
Abstract
Drug repositioning is commonly done within the drug discovery process in order to adjust or expand the application line of an active molecule. Previous computational methods in this domain mainly focused on shared genes or correlations between genes to construct new drug-disease associations. We propose a method that can not only handle drugs or diseases with or without related genes but consider the network modularity. Our method firstly constructs a drug network and a disease network based on side effects and symptoms respectively. Because similar drugs imply similar diseases, we then cluster the two networks to identify drug and disease modules, and connect all possible drug-disease module pairs. Further, based on known drug-disease associations in CTD and using local connectivity of modules, we predict potential drug-disease associations. Our predictions are validated by testing their overlaps with drug indications reported in published literatures and CTD, and KEGG enrichment analysis are also made on their related genes. The experimental results demonstrate that our approach can complement the current computational approaches and its predictions can provide new clues for the candidate discovery of drug repositioning.
Similar content being viewed by others

Introduction
Traditionally, drug discovery process mainly consists of three stages: discovery, preclinical stage, and clinical development1. In the discovery stage, new drugs are screened and identified. Then, the new drugs are tested in vitro and in animal models in the preclinical stage. Finally, the drug candidates are tested in human beings as clinical trials in the clinical development stage. The whole process is time-consuming, costly, and often inefficient. It is conservatively estimated that the time for bring a drug to market is about 15 years2, and the cost is $800~1000 million3. Drug repositioning, which refers to identify and use the known drugs that can treat diseases other than those for which they were originally designed4, is an increasingly attractive mode of therapeutic discovery. This strategy certainly has the potential of being the most efficient technique for drug development since it does not need the initial six to nine years, thus reducing risk and costs5. There are a number of drug examples that have been successfully repositioned such as sildenafil citrate (brand name: Viagra), which was repositioned from a common angina drug to a therapy for erectile dysfunction and pulmonary hypertension6, and minoxidil which was originally tested for hypertension and now was indicated for hair loss7.
With the dramatic expansion of available high-throughput datasets, many approaches to discover new drug indications have been developed. Some are mainly focused on small-scale applications to analyze specific classes of drugs or drugs for specific diseases8,9,10. For example, based on chemical similarity, Noeske et al. considered the pharmacophore descriptors of drugs to cluster drugs8. In 2010, Kotelnikova et al. firstly constructed signaling pathways related to glioblastoma based on data got from scientific literature and ResNet database. Then using Sub-Network Enrichment Analysis (SNEA), they analyzed the differential expression in glioblastoma patients9. There are also a few examples involving a relatively large number of drugs and diseases11,12,13,14,15. Lamb et al.11 created the first installment of a reference collection of gene-expression profiles to discovery functional connections among diseases, genetic perturbation, and drug action. Based on molecular activity similarity, the researchers constructed a drug network12 and they partitioned the network into densely interconnected groups. The drugs in the same group are significantly enriched for compounds with similar mode of action, or in the same pathway, which can be used to identify the compound-targeted biological pathways. Some methods also predicted drug-target interactions for drug repositioning16,17 and microRNA-disease associations based on social network analysis methods18,19. In 2014, Ye et al. constructed drug-drug interaction through side effect similarities and predicted the indications of a drug by the functions of its neighboring drugs20. PREDICT is based on the observation that similar drugs are indicated for similar diseases, and utilizes multiple drug-drug and disease-disease similarity measures for the prediction of drug-disease associations21. It allows easy integration of additional similarity measures among diseases and drugs.
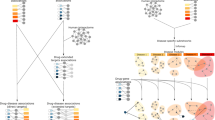
Here, we propose a method for predict potential drug-disease associations that can not only handle drugs or diseases with or without related genes but consider the network modularity. The main framework of our method is shown in Fig. 1. Based on side effects of drugs and symptoms of diseases, drug-drug and disease-disease weighted networks are firstly constructed. Then, we cluster the two networks and get drug-module and disease-module sets. Further, according to the known associations in Comparative Toxicogenomics Database (CTD; http://ctd.mdibl.org)22 between drugs and diseases, we correlate drug and disease modules with scores. Finally, we rank the drug-disease module pairs according to their scores and select the top-3 drug-disease module pairs for further analysis. For each selected drug-disease module pair, we score the connection between each drug and each disease in the pair. The larger the score, the greater the degree of reliability, thus the greater the possibility of drug relation to disease. We evaluate our predictions with drug-disease associations that are reported in published literature and CTD benchmark, and also make Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis on their related genes. The results demonstrate that our predictions can provide new clues for drug repositioning based on drug side effects, disease symptoms and network modularity.

