.05. Each of the phylogenetic, SEED and KEGG classifications were subjected to such filtering using a custom script that utilized PERL interface to R40 base functions. Differences between datasets were visualized as heat maps of log2(x + 1)-transformed, standardized to z-scores, and hierarchically clustered data. The z-scores were uniformly scaled from 0 to 1. The heat maps were generated using the “enhanced heat map function” from the R package gplots41. Two-way complete linkage hierarchical clustering was performed with 1,000 bootstraps by pvclust R package42 modified to use a Spearman’s correlation matrix generated from the standardized z-scores.
In addition, different datasets were compared based on phylogenetic, SEED and KEGG classifications. Statistical significant differences were identified and visualized using the Statistical Analysis of Metagenomic Profiles (STAMP) software43. The following two-group comparisons were considered: the total unfiltered datasets vs. sediment specific ones, and ATIID-1 sections vs. all other brine pool sediment sections. The two-group comparisons used White’s non-parametric t-test44 for detecting significant differences in mean relative proportions and were set to 10,000 permutations with Storey’s false discovery rate correction45 and a 0.05 p-value cut-off. Comparisons between total datasets and the corresponding sediment specific datasets were further investigated using the two sample comparison Fisher’s Exact Test38,39 and the more conservative Bonferroni correction with a 0.05 p-value cut-off.
Identifying Mobile Genetic Element reads
Abundance and diversity of plasmids, insertion sequences (IS) and integrons were assessed by matching the metagenomic data to the RefSeq database (ftp://ftp.ncbi.nlm.nih.gov/refseq/release/plasmid/, downloaded on January 26, 2014), INTEGRALL database46 (http://integrall.bio.ua.pt/, downloaded on January 29, 2014) and IS-Finder database47 (https://www-is.biotoul.fr/, downloaded on July 9, 2014). BLASTN was used to align sequence reads to the former databases using BLASTn (E-value threshold = 10−5). A read was annotated as a plasmid-like read if it aligned with >95% identity, over at least 90 nucleotides48. On the other hand, a read was assigned to either integrons or IS if it aligned with members of the respective database over 50 nucleotides with more than 90% identity48,49. MGE reads were normalized to calculate plasmid abundance index (PAI), abundance index (IAI) and IS abundance index (ISAI).

All the samples in this study were submitted to NCBI SRA under bio-project accession PRJNA299097 - SRP064947 titled “Metagenomes of Sediments from Red Sea Atlantis II, Discovery and Chain Deep Brine Pools”.
Additional Information
How to cite this article: Adel, M. et al. Viruses-to-mobile genetic elements skew in the deep Atlantis II brine pool sediments. Sci. Rep. 6, 32704; doi: 10.1038/srep32704 (2016).
References
Antunes, A., Ngugi, D. K. & Stingl, U. Microbiology of the Red Sea (and other) deep-sea anoxic brine lakes. Environmental Microbiology Reports 3, 416–433, 10.1111/j.1758-2229.2011.00264.x (2011).
Blanc, G., Anschutz, P. & Pierret, M. C. In Sedimentation and Tectonics in Rift Basins Red Sea:- Gulf of Aden (eds BruceH, Purser & Bosence, DanW J. ) Ch. 27, 505–520 (Springer: Netherlands, 1998).
Gurvich, E. G. In Metalliferous Sediments of the World Ocean Ch. 3, 127–210 (Springer: Berlin Heidelberg, 2006).
Swallow, J. C. & Crease, J. Hot Salty Water at the Bottom of the Red Sea. Nature 205, 165–166 (1965).
Bosworth, W., Huchon, P. & McClay, K. The Red Sea and Gulf of Aden Basins. Journal of African Earth Sciences 43, 334–378, 10.1016/j.jafrearsci.2005.07.020 (2005).
Kastner, M. Oceanic minerals: Their origin, nature of their environment, and significance. Proceedings of the National Academy of Sciences 96, 3380–3387, 10.1073/pnas.96.7.3380 (1999).
Winckler, G. et al. Sub sea floor boiling of Red Sea brines: new indication from noble gas data. Geochimica et Cosmochimica Acta 64, 1567–1575, 10.1016/S0016-7037(99)00441-X (2000).
Edwards, A. J. Red Sea. (Elsevier, 2013).
Swift, S. A., Bower, A. S. & Schmitt, R. W. Vertical, horizontal, and temporal changes in temperature in the Atlantis II and Discovery hot brine pools, Red Sea. Deep Sea Research Part I: Oceanographic Research Papers 64, 118–128, 10.1016/j.dsr.2012.02.006 (2012).
Abdallah, R. Z. et al. Aerobic methanotrophic communities at the Red Sea brine-seawater interface. Frontiers in Microbiology 5, 10.3389/fmicb.2014.00487 (2014).
