
Abstract
A series of novel peptidomimetics bearing dehydroepiandrosterone moiety were designed, synthesized, and evaluated for their inhibition activities against cell proliferation. According to the preliminary studies on inhibitory activities, some of the newly prepared compounds indicated significantly inhibition activities against human hepatoma cancer (HepG2), human lung cancer (A549), human melanoma (A875) cell lines compared with the control 5-fluorouracil. Especially, compounds Ii (IC50 < 14 μM) and Ik (IC50 < 13 μM) exhibited obvious inhibition activities against all tested cell lines. The highly potential compound Ik induced apoptosis in HepG2 cells were analyzed by flow cytometry, and the apoptotic effects of compound Ik were further evaluated using Annexin V-FITC/propidium iodide dual staining assay, which revealed these highly potential compounds induced cell death in HepG2 cells at least partly by apoptosis.
Similar content being viewed by others


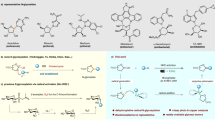
Introduction
Cancers is the leading cause of morbidity and mortality worldwide, with approximately 14 million new cases and 8.2 million cancer related deaths in 20121. The number of new cases is expected to rise by about 70% over the next 2 decades2. Searching and developing effective anticancer drugs is more and more important. Peptides and their derivatives are important molecules with versatile pharmacological properties3, and which are normally designed to mimic a natural protein or peptide. Nevertheless, stability and bioavailability of peptides and mimetics can be improved by several modifications4. In addition, some dipeptide derivatives have exhibited potent inhibition activities against human tumors cells5.
Besides that, steroids are a class of typical lipids found in living world that have broad biological activities6, and which have been widely used in medicine as essentials of anti-inflammatory, anabolic, anticancer and contraceptive drugs7. Recent years, the modifications of natural steroid have become a focus of research probably on account of the various advantages associated with steroid based chemotherapeutics. Dehydroepiandrosterone (DHEA) is a major steroid secreted by the adrenal gland and the most abundant steroid in humans8. Furthermore, several steroidal derivatives have been investigated as potential anti-cancer agents for the treatment of breast cancer, prostate cancer, ovary cancer, lung cancer, gastric cancer, esophageal cancer, heptoma cancer, melanoma cancer, multiple myeloma9,10,11,12,13,14,15,16,17,18. On the other hand, structural modifications carried out at positions 17 of DHEA have exhibited a broad range biological activities as potent antimicrobial agents and anticancer agents10,19.
Recently, during the course of our research for high active compounds, three series of novel peptidomimetics bearing natural tryptamine moiety were designed, synthesized, and evaluated for their inhibition activities against cell proliferation20. Some of the prepared compounds exhibited significant inhibition activities against human hepatoma cancer (HepG2 and Huh-7), human melanoma (A875) cell lines compared with the control 5-fluorouracil. The results from these investigations inspired us to further investigate the novel amino acid-conjugates of dehydroepiandrosterone, which adopt the natural DHEA scaffold to replace the natural tryptamine moiety (Fig. 1). To study the possible structure-activity relationships, several efforts in structure modifications of such type of compounds were designed, and the synthesis of target compounds is simple and convenient as shown in Fig. 2. Besides, their inhibition activities against various cancer cell lines (HepG2, A549, and A875) were also evaluated by MTT method, and the possible mechanism of action for the highly potential compounds were also evaluated using Annexin V-FITC/propidium iodide dual staining assay.

Design strategy of novel amino acid-conjugates of dehydroepiandrosterone.

Synthetic route and conditions for target compounds.
(1) (Boc)2O or CbzCl, NaOH, 0 oC to rt, yields 65–80%; (2) NH2NH2·H2O, EtOH (X = NH) or NH2OH·HCl, CH3COONa, EtOH (X = O), reflux, yields 80–85%; (3) CDI, Et3N, MeCN, rt, yields 50–80%.
Results and Discussion
Chemistry
In the present study, a series of peptidomimetics including steroids groups were designed and synthesized in a simple and convenient route. The general synthetic method for all compounds is outlined in Fig. 2.