The framework of our method.
Results
Based on the top-3 drug-disease module pairs, shown in Table 1, we get three ranking lists of drug-disease association and predict new drug indications. The top associations in each list are reliable. Therefore, we evaluate the top-20 predicted associations in each list by their overlap with drug indications that are reported in published literature and Comparative Toxicogenomics Database (CTD; http://ctd.mdibl.org) benchmark, and we also make Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis on them.
CTD benchmark and literature verification
Table 2, 3, 4 respectively show three lists of top-20 drug-disease associations corresponding to the three drug-disease module pairs in Table 1 (Rank = 1 to 3). In these tables, the predicted results are divided into two categories: known and potential associations. If there is direct evidence for a chemical-disease association marked as “therapeutic” or “marker/mechanism” in CTD database, we take it as a known association. Otherwise, it is considered as a potential drug-disease association (marked as bold italic items), such as prochlorperazine and Tremor (ID = 9) in Table 2. In the following section, we abbreviate “therapeutic” to “T”, and “marker/mechanism” to “M”.
Verification of top-20 drug-disease associations in module pair while Rank = 1
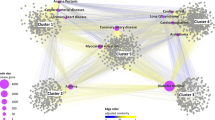
Figure 2 shows the network topology of the first drug-disease module pair (Rank = 1 in Table 1). In the figure, internal connections within a module are labelled by blue, and external connections between two modules are labelled by purple. Green circle and red diamond nodes represent drugs and diseases, respectively. The purple edges represent the reliably curated drug-disease associations in CTD, which are also shown in Table 2 (CTD mark are “M” or “M&T”).

Internal connections within a module are labelled by blue, and external connections between two modules are labelled by purple. Green circle and red diamond nodes represent drugs and diseases respectively. The purple edges represent the reliably curated drug-disease associations in CTD (CTD mark is “M” or “M&T”).
In Table 2, we find 18 of 20 are known associations. The percentage reaches up to 90%. Two results are new predictions and labelled by bold italic. The association between fluphenazine and Parkinsonian Disorders (ID = 19) is inferred by CTD (Inference Score = 7.38). The inference score23 reflects the degree of similarity between CTD chemical-gene-disease networks and a similar scale-free random network, which is computed as shown below:

where Y represents the inference score; P represents the probability that a vertex in a large network interacts with another vertex decays according to a power law24; G, C, and D represent a gene, chemical, and disease respectively; k represents the number of connection between G, C, or D; nG represents a gene set. The higher the Y score, the more likely the inference network has atypical connectivity25.
We also find the inferred association (ID = 19) is based on three genes: NGF26, DRD227, and PRL28. Moreover, the two results (in bold italic) are also supported by external literature. Studies29 have shown that prochlorperazine can cause Tremor (ID = 9). People possibly have tremor while taking prochlorperazine from FDA (Food and Drug Administration) and social media. Fluphenazine (ID = 19) is a drug used to treat psychotic disorders, agitation, and dementia30. And its use may lead to the development of symptoms that resemble Parkinson’s disease31.
Verification of top-20 drug-disease associations in module pair while Rank = 2
Supplementary Fig. S1 shows the relationships of the second drug-disease module pair (Rank = 2 in Table 1). Table 3 gives the details of top-20 drug and disease associations. In this table, sixteen associations are known and the remaining four ones marked as bold italic are new predicted results. The association between thorazine and Lewy Body Disease (ID = 16) is inferred associations in CTD (Inference Score = 3.77). The relationship is based on the gene “MAG”. Evidences show that thorazine affects the expression of MAG mRNA32 and that MAG expression significantly relates to Lewy Body Disease33. Thorazine is an antipsychotic medication, which is primarily used to treat psychotic disorders such as schizophrenia. Lewy body dementia (LBD) is a type of progressive dementia that leads to a decline in thinking, reasoning and independent function because of abnormal microscopic deposits that damage brain cells over time34. LBD is found among people who take thorazine, especially for people who are male, more than 60 years old, also take medication protonix, and have insomnia35. The researchers found thorazine had shown better capacity to control hyperkinetie manifestations, and had not shown any undesirable side effects36,37.
