
Abstract
The highly anticipated transition from next generation sequencing (NGS) to third generation sequencing (3GS) has been difficult primarily due to high error rates and excessive sequencing cost. The high error rates make the assembly of long erroneous reads of large genomes challenging because existing software solutions are often overwhelmed by error correction tasks. Here we report a hybrid assembly approach that simultaneously utilizes NGS and 3GS data to address both issues. We gain advantages from three general and basic design principles: (i) Compact representation of the long reads leads to efficient alignments. (ii) Base-level errors can be skipped; structural errors need to be detected and corrected. (iii) Structurally correct 3GS reads are assembled and polished. In our implementation, preassembled NGS contigs are used to derive the compact representation of the long reads, motivating an algorithmic conversion from a de Bruijn graph to an overlap graph, the two major assembly paradigms. Moreover, since NGS and 3GS data can compensate for each other, our hybrid assembly approach reduces both of their sequencing requirements. Experiments show that our software is able to assemble mammalian-sized genomes orders of magnitude more quickly than existing methods without consuming a lot of memory, while saving about half of the sequencing cost.
Similar content being viewed by others



Introduction
The Human Genome Project (HGP), which is perhaps the largest biomedical research project humans have ever undertaken, is responsible for greatly accelerating the advancement of DNA sequencing technologies1. Three generations of DNA sequencing technologies have been developed in the last three decades and we are at the crossroads of the second and third generations of the sequencing technologies. Third generation sequencing (3GS) technology promises to significantly improve assembly quality and expand its applications in biomedical research and biotechnology development. However, lack of efficient and effective genome assembly algorithms has arguably been the biggest roadblock to the widespread adoption of 3GS technologies. 3GS long reads (averaging up to 5–20 kb per run at this time) usually have high error rates: ~15% with PacBio sequencing2 and as high as ~40% with Oxford Nanopore sequencing3. These high error rates make the assembly of 3GS sequences seem disproportionally complex and expensive compared to the assembly of NGS sequences. As a comparison, the whole genome assembly of a human genome using 3GS data was first reported to have taken half a million CPU hours4 compared to ~24 hours with Illumina NGS sequencing data5. Consequently, in practice, many applications of 3GS technology have been limited to re-sequencing bacteria and other small genomes6. While software for 3GS assembly has made important improvements2,7,8,9,10,11,12,13,14,15,16,17, especially for high coverage data, the software is still quite slow and not ideally suited for modest coverage data. Another major issue is that the sequencing cost of 3GS technology, while decreasing with time, is still at least an order of magnitude more expensive than the popular Illumina NGS sequencing at the time of this work.
While the evolution of genome assembly software solutions has been influenced by multiple factors, the most significant one has been the length of the sequences18. Although increasing sequence lengths may simplify the assembly graph6, the sequence length also has a critical impact on the computational complexities of genome assembly. Computational biologists have historically formulated the genome assembly problem as a graph traversal problem18,19,20, i.e., searching for a most likely genome sequence from the overlap graph of the sequence reads in the case of the first generation sequencing technology. The string graph and the best overlap graph are specific forms of the Overlap-Layout-Consensus (OLC) paradigm that are more efficient by simplifying the global overlap graph19,21,22. The read-based algorithms, aiming to chain the sequencing reads in the most effective way, are computationally expensive because pair-wise alignment of the sequences is required to construct the overlap graph. This issue was tolerable for the relatively low amount of sequences produced from the low-throughput first generation sequencing technologies, but quickly became overwhelming with the enormous amount of short reads produced by high-throughput NGS data. The strategy of chopping the sequencing reads into shorter and overlapping k-grams (so-termed k-mers) and building links between the k-mers, was developed in the de Bruijn graph (DBG) framework to simplify NGS assembly. Assembly results are extracted from the linear (unbranched) regions of the k-mer graph in this approach20.