Roder, C. et al. First biological measurements of deep-sea corals from the Red Sea. Sci. Rep. 3, 10.1038/srep02802 (2013).
Wang, Y. et al. Autotrophic Microbe Metagenomes and Metabolic Pathways Differentiate Adjacent Red Sea Brine Pools. Sci. Rep. 3, 10.1038/srep01748 (2013).
Anschutz, P. & Blanc, G. Heat and salt fluxes in the Atlantis II Deep (Red Sea). Earth and Planetary Science Letters 142, 147–159, 10.1016/0012-821X(96)00098-2 (1996).
Hartmann, M., Scholten, J. C., Stoffers, P. & Wehner, F. Hydrographic structure of brine-filled deeps in the Red Sea—new results from the Shaban, Kebrit, Atlantis II, and Discovery Deep. Marine Geology 144, 311–330, 10.1016/S0025-3227(97)00055-8 (1998).
Siam, R. et al. Unique Prokaryotic Consortia in Geochemically Distinct Sediments from Red Sea Atlantis II and Discovery Deep Brine Pools. Plos One 7, e42872 (2012).
Wang, Y. et al. Bacterial Niche-Specific Genome Expansion Is Coupled with Highly Frequent Gene Disruptions in Deep-Sea Sediments. Plos One 6, e29149 (2011).
Aziz, R. K. et al. SEED Servers: High-Performance Access to the SEED Genomes, Annotations, and Metabolic Models. Plos One 7, e48053, 10.1371/journal.pone.0048053 (2012).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic acids research 28, 27–30 (2000).
Qian, P.-Y. et al. Vertical stratification of microbial communities in the Red Sea revealed by 16S rDNA pyrosequencing. ISME J 5, 507–518, 10.1038/ismej.2010.112 (2011).
Acinas, S. G., Sarma-Rupavtarm, R., Klepac-Ceraj, V. & Polz, M. F. PCR-Induced Sequence Artifacts and Bias: Insights from Comparison of Two 16S rRNA Clone Libraries Constructed from the Same Sample. Applied and Environmental Microbiology 71, 8966–8969, 10.1128/aem.71.12.8966-8969.2005 (2005).
Mann, N. H. Phages of the marine cyanobacterial picophytoplankton. FEMS Microbiology Reviews 27, 17–34, 10.1016/s0168-6445(03)00016-0 (2003).
Anderson, R. E., Brazelton, W. J. & Baross, J. A. The deep viriosphere: assessing the viral impact on microbial community dynamics in the deep subsurface. Rev Mineral Geochem. 75, 649–675 (2013).
Schauer, R. et al. Bacterial sulfur cycling shapes microbial communities in surface sediments of an ultramafic hydrothermal vent field. Environmental Microbiology 13, 2633–2648, 10.1111/j.1462-2920.2011.02530.x (2011).
Corinaldesi, C., Tangherlini, M., Luna, G. M. & Dell’Anno, A. Extracellular DNA can preserve the genetic signatures of present and past viral infection events in deep hypersaline anoxic basins. Proceedings of the Royal Society B: Biological Sciences 281, 10.1098/rspb.2013.3299 (2014).
Ortmann, A. C. & Suttle, C. A. High abundances of viruses in a deep-sea hydrothermal vent system indicates viral mediated microbial mortality. Deep Sea Research Part I: Oceanographic Research Papers 52, 1515–1527, 10.1016/j.dsr.2005.04.002 (2005).
Orcutt, B. N., Sylvan, J. B., Knab, N. J. & Edwards, K. J. Microbial Ecology of the Dark Ocean above, at, and below the Seafloor. Microbiology and Molecular Biology Reviews 75, 361–422, 10.1128/mmbr.00039-10 (2011).
Mustafa, G. A., Abd-Elgawad, A., Abdel Haleem, A. M. & Siam, R. Egypt’s Red Sea Coast: Phylogenetic analysis of cultured microbial consortia in industrialized sites. Frontiers in microbiology 5, 10.3389/fmicb.2014.00363 (2014).
Huse, S., Huber, J., Morrison, H., Sogin, M. & Welch, D. Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biology 8, R143 (2007).
Schmieder, R. & Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864, 10.1093/bioinformatics/btr026 (2011).
Gomez-Alvarez, V., Teal, T. K. & Schmidt, T. M. Systematic artifacts in metagenomes from complex microbial communities. ISME J 3, 1314–1317 (2009).
Niu, B., Fu, L., Sun, S. & Li, W. Artificial and natural duplicates in pyrosequencing reads of metagenomic data. BMC Bioinformatics 11, 187 (2010).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. Journal of Molecular Biology 215, 403–410 (1990).