For the association (ID = 10), thioridazine is a typical antipsychotic drug used in the treatment of Psychoses (ID = 10)38. Prochlorperazine (ID = 18) belongs to a group of medicines called “phenothiazines”. It is prescribed for a variety of unrelated conditions, including problems with balance and dizziness, sickness, agitation and restlessness, and schizophrenia39. Bipolar Disorder (ID = 18) is found among people who take prochlorperazine, especially for people who are female, more than 60 years old, also take medication klonopin, and have nausea40. Symptoms typical of alcohol withdrawal (ID = 20) include agitation, seizures, and delirium tremens41. Laties and colleagues42 determined that promazine and chlorpromazine (ID = 20) were equally efficacious in the treatment of delirium tremens. Chlorpromazine, promazine, and other low-potency typical antipsychotic agents have been reported43 to have the greatest effect on lowering seizure threshold.
Verification of top-20 drug-disease associations in module pair while Rank = 3
The network topology of the third drug-disease module pair (Rank = 3 in Table 1) is shown in Supplementary Fig. S2. From Table 4, we find 2 associations (in bold italic) are new predictions by our method. The other 18 associations are all known in CTD. The percentage reaches up to 90%. Though, two other associations (ID = 15 and ID = 20) are not existed in CTD at present, their associations are supported by features of their ancestors and literatures. Circadian rhythms (“body clocks”) are controlled by a biological clock and work on a daily time scale. Circadian rhythm sleep disorders (CRSD) (ID = 15) are a family of sleep disorders. People with circadian rhythm sleep disorders are unable to sleep and wake at the times required for normal work, school, and social needs. We find amitriptyline (ID = 15) has strong connections with sleep disorders in CTD (marked as “M&T”). They are related by the gene “CHRNB2”: amitriptyline results in the decreased expression of CHRNB2 mRNA44 and CHRNB2 protein results in the increased susceptibility to amitriptyline45. Hence, it is reasonable to infer that circadian rhythm sleep disorders and amitriptyline probably have close connection. Moreover, amitriptyline is found useful as a sleep aid continues46. Postpartum depression (ID = 20) is moderate to severe depression in a woman after she has given birth, which is a descendant of depressive disorder. The relationship between imipramine (ID = 20) and depressive disorder is marked as “therapeutic” and “marker/mechanism”, which is based on five genes: ALB47, BDNF48, CRH49, POMC50, SLC6A451. Therefore, it is likely that postpartum depression has connection with imipramine. Furthermore, Cohen and Rosenbaum52 pointed out the use of tricyclic antidepressants should not pose a risk when used in pregnancy or in the postpartum period. They maintained that the safest medications to use at this point in time are nortriptyline, imipramine, and fluoxetine.
KEGG pathway enrichment analysis
In this section, for the predicted drug-disease associations shown in bold italic in above tables, we perform KEGG pathway enrichment analysis on their related gene sets with the functional annotation tool of DAVID53. For DAVID, EASE Score, a modified Fisher Exact P-Value, is used as a threshold for gene-enrichment analysis54. It ranges from 0 to 1. When Fisher Exact P-Value is 0, it represents perfect enrichment. We set it as 0.01.
For each drug, we combine the genes obtained from Drugbank database55, which combines detailed drug data with comprehensive drug target information, and the top interacting genes got from CTD database. Similarly, the genes related to each disease are got from OMIM database56, which is a comprehensive, authoritative compendium of human genes and genetic phenotypes.
For each predicted drug and disease pair, we respectively put their corresponding genes into DAVID and examine whether the drug and the disease have overlapped KEGG pathways. If they have more overlapped pathways, they are more relevant, that is the drug has strong correlation with the disease. For example, prochlorperazine and tremor (Rank = 9 in Table 2) has one overlapped pathway: “Neuroactive ligand-receptor interaction” (p-value = 1.2E-3). We find thioridazine (Rank = 10 in Table 3) has two pathways overlapping with psychoses, substance-induced: “neuroactive ligand-receptor interaction” and “calcium signaling pathway”. Their corresponding p-values are 2.14E-05 and 2.81E-04 respectively. Lower p-values indicate the predicted associations are reliable. Especially, the pathway overlapped between amitriptyline and dyssomnias (Rank = 16 in Table 4) is “neuroactive ligand-receptor interaction”, which has a very low p-value: 8.66E-29. These good results show their high reliability. Because of the incompleteness of data, the numbers of genes related to some drugs and diseases are all small, such as prochlorperazine and bipolar disorder (Rank = 18) in Table 3. Therefore, it is hard to find pathways related to prochlorperazine and bipolar disorder using DAVID tool at present. With the improvement of data, the performance of our method will be more effectively.
Comparison with other method based on CTD benchmark
To evaluate the performance of our method, we compare it with PREDICT21. PREDICT integrates multiple data sources, including chemical structures, drug side effects, drug target protein sequences and target protein interactions and phenotype data. It applies network analysis for target protein distance calculation, applies text mining to identify disease phenotypes and use machine learning algorithms to classify true and false drug-disease associations2.