The overlap graph model or the OLC-based software packages, such as Celera Assembler1, AMOS23 and ARACHNE24, originally used for assembling the first generation sequence data, were also adopted for the NGS assembly before DBG-based approaches became the de facto standard. Newer 3GS technologies, including single-molecule, real-time sequencing (SMRT) and Oxford Nanopore sequencing, produce much longer reads than NGS. The longer reads from 3GS technology make the OLC approaches, which were originally used in the first generation genome assembly, feasible again. Nevertheless, the high error rates of current 3GS technologies render the existing OLC-based assemblers developed for relatively accurate sequences unusable. Similarly, the error-prone long reads make the DBG full of branches and therefore unsuitable for 3GS assembly. Faced with these challenges, the developers of 3GS technology have resorted to using error correction techniques2,7,9,10,13,17 to create high quality long reads and reusing the algorithms originally developed for the first generation sequence assembly. However, error correction for these long reads require extensive computational resources, even for small microbial genomes. Moreover, the high sequencing depth (usually 50x–100x) required by existing 3GS genome assemblers increases sequencing cost significantly, especially for large genomes. These issues have put 3GS technology at a severe disadvantage when competing against widely used NGS technology. In this article, we introduce algorithmic techniques that effectively resolve many of these issues. But first, we present a brief account of the existing genome assembly software technologies to put our contribution in proper context.
Researchers began with scaffolding approaches such as AHA16, PBJelly15 and SSPACE-LongRead11 to patch the gaps between high quality assembly regions, i.e., first build a scaffold by aligning reads to the contigs and then use reads that span multiple contigs as links to build a scaffold graph. In ALLPATHS-LG14 and Cerulean12, long reads are used to find the best path in the de Bruijn graph that bridges the gaps between large contigs. Although these software packages have indeed achieved important advances for 3GS genome assembly, resolving intricate ambiguities is inherently difficult and can lead to structural errors. Furthermore, the underlying graph search algorithms usually have exponential complexity with respect to the search depth and thus, scales poorly; highly repeating regions (such as long repeats of simple sequences) will lead to large search depths and are not resolvable. In addition, the more powerful read overlap graph structure (of the long reads) was not fully explored in all these approaches. Often these algorithms rely on heuristics such as contig lengths and require iterations12,14. To circumvent these important issues associated with the hybrid approach, a Hierarchical Genome-Assembly Process (HGAP)13 was developed using a non-hybrid strategy to assemble PacBio SMRT sequencing data, which does not use the NGS short reads. HGAP contains a consensus algorithm that creates long and highly accurate overlapping sequences by correcting errors on the longest reads using shorter reads from the same library. This correction approach was proposed earlier in the hybrid setting and is widely used in assembly pipelines2,9,10,17. Nonetheless, this non-hybrid, hierarchical assembly approach requires relatively high sequencing coverage (50x–100x) and substantial error correction time to obtain satisfactory results. It is noteworthy that most of the algorithms we reviewed here were originally designed for bacterial-sized genomes. Though recent advancements in aligning erroneous long reads6,25 have also shortened the computational time of 3GS assembly, running these programs on large genomes, especially mammalian-sized genomes, usually imposes a large computational burden (sometimes up to 105 or 106 CPU hours) more suited to large computational clusters and well beyond the capability of a typical workstation.
In this study, we design algorithms to enable efficient assembly of large mammalian-sized genomes. We observe that per-base error correction of each long erroneous reads and their pair-wise alignment takes a significantly large portion of time in existing pipelines, but neither of these is necessary at an initial assembly stage. If all sequencing reads are structurally correct (non-chimeic), one can produce a structurally correct draft genome and improve the base-level accuracy in the final stage, as was originally done in the OLC approach. Taking advantage of this observation, we develop a base-level correction-free assembly pipeline by directly analyzing and exploiting overlap information in the long reads. Unlike previous approaches, we use the NGS assembly to lower the computational burden of aligning 3GS sequences rather than just polishing 3GS data. This allows us to take advantage of the cheap and easily accessible NGS reads, while avoiding the issues associated with existing hybrid approaches mentioned previously. Meanwhile, since NGS and 3GS are independent of each other, the sequencing gaps in one type of data may be covered by the data from the other. The utilization of NGS data also lowers the required sequencing depth of 3GS and the net result is reduced sequencing cost. Hence, we get the best of both worlds of hybrid and non-hybrid assembly approaches. Specifically, we map the DBG contigs from NGS data to the 3GS long reads to create anchors for the long reads. Each long read is (lossily) compressed into a list of NGS contig identifiers. Because the compressed reads are often orders of magnitude shorter than the original reads, finding candidate overlaps between them becomes a simple bookkeeping problem and the approximate alignments and overlaps can be calculated cheaply with the help of the contig indentifiers. An overlap graph is constructed by chaining the best overlapped-reads in the compressed domain. The linear unbranched regions of the overlap graph are extracted and uncompressed to construct the draft assembly. Finally, we polish the draft assembly at the base-level with a consensus module to finish the assembly. Overall, compared with the existing approaches, our algorithm offers an efficient algorithmic solution for assembling large genomes with 3GS data in terms of computational resources (time and memory) and required sequencing coverage while also being robust to sequencing errors. Furthermore, our pipeline utilizes the reads overlap information directly and provides an efficient solution to the traditional read threading problem, which is valuable both theoretically and practically even for the NGS assembly20,26.