The easily available amino acids 1a-g was selected as staring materials, and which were transferred to the corresponding N-(tert-butoxycarbonyl)-amino acids and N-benzyloxycarbonyl-amino acids 2a-m by electrophilic substitution reactions. Meanwhile, compounds dehydroepiandrosterone-17 hydrazone 4 and dehydroepiandrosterone-17 oxime 5 were conveniently prepared from dehydroepiandrosterone 3 by nucleophilic addition elimination reactions. Then the desired peptidomimetics Ia-m were obtained from N-protected amino acids 2a-m and dehydroepiandrosterone-17 hydrazone 4 by nucleophilic substitution. Similarly, compounds IIa-l were also obtained from N-protected amino acids 2a-m with dehydroepiandrosterone-17 oxime 5 by nucleophilic substitution as well. All the compounds gave satisfactory chemical analyses, and the chemical structures and physiochemical properties of the synthesized compounds were summarized in Table 1.
Although the condensation reactions between carboxylic acid and RNH2/ROH can be generated by a lot of catalysts, we wish to develop convenient and effective methods for our own syntheses. First, compound dehydroepiandrosterone-17 hydrazone 4 and N-cbz-L-valine was chosen as a model system (Fig. 3). Six kinds of common and appropriate catalyst composition were examined and screened, dicyclohexylcarbodiimide and 4-dimethylaminopyridine (DCC/DMAP), dicyclohexylcarbodiimide and 4-methylmorpholine (DCC/NMM), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride and 4-dimethylaminopyridine (EDCI/DMAP), O-(benzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium tetrafluoroborate and triethylamine (TBTU/Et3N), trimethyl borate (B(OCH3)3), N,N’-carbonyldiimidazole and triethylamine (CDI/Et3N) (Fig. 3, Entries 1–6). CDI and Et3N composition was found to give the best conversion. Besides, we found high temperature cannot improve the conversion (Fig. 3, Entries 6, 7). Different solvents were screened in order to increase the conversion (Fig. 3, Entries 6, 8, 9). Acetonitrile was found to give the best conversion among the three solvents. Without triethylamine, the conversion has a little decrease (Fig. 3, Entry 10). Furthermore, different molar ratios for the substrates were also examined to increase the conversion (Fig. 3, Entries 6, 11, 12, and 13). The best condition was shown in Fig. 3 as entry 6 in summary.

Reaction conditions screening.
Inhibitory effects of compounds on the proliferation of various cancer cells
The newly prepared peptidomimetics derivatives were evaluated for their in vitro cytotoxic effects against HepG2 (hepatocellular liver carcinoma), A549 (Human lung cell line), A875 (human melanoma cell line) by the standard MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) assay21 using 5-FU (5-Fluorouracil) as a positive control. The preliminary results were summarized in Fig. 4 and Table 2. The IC50 value represents the drug concentration required to inhibit cell growth by 50%.

Inhibition activities against cell proliferation for target compounds at 40 μg/mL.
Abbreviations: HepG2 - Human hepatocellular liver carcinoma cell line; A549 -Human lung cell line; A875 - Human melanoma cell line; 5-FU - 5-Fluorouracil, used as a positive control.
Generally, as shown in Fig. 4, the prepared peptidomimetics derivatives (1–26) showed moderate to good inhibition activities against the three tested human cancer cell lines. Most compounds displayed better inhibition activities than 5-FU. Notably, the compounds Ia, Ib, Ii, Ij, Ik, Il, and IId, IIk, IIl exhibited significant inhibitory activities against all three tested cell lines with 70.1–86.4% growth inhibition at 40 μg/mL concentration compared to the positive control 5-FU (56.6–65.3%). Also, it is interesting to note that compound IIe showed selective cytotoxicity to A549 cell line and A875 cell line with 62.1% and 62% growth inhibition respectively, and with 31% inhibitions to HepG2 cell lines.