Based on CTD benchmark, we make a comparison between our method and PREDICT21. We choose the top-20 drug-disease associations of PREDICT for analysis. They are shown in Table 5. In the table, the column “Disease name” sometimes includes more than one disease name. As long as one disease is found related to a drug in CTD, the association is marked as “T”, “M”, “M&T” or “inferred”. Taking the fourth association (Rank = 4) as an example, its “Disease name” = “Pyogenic Sterile Arthritis, Pyoderma Gangrenosum, And Acne” (Rank = 4). We find “Arthritis” is related to “Gonadorelin” in CTD and it is an inferred association. Thus the association is marked as “inferred”, i.e. CTD mark = “inferred”. Finally, there are 14 drug-disease connections are found in CTD benchmark, but only 5 of them are known associations (CTD mark = “T” or “M&T” and marked as bold italic in Table 5) and the other 9 associations are inferred in CTD (CTD mark = “inferred”). Its precision is 5/20 = 0.25, which is lower than that of our method. The precision of our top-20 associations in each list is more than 0.8. From Table 2, 3, 4, they are 0.9, 0.8, and 0.9 respectively.
Discussion
Based on side effects of drugs and symptoms of diseases, we construct drug and disease networks firstly. Then we cluster them to get two types of modules: drug module and disease module. According to the known drug-disease associations in CTD database, we score each pair of drug-disease module and reserve the top-3 drug-disease module pairs. For each selected drug-disease module pairs, we construct a drug-disease bipartite graph. We calculate all the connections to predict potential drug-disease associations. The significant enrichments of our predictions in the biomedical literature, clinical trials and KEGG pathways demonstrate that our approach can effectively identify new indications as an indicator for the mode of action. The success of our methods PDCIM can be attributed as follows: First, we integrate clinical data into our model, such as disease symptoms and drug side effects. Second, our approach is based on clustering and known drug-disease associations. Finally, we combine the neighborhood information of nodes in drug modules and disease modules. We believe that the combination of clinical data, network clustering and subnetwork connectivity could help us to predict new hypotheses to infer the drug-disease relationship and even improve the drug development. Moreover, our predicted drug-disease associations are not generated using the genes associated with diseases or drugs, so we can find some drug-disease associations that these drugs or diseases have less or no related genes. However, the limitation is the difficulty in distinguishing the positive and negative associations between drugs and diseases. In the future, we can choose different methods to calculate the similarity between drugs and diseases to enhance the reliability of our constructed networks. On the other hand, we can integrate various data sources such as pharmacological data, therapeutic/toxicological expression profiles and DNA methylation data to try to distinguish the positive and negative associations between drug and disease.
Methods
Data Source
Human symptom-disease data
The human symptom-disease data is got from57. Based on 322 disease symptoms, a weighted disease network is constructed, which contains 133,106 interactions between 1,596 distinct diseases (see Supplementary Table S1). The disease network combines phenotypic relations with shared molecular mechanisms.
Drug-side effect data
The drug-side effect data is downloaded from SIDER (Side Effect Resource) version 258. SIDER contains information on 996 marketed drugs, corresponding 4,192 recorded adverse drug reactions, and 99,423 drug-side effect pairs (see Supplementary Table S2). The information is extracted from public documents and package inserts. The available information include side effect frequency, drug and side effect classifications as well as links to further information, for example drug–target relations.
Drug-Disease data
We obtain the drug–disease associations from Comparative Toxicogenomics Database (CTD) in December 201522. CTD contains curated and inferred chemical–disease associations. They can help researchers develop hypotheses about environmental diseases and their underlying mechanisms. We only preserve the curated associations with marker “therapeutic” or “marker/mechanism” as known associations. “Therapeutic” represents that a chemical has a known or potential therapeutic role in a disease (e.g., chemical X is used to treat leukemia). “Marker/mechanism” represents that a chemical correlates with a disease (e.g., increased abundance in the brain of chemical X correlates with Alzheimer disease) or may play a role in the etiology of a disease (e.g., exposure to chemical X causes lung cancer)22. Finally, we get 82,858 chemical–disease associations in all (see Supplementary Table S3).