Darling, A., Carey, L. & Feng, W.-c. The design, implementation, and evaluation of mpiBLAST. Proceedings of ClusterWorld 2003 (2003).
Ferreira, A. J. S. et al. Core Microbial Functional Activities in Ocean Environments Revealed by Global Metagenomic Profiling Analyses. PLoS ONE 9, e97338 (2014).
Angly, F. E. et al. The GAAS Metagenomic Tool and Its Estimations of Viral and Microbial Average Genome Size in Four Major Biomes. Plos Computational Biology 5, e1000593, 10.1371/journal.pcbi.1000593 (2009).
Aziz, R. K., Dwivedi, B., Akhter, S., Breitbart, M. & Edwards, R. A. Multidimensional metrics for estimating phage abundance, distribution, gene density, and sequence coverage in metagenomes. Frontiers in microbiology 6, 381, 10.3389/fmicb.2015.00381 (2015).
Huson, D. H., Mitra, S., Ruscheweyh, H.-J., Weber, N. & Schuster, S. C. Integrative analysis of environmental sequences using MEGAN4. Genome Research 21, 1552–1560, 10.1101/gr.120618.111 (2011).
Agresti, A., Mehta, C. R. & Patel, N. R. Exact inference for contingency tables with ordered categories. Journal of the American Statistical Association 85, 453–458 (1990).
Rivals, I., Personnaz, L., Taing, L. & Potier, M.-C. Enrichment or depletion of a GO category within a class of genes: which test? Bioinformatics 23, 401–407 (2007).
R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, Vienna, Austria, 2013).
gplots: Various R programming tools for plotting data v. R package version 2.12.1 (2013).
pvclust: Hierarchical Clustering with P-Values via Multiscale Bootstrap Resampling v. R package version 1.2-2 (2011).
Parks, D. H. & Beiko, R. G. Identifying biologically relevant differences between metagenomic communities. Bioinformatics 26, 715–721, 10.1093/bioinformatics/btq041 (2010).
White, J. R., Nagarajan, N. & Pop, M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. Plos computational biology 5, e1000352 (2009).
Storey, J. D. & Tibshirani, R. Statistical significance for genomewide studies. Proceedings of the National Academy of Sciences 100, 9440–9445 (2003).
Moura, A. et al. INTEGRALL: a database and search engine for integrons, integrases and gene cassettes. Bioinformatics (Oxford, England) 25, 1096–1098, 10.1093/bioinformatics/btp105 (2009).
Siguier, P., Perochon, J., Lestrade, L., Mahillon, J. & Chandler, M. ISfinder: the reference centre for bacterial insertion sequences. Nucleic acids research 34, D32–D36, 10.1093/nar/gkj014 (2006).
Chen, B. et al. Metagenomic Profiles of Antibiotic Resistance Genes (ARGs) between Human Impacted Estuary and Deep Ocean Sediments. Environmental Science & Technology 47, 12753–12760, 10.1021/es403818e (2013).
Chao, Y. et al. Metagenomic analysis reveals significant changes of microbial compositions and protective functions during drinking water treatment. Sci. Rep. 3, e3550, 10.1038/srep03550 (2013).
Acknowledgements
This work was initially supported by King Abdullah University for Science and Technology Global Collaborative Partners (GCR) program (Award Number: SA-C0039). The work was funded by an American University in Cairo Faculty (Research) Support Grant to RS. We thank the crew and scientists on board the KAUST Red Sea Expedition in spring 2010, in particular chief scientist Dr. Abdulaziz Al-Suwailem. We acknowledge Professor Hamza El-Dorry, of the American University in Cairo, for help with the initial study design. We also thank Amged Ouf and Dr. Ahmed Abdelaziz of the American University in Cairo for DNA preparation.
Author information
Authors and Affiliations
Contributions
M.A., R.K.A. and R.S. designed experiment; M.A., A.H.A.E. and S.K.A. executed and analyzed data; M.A., A.H.A.E., S.K.A., R.K.A., H.-P.G. and R.S. wrote and approved the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Adel, M., Elbehery, A., Aziz, S. et al. Viruses-to-mobile genetic elements skew in the deep Atlantis II brine pool sediments. Sci Rep 6, 32704 (2016). https://doi.org/10.1038/srep32704
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep32704
This article is cited by
-
Discovery of the deep-sea NEOM Brine Pools in the Gulf of Aqaba, Red Sea
Communications Earth & Environment (2022)
-
The association of group IIB intron with integrons in hypersaline environments
Mobile DNA (2021)
-
Diversity, evolutionary contribution and ecological roles of aquatic viruses
Science China Life Sciences (2018)
-
Insertion sequences enrichment in extreme Red sea brine pool vent
Extremophiles (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