Moreover, the preliminary bioassay indicated that most of the target compounds (such as Ia, Ib, Ii, Ij, Ik, and IId, IIk and IIl) displayed good inhibitory activities compared to 5-FU, so in order to investigate the potential activities, the IC50 values were further evaluated. The inhibitory activities expressed as IC50 values for the target compounds are presented in Table 2. The results also testify that some of the designed peptidomimetics derivatives exhibited higher inhibition activity than the control 5-FU under the same conditions. As indicated in Table 2, compound IId showed the strongest inhibitory effect against HepG2, with an IC50 value of 7 μM; compound Ik showed the strongest inhibitory effect against A549 and A875 with an IC50 value of 6 and 13 μM, respectively. We also can find that compound Ib have the same inhibition activities trend as 5-FU against the three cancer cell lines. Especially, compounds Ib, Ic, Ii, Ik, Il, Im, IId and IIk exhibited significant inhibition against all tested cancer cell lines compared to the positive control 5-FU.
Furthermore, the dose-response analysis of cell growth inhibition activity for representative compounds Ii, Ik, IId and 5-FU has been displayed in Fig. 5, which revealed that the cytotoxic effects on cell lines of target compounds indicated obvious concentration-dependent manner.

Dose–response analysis of cell growth inhibitory activity for representative compounds Ii, Ik, IId and 5-FU (positive control) against HepG2 (left), A549 (middle) and A875(right) cell lines.
Results of Annexin V-FITC assay for apoptosis
Potential of the investigated compound Ik to induce apoptosis in HepG2 cells was analyzed by flow cytometry, following treatment with IC50 or 2 × IC50 concentrations for 24 h. The apoptotic effect of compound Ik was evaluated using Annexin V-FITC/PI dual staining assay, which can examine the occurrence of phosphatidylserine (PS) externalization as well as understand whether it is due to physiological apoptosis or nonspecific necrosis22. All data obtained in this study are presented in Figs 6 and 7, as a percentages of an early apoptotic cells, FITC(+)PI(−); late apoptotic cells, FITC(+)/PI(+); and necrotic cells, FITC(−)/PI(+); presenting intact cells, FITC(−)/PI(−).

Annexin V-FITC flow cytometry.
Cell cycle progression (A–C) and annexin V-FITC/PI staining was monitored in HepG2 cells following 24 h treatment with compound Ik at concentrations corresponding to their IC50 or 2 × IC50 (E and F). Representative dot plots of three independent experiments are given, presenting intact cells at lower-left quadrant, FITC(−)/PI(−); early apoptotic cells at lower-right quadrant, FITC(+)PI(−); late apoptotic or necrotic cells at upper-right quadrant, FITC(+)/PI(+); necrotic cells at upper-left quadrant, FITC(−)/PI(+).

Apoptotic effect of compound.
Ik was evaluated after 24 h treatment; bar graphs represent mean ± SD in at least three independent experiments.
Results revealed that compound Ik induced apoptotic changes following 24 h treatment. As shown in Figs 6 and 7, compound Ik initiated excellent apoptosis, in terms of FITC(+)PI(−) staining, compared to the control. Compound Ik exhibited 17.2% of apoptosis at IC50 and 51.5% of apoptosis at 2 × IC50 concentrations, whereas 2.5% of apoptosis was observed in control (0.1% DMSO). Besides, Compound Ik existed 75.7% of presenting intact cells at IC50 and 47.7% of presenting intact cells 2 × IC50 concentrations, while 95.6% of presenting intact cells was observed in control. From this experiment it was observed that the highly potential compound induced cell death in HepG2 cells at least partly (initially), by apoptosis. However, the precise mechanisms of cell death induction by tested compound still remain to be further explored.