Construct drug similarity network based on side effects
The relationship between drug j and side effects is treated as a feature vector dj:

where wi,j quantifies the strength of the association between side effect i and drug j. The prevalence of the different side effects and diseases is very different. For example, there are highly abundant side effects like abdominal pain, and publication biases towards certain drugs. To account for this heterogeneity, we do not use the absolute co-occurrence Wi,j to measure the strength of an association between side effect i and drug j, but the term frequency-inverse document frequency59wi,j:

where N denotes the number of all drugs in the dataset and ni denotes the number of drugs in which side effect i appears. Wi,j is equal to 1, if drug j displays side effect i, otherwise, it will be 0. Since all side effects in our data have at least one associated drug, the potential problem of dividing by zero does not arise. For the 996 drugs with recorded clinical side effect data, each will be assigned a 4192-dimension vector.
We use the cosine similarity60 to measure the similarity between the vectors di and dj of two drugs i and j. The formula is shown as:

where the cosine similarity ranges from 0 (no shared side effects) to 1 (identical side effects).
Cluster constructed networks using ClusterONE
After filtering drug and disease networks, we respectively cluster drug network and disease network using ClusterONE (Clustering with Overlapping Neighborhood Expansion)61 and obtain two kinds of modules: drug module and disease module. ClusterONE is a graph clustering algorithm that is able to handle weighted graphs. Owing to these properties, ClusterONE is especially useful for detecting modules in networks with associated confidence values.
Cluster drug similarity network
In order to improve the reliability of the drug network, we filter the edges with lower similarity. To remain relatively more reliable edges, the cutoff is set to be 0.4 and we get a new drug network with 248 nodes and 379 edges, as shown in Supplementary Table S4. We run ClusterONE with default parameter values in the filtered drug network and get 27 modules (See Supplementary Table S5). The p-values of 26 modules are lower than 0.05. The network topologies of 27 modules are also shown in Fig. 3 (nodes representing drugs). Rectangles and diamonds represent nodes in clusters and overlap between different clusters, respectively.

The network topologies of 27 modules, here nodes representing drugs; rectangles represent nodes in clusters; and diamonds represent overlap nodes between different clusters.
Cluster disease similarity network
When the scores of edges in disease network are not lower than 0.5, we can obtain more meaningful modules (p–value ≤ 0.05). Therefore, we discard the edges with scores lower than 0.5. After filtering, the disease network includes 1,367 nodes and 9,792 edges, as shown in Supplementary Table S6. We also run ClusterONE with default parameter values in disease network and get 145 modules (see Supplementary Table S7). The p-values of 64 modules are lower than 0.05.
Construct connections between disease and drug modules based on CTD
Based on reliable chemical-disease associations got from CTD database, we construct connections between disease and drug modules. As an example shown in Fig. 4, we assume drug module i includes four drugs (drug1 to drug4) and disease module j includes four diseases (disease1 to disease4). The strength of the association between drug module i and disease module j, w(i, j), is defined as:

An example of calculating the correlation between drug module i and disease module j based on the drug-disease associations verified by CTD (marked as “M” or “M&T”).

where Mij denotes the number of chemical-disease associations verified by CTD (marked as T or M/T) between drug module i and drug module j; N(i) and N(j) denote the number of elements in drug module i and drug module j, respectively. In Fig. 4, Mij = 5 and N(i) = N(j) = 4, so  . In this way, we can get the correlation between any drug-disease module pair.
. In this way, we can get the correlation between any drug-disease module pair.
Finally, we obtain 1,180 drug-disease module pairs whose score are not zero. According to the definition of the association between a drug-disease module pair (see formula (5)), the drug-disease module pairs with higher score will be preserved. There are 27 drug-disease module pairs whose scores are all not lower than 0.2, which are shown in Supplementary Table S8. In order to analyze our results more targeted and find more valuable associations, we focused on the top-3 drug-disease module pairs for further analysis. The top-3 drug-disease module pairs whose correlations are all not lower than 0.36. Their details are shown in Table 1. In Results section, we will analyze the top-3 drug-disease module pairs to predict new drug indications.
Predict drug-disease associations based on top-3 drug-disease module pairs
In this section, we predict novel drug-disease associations based on the neighborhood partnerships of drugs and diseases in their own modules. Table 1 shows the information of top-3 drug-disease module pairs. We can find these modules are dense, that is to say, the nodes in a same module interact frequently. We know that the stronger the interactions between nodes within a same module, the more similar their functions. Therefore, based on the strong correlations between dense drug and disease modules, we can predict potential associations between drugs and diseases. The details are as following:
Step 1: Recalculate the similarity of each pair of nodes in a module based on subgraph topology of the module. Given a and b is a pair of nodes in a drug module or a disease module, there may be many different paths between a and b . Suppose pathi represents a path between a and b with i edges; sum ( pathi) represents the weight of pathi, which equals to the sum of weight on each edge in it; avg ( pathi) represents the average weight of pathi: avg ( pathi) = sum ( pathi)/ i . Then, the similarity between a and b is defined as:

where n represents the number of nodes in the module. If there does not exist a path with i edges, we set avg ( pathi) = 0. In this way, we score all the correlations between each pair of nodes in a module.
From the above steps, we know drugs or diseases have similar properties are clustered in a same group. The indications of a drug may be inferred by the enriched FDA-approved functions of its neighboring drugs in a same drug subgraph. Relationships in strong similar edges and connected components provide potential candidates for the previously unknown therapeutic effects of drugs. In the same way, the treatments of a disease may be inferred by its neighboring diseases in a same disease subgraph. Therefore, for a drug-disease module pair, we predict new drug-disease associations based on the subgraph topology of modules.
Step 2: Recalculate the association of each pair of nodes belong to two different modules based on Step 1. Based on known drug-disease associations in CTD database, and correlations of drug-drug and disease-disease obtained from Step 1, we get a drug-disease heterogeneous network, which includes two types of nodes: drug and disease, and two types of edges: internal connections and external connections. Then, based on the heterogeneous network, we score the correlations between each pair of drug and disease nodes. Given v represents a node in disease module DI and w represents a node in drug module DR , that is v ∈ DI and w ∈ DR . Their relation corr ( v , w ) is defined as:

The direct neighbors of v in DI , which have connections with nodes in DR , are stored in NDI( v ). The direct neighbors of w in DR , which have connections with nodes in DI , are stored in NDR( w ). sim ( v , p ) and sim ( w , q ), defined as Equation (6), denote the similarities between v and p , w and q , respectively.
After the process of Step 1 and Step 2, we will score all the drug-disease associations between a drug-disease module pair and rank them in descending order. We do the same process for the top-3 drug-disease module pairs and get three ranking lists of drug-disease associations.
Additional Information
How to cite this article: Yu, L. et al. Prediction of new drug indications based on clinical data and network modularity. Sci. Rep.6, 32530; doi: 10.1038/srep32530 (2016).
Change history
24 February 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41598-022-07363-5
References
Wilson, J. F. Alterations in processes and priorities needed for new drug development. Ann. Intern. Med.145, 793–796 (2006).
Li, J. et al. A survey of current trends in computational drug repositioning. Brief Bioinform.17, 2–12 (2016).
Adams, C. P. & Brantner, V. V. Estimating the cost of new drug development: is it really $802 million? Health Aff.25, 420–428 (2006).
Ekins, S., Williams, A. J., Krasowski, M. D. & Freundlich, J. S. In silico repositioning of approved drugs for rare and neglected diseases. Drug Discov. Today16, 298–310 (2011).
Ashburn, T. T. & Thor, K. B. Drug repositioning: identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov.3, 673–683 (2004).
Novac, N. Challenges and opportunities of drug repositioning. Trends Pharmacol. Sci.34, 267–272 (2013).
Varothai, S. & Bergfeld, W. F. Androgenetic alopecia: an evidence-based treatment update. Am. J. Clin. Dermatol.15, 217–230 (2014).
Noeske, T. et al. Predicting compound selectivity by self-organizing maps: cross-activities of metabotropic glutamate receptor antagonists. ChemMedChem1, 1066–1068 (2006).
Kotelnikova, E., Yuryev, A., Mazo, I. & Daraselia, N. Computational approaches for drug repositioning and combination therapy design. J. Bioinform. Comput. Biol.8, 593–606 (2010).
Li, J., Zhu, X. & Chen, J. X. Building disease-specific drug-protein connectivity maps from molecular interaction networks and PubMed abstracts. PLoS Comput. Biol.5, e1000450 (2009).
Lamb, J. et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science313, 1929–1935 (2006).
Iorio, F. et al. Discovery of drug mode of action and drug repositioning from transcriptional responses. Proc. Natl. Acad. Sci. USA107, 14621–14626 (2010).
Sirota, M. et al. Discovery and preclinical validation of drug indications using compendia of public gene expression data. Sci. Transl. Med.96, 96–77 (2011).
Zou, Q. et al. Survey of MapReduce frame operation in bioinformatics. Brief Bioinform.15, 637–647 (2014).
Zeng, X., Zhang, X. & Zou, Q. Integrative approaches for predicting microRNA function and prioritizing disease-related microRNA using biological interaction networks. Brief Bioinform.17, 193–203 (2016).
Ekins, S., Mestres, J. & Testa, B. In silico pharmacology for drug discovery: methods for virtual ligand screening and profiling. Br. J. Pharmacol.152, 9–20 (2007).