Structure and activity relationship (SAR)
Within the limits of experimental error, for the present series of compounds Ia-f and IIa-f, of which the N-protected group is carbobenzoxy, the compounds Ia, Ib, Id, IIa, IIb and IId displayed better antiproliferation activities. In terms of these compounds, the compounds with lower molecular weight indicated better inhibitory activities against the cell lines. On the contrary, when the N-protected group is tert-butoxycarbonyl, the other compounds Il, Im, IIk and IIl showed better inhibitory activities. It can be speculated, when steric hindrance is smaller, compounds bearing aromatic ring displayed better antiproliferation activities, otherwhise, compounds with lower molecular weight displayed better antitumor activities. As far as glycine and proline derivatives are concerned, N-carbobenzoxy protected target compounds displayed better inhibitory activities compared to N-tert-butoxycarbonyl-protected compounds. However, for phenylalanine and tryptophan derivatives, N-tert-butoxycarbonyl-protected compounds indicated better inhibitory activities. Meanwhile, we also can found that the compounds containing oxime unit presented poor antiproliferation activities than that of the compounds bearing hydrazine moiety, which perhaps to prove the importance of amide bond.
Conclusion
In the present study, twenty-five novel peptidomimetics derivatives containing natural steroid moiety have been conveniently synthesized, and their potential antitumor activities have also been evaluated in vitro. The preliminary bioassay results indicated that some of the compounds displayed obviously good inhibition activities against human cancer cell lines including HepG2, A549 and A875, especially, compounds Ii and Ik exhibited high cytotoxic activities against the three cancer cell lines, which might be developed as novel lead scaffold for potential antitumor agents. The further annexin V-FITC assay revealed these highly potential compounds induced cell death in HepG2 cells at least partly by apoptosis.
Experimental
Synthesis of target compounds
The instrumentation, chemicals, synthetic procedures and characterization were provided in supplementary data.
In vitro cytotoxicity assays
The in vitro cytotoxicity of the synthesized compounds against different human cancer cell lines (HepG2, A549, A875) was measured with the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay21. All the data of the experiment were analyzed with SPSS software, and the 50% inhibitory concentrations (IC50) of each compound for the different cell lines were determined. A control was run for each test, and all assays were performed in triplicate on three independent experiments, and measurement data were expressed as the mean ± S.D.
Flow cytometric analysis of apoptosis
Quantitative analysis of apoptotic and necrotic cell death induced by the test compounds was performed by Annexin V-FITC apoptosis detection kit according to the manufacturer’s instructions (BD Biosciences). Briefly, 2 × 105 HepG-2 cells were seeded in 6-well plates and grown overnight. After removal of the growth medium, cells were treated with compound Ik for 24 h, at concentrations corresponding to their IC50s or 2 × IC50s. Cells treated with 0.1% DMSO were served as solvent control. Following treatment cells were harvested, washed twice with ice-cold PBS and resuspended in binding buffer. Then the cells were stained by adding 5 μL of Annexin V-FITC and 5 μL of propidium iodide, sit for 15 min at room temperature in the dark and analyzed by flow cytometry (Beckman coulter FC500).
Additional Information
How to cite this article: Wang, X. et al. Peptidomimetics Based On Dehydroepiandrosterone Scaffold: Synthesis, Antiproliferation Activity, Structure-Activity Relationship, and Mechanisms. Sci. Rep. 6, 32654; doi: 10.1038/srep32654 (2016).
References
Stewart, B. W. & Wild, C. P. World Cancer Report 2014, World Health Organization. (2014).
Caner, Fact sheet N°297. World Health Organization. (2015).
Shivamallu, C. et al. Pseudo-peptides as novel antileptospiral agents: Synthesis and spectral characterization. Spectrochim. Acta A. 118, 1152–1157 (2014).
Adessi, C. & Soto, C. Converting a peptide into a drug: strategies to improve stability and bioavailability. Curr. Med. Chem. 9, 963–978 (2002).
Huang, X. et al. Synthesis and antitumor activities of novel dipeptide derivatives derived from dehydroabietic acid. Bioorg. Med. Chem. Lett. 24, 1511–1518 (2014).
Salvador, J. A. R. et al. Anticancer steroids: linking natural and semi-synthetic compounds. Nat. Prod. Rep. 30, 324–374 (2013).
Gan, C. et al. Synthesis and antiproliferative activity of some steroidal thiosemicarbazones, semicarbazones and hydrozones. Steroids 87, 99–107 (2014).