Li, Y. & Agarwal, P. A pathway-based view of human diseases and disease relationships. PLoS One4, e4346 (2009).
Zeng, X., Liao, Y., Liu & Y. & Zou, Q. Prediction and validation of disease genes using HeteSim Scores. IEEE/ACM Trans. Comput. Biol. Bioinform. 10.1109/TCBB.2016.2520947 (2016).
Zou, Q. et al. Prediction of microRNA-disease associations based on social network analysis methods. Biomed. Res. Int.810514 (2015).
Ye, H., Liu, Q. & Wei, J., Construction of drug network based on side effects and its application for drug repositioning. Plos ONE9, e87864 (2014).
Gottlieb, A., Stein, G. Y., Ruppin, E. & Sharan, R. PREDICT: a method for inferring novel drug indications with application to personalized medicine. Mol. Syst. Biol.7, 496 (2011).
Davis, A. P. et al. The Comparative Toxicogenomics Database’s 10th year anniversary: update 2015. Nucleic Acids Res.43, D914–D920 (2014).
Li, H. & Liang S. Local network topology in human protein interaction data predicts functional association. Plos ONE4, e6410 (2009).
Barabasi, A. L. & Albert, R. Emergence of scaling in random networks. Science286, 509–512 (1999).
King, B. L., Davis, A. P., Rosenstein, M. C., Wiegers, T. C. & Mattingly, C. J. Ranking transitive chemical-disease inferences using local network topology in the comparative toxicogenomics database. PLoS One7, e46524 (2012).
Kurakhmaeva, K. B. et al. Brain targeting of nerve growth factor using poly(butyl cyanoacrylate) nanoparticles. J. Drug Target17, 564–574 (2009).
Briani, C. et al. Thiethylperazine-induced parkinsonism: in vivo demonstration of dopamine D2 receptors blockade. Eur. J. Neurol.11, 709–710 (2004).
Kolakowska, T., Wiles, D. H., McNeilly, A. S. & Gelder, M. G. Correlation between plasma levels of prolactin and chlorpromazine in psychiatric patients. Psychol. Med.5, 214–216 (1975).
Available at: http://www.druglib.com/reported-side-effects/prochlorperazine/reaction_tremor/ (Accessed: 4th January 2016).
Joint Formulary Committee. British National Formulary (BNF) (65 ed.) 237–238 (London, UK: Pharmaceutical Press 2013).
PRODUCT INFORMATION MODECATE (Fluphenazine Decanoate Oily Injection). TGA eBusiness Services. Bristol-Myers Squibb Australia Pty Ltd. 1 November 2012 (Retrieved 9 December 2013).
Driessen, M. et al. Gene expression markers in the zebrafish embryo reflect a hepatotoxic response in animal models and humans. Toxicol. Lett.230, 48–56 (2014).
Tong, M., Dong, M. & de la Monte, S. M. Brain insulin-like growth factor and neurotrophin resistance in Parkinson’s disease and dementia with Lewy bodies: potential role of manganese neurotoxicity. J. Alzheimers Dis.16, 585–599 (2009).
Van, G. & Jay A. Assn, Lewy Body Dementia, New Trends in Lewy Body Dementia, from “The Many Faces of Lewy Body Dementia” series at Coral Springs Medical Center, FL, YouTube (2007).
Available at: http://www.ehealthme.com/ds/thorazine/lewy+body+dementia (Accessed: 4th January 2016).
Kaim, S. C., Klett, C. J. & Rothfeld, B. Treatment of the acute alcohol withdrawal state: a comparison of four drugs. Am. J. Psychiatry.125, 1640–1646 (1969).
Stern, T. A., Gross, A. F., Stern, T. W., Nejad, S. H. & Maldonado, J. R. Current approaches to the recognition and treatment of alcohol withdrawal and delirium tremens: “old wine in new bottles” or “new wine in old bottles”. Prim. Care Companion. J. Clin. Psychiatry.12 (2010).
Buschmann, H. et al. Antidepressants, Antipsychotics, Anxiolytics: From Chemistry and Pharmacology to Clinical Application (Weinheim: Wiley-VCH. p. 520. ISBN 3-527-31058-4 2007).
Husseini, A. & Gianakos, D. The 15-minute visit. Patient Care40, 9–10 (2006).
Available at: http://www.ehealthme.com/ds/compazine/bipolar+i+disorder (Accessed: 4th January 2016).
Hughes, J. R. Alcohol withdrawal seizures. Epilepsy Behav.15, 92–97 (2009).