Liu, X. et al. Synthesis and antitumor activity of dehydroepiandrosterone derivatives on Es-2, A549, and HepG2 Cells in vitro. Chem. Biol. Drug Des. 79, 523–529 (2012).
Huang, Y. et al. Synthesis and antitumor evaluation of novel hybrids of phenylsulfonylfuroxan and epiandrosterone/dehydroepiandrosteron derivatives. Steroids 101, 7–14 (2015).
Ke, S. et al. Synthesis of novel steroid derivatives derived from dehydroisoandrosterone as potential anticancer agents. Anti-Cancer Agents Med. Chem. 13, 1291–1298 (2013).
Ni, S. et al. The DHEA metabolite 7b-hydroxy-epiandrosterone exerts anti-estrogenic effects on breast cancer cell lines. Steroids 77, 542–551 (2012).
Zhang, B. et al. Synthesis and biological evaluation of dehydroepiandrosterone-fused thiazole, imidazo[2,1-b]thiazole, pyridine steroidal analogues. Steroids 80, 92–101 (2014).
Kvasnica, M. et al. Platinum(II) complexes with steroidal esters of L-methionine and L-histidine: Synthesis, characterization and cytotoxic activity. Bioorg. Med. Chem. 16, 3704–3713 (2008).
Garrido, M. et al. Cytotoxic effect of novel dehydroepiandrosterone derivatives on different cancer cell lines. Eur. J. Med. Chem. 68, 301–311 (2013).
Bratoeff, E. et al. Effect of dehydroepiandrosterone derivatives on the activity of 5a-reductase isoenzymes and on cancer cell line PC-3. Bioorg. Med. Chem. 22, 6233–6241 (2014).
Ke, S., Shi, L. & Yang, Z. Discovery of novel isatin–dehydroepiandrosteron conjugates as potential anticancer agents. Bioorg. Med. Chem. Lett. 25, 4628–4631 (2015).
Vosooghi, M. et al. Synthesis and in vitro cytotoxic activity evaluation of (E)-16-(substituted benzylidene) derivatives of dehydroepiandrosterone. J. Pharm. Sci. 21, 34–41 (2013).
Schneider, G. et al. Stereocontrolled synthesis of the four 16-hydroxymethyl-19-nortestosterone isomers and their antiproliferative activities. Steroids 105, 113–120 (2016).
Handratta, V. D. et al. Novel C-17-heteroaryl steroidal CYP17 inhibitors/antiandrogens: Synthesis, in vitro biological activity, pharmacokinetics, and antitumor activity in the LAPC4 human prostate cancer xenograft model. J. Med. Chem. 48, 2972–2984 (2005).
Wang, X., Su, H., Chen, C. & Cao, X. Synthesis and biological evaluation of peptidomimetics containing tryptamine moiety as potential antitumor agents. RSC Adv. 5, 15597–15602 (2015).
Alley, M. C. et al. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res. 48, 589–601 (1988).
Browne, L. J. et al. Fadrozole hydrochloride: a potent, selective, nonsteroidal inhibitor of aromatase for the treatment of estrogen-dependent disease. J. Med. Chem. 34, 725–736 (1991).
Acknowledgements
We gratefully acknowledge the support of this work by the Fundamental Research Funds for the Central Universities (2662015PY034, 2662016PY112).
Author information
Authors and Affiliations
Contributions
X.C. initiated the idea and designed the research; X.W. performed the chemical synthesis and characterization; H.S. and W.W. performed the assays and data analysis; X.W., C.C. and X.C. analyzed the results and wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wang, X., Su, H., Wang, W. et al. Peptidomimetics Based On Dehydroepiandrosterone Scaffold: Synthesis, Antiproliferation Activity, Structure-Activity Relationship, and Mechanisms. Sci Rep 6, 32654 (2016). https://doi.org/10.1038/srep32654
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep32654
This article is cited by
-
Peptidomimetics in cancer targeting
Molecular Medicine (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