Laties, V. G., Lasagna, L., Gross, G. M., Hitchman, I. L. & Flores, J. A controlled trial on chlorpromazine and promazine in the management of delirium tremens. Q. J. Stud. Alcohol.19, 238–243 (1958).
Hyman, S. E., Arana, G. W. & Rosebaum, J. F. Handbook of psychiatric drug therapy (3rd ed). (Philadelphia, Pa: Lippincott Williams & Wilkins, 1995).
McGeary, J. E., Gurel, V., Knopik, V. S., Spaulding, J. & McMichael, J. Effects of nerve growth factor (NGF), fluoxetine, and amitriptyline on gene expression profiles in rat brain. Neuropeptides45, 317–322 (2011).
Liu, J. et al. Discovery of isoxazole analogues of sazetidine-A as selective α4β2-nicotinic acetylcholine receptor partial agonists for the treatment of depression. J. Med. Chem.54, 7280–7288 (2011).
Available at: http://www.insomnia.net/medications/amitriptyline/ (Accessed: 4th January 2016).
Yoo, M. J. & Hage, D. S. Use of peak decay analysis and affinity microcolumns containing silica monoliths for rapid determination of drug-protein dissociation rates. J. Chromatogr. A.1218, 2072–2078 (2011).
Xu, Y. et al. Curcumin reverses impaired hippocampal neurogenesis and increases serotonin receptor 1A mRNA and brain-derived neurotrophic factor expression in chronically stressed rats. Brain Res.1162, 9–18 (2007).
Grigoriadis, D. E., Pearsall D. & De Souza, E. B. Effects of chronic antidepressant and benzodiazepine treatment on corticotropin-releasing-factor receptors in rat brain and pituitary. Neuropsychopharmacology2, 53–60 (1989).
Gómez-Gil, E. et al. Hormonal responses to the 5-HT1A agonist buspirone in remitted endogenous depressive patients after long-term imipramine treatment. Psychoneuroendocrinology35, 481–489 (2010).
Dutta, A. K., Ghosh, B., Biswas, S. & Reith, M. E. D-161, a novel pyran-based triple monoamine transporter blocker: behavioral pharmacological evidence for antidepressant-like action. Eur. J. Pharmacol.589, 73–79 (2008).
Christa, A. F. A Review of Postpartum Depression. Prim. Care Companion. J. Clin. Psychiatry.1, 9–14 (1999).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc.4, 44–57 (2008).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Statist. Soc. B.57, 289–300 (1995).
Law, V. et al. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res.42, D1091–D1097 (2014).
Ada, H., Scott, A. F., Joanna, A., Bocchini, C. & McKusick, V. A. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res.33, D514–D517 (2005).
Zhou, X. Z., Menche, J., Barabási, A. L. & Sharma, A. Human symptoms-disease network. Nat. Commun.5, 4212 (2014).
Kuhn, M., Campillos, M., Letunic, I., Jensen, L. J. & Bork, P. A side effect resource to capture phenotypic effects of drugs. Mol. Syst. Biol.6, 343 (2010).
Salton, G., Wong, A. & Yang, C. S. A vector space model for automatic indexing. Commun. ACM18, 613–620 (1975).
Singhal, A. Modern information retrieval: a brief overview. Bulletin of the IEEE Computer Society Technical Committee on Data Engineering24, 35–43 (2001).
Nepusz, T., Yu, H. & Paccanaro, A. Detecting overlapping protein complexes in protein-protein interaction networks. Nat. Methods.9, 471–472 (2012).
Acknowledgements
This work was supported in part by the National Natural Science Foundation of China (Nos 61672406, 61532014, 91530113, 61502363, 61303118, 61303122, and 61402349), the Natural Science Basic Research Plan in Shaanxi Province of China (No. 2016JQ6057).
Author information
Authors and Affiliations
Contributions
L.Y. designed and performed experiments, analyzed data and wrote the paper; X.M. and L.Z. designed experiments and wrote the paper; J.Z. and L.G. designed experiments and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Yu, L., Ma, X., Zhang, L. et al. Prediction of new drug indications based on clinical data and network modularity. Sci Rep 6, 32530 (2016). https://doi.org/10.1038/srep32530
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep32530
This article is cited by
-
A Visual Phenotype-Based Differential Diagnosis Process for Rare Diseases
Interdisciplinary Sciences: Computational Life Sciences (2022)
-
RDmap: a map for exploring rare diseases
Orphanet Journal of Rare Diseases (2021)
-
Drug prioritization using the semantic properties of a knowledge graph
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