Methods and Implementations
Our algorithm starts with linear unambiguous regions of a de Bruijn graph (DBG) and ends up with linear unambiguous regions in an overlap graph (used in the Overlap-Layout-Consensus framework). Due to this property, we dub our software DBG2OLC. The whole algorithm consists of the following five procedures and we implement them as a pipeline in DBG2OLC. Each piece of the pipeline can be carried out efficiently.
-
1
Construct a de Bruijn graph (DBG) and output contigs from highly accurate NGS short reads.
-
2
Map the contigs to each long read to anchor the long reads. The long reads are compressed into a list of contig identifiers to reduce the cost of processing the long reads (Fig. 1A).
Figure 1 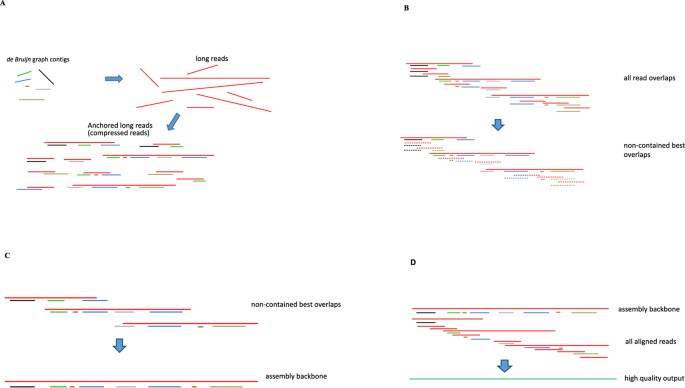
(A) Map de Bruijn graph contigs to the long reads. The long reads are in red, the de Bruijn graph contigs are in other colors. Each long read is converted into an ordered list of contigs, termed compressed reads. (B) Calculate overlaps between the compressed reads. The alignment is calculated using the anchors. Contained reads are removed and the reads are chained together in the best-overlap fashion. (C) Layout: construct the assembly backbone from the best overlaps. (D) Consensus: align all related reads to the backbone and calculate the most likely sequence as the consensus output.
-
3
Use multiple sequence alignment to clean the compressed reads and remove reads with structural errors (or so-called chimeras) (Fig. 2).
Figure 2 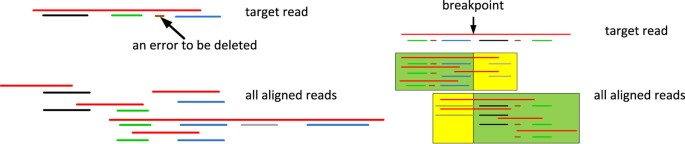
Reads correction by multiple sequence alignment.
The left portion shows removing a false positive anchoring contig (brown) that appears only once in the multiple alignment. The right portion shows detection of a chimeric read by aligning it to multiple reads. A breakpoint is detected as all the reads can be aligned with the left portion of the target read are not consistent with all the reads that can be aligned with the right portion of the target read.
-
4
Construct a best overlap graph using the cleaned compressed long reads (Fig. 1B).
-
5
Uncompress and chain together the long reads (Fig. 1C) and resort to a consensus algorithm to convert them into DNA sequences (Fig. 1D).


Details for procedure (2–5) are explained below. The explanation of procedure (1) can be found in our previous SparseAssembler for NGS technology5 and is omitted here.
Availability
The source code and a compiled version of DBG2OLC is available in the following site: https://github.com/yechengxi/DBG2OLC.
Reads Compression
We use a simple k-mer index technique to index each DBG contig and map the pre-assembled NGS contigs back to the raw sequencing reads as anchors. The k-mers that appear in multiple contigs are excluded in our analysis to avoid ambiguity. Empirically for PacBio reads, we found that using k = 17 were adequate for all our experiments. For each 3GS long read, we report the matching contig identifiers as an ordered list. A contig identifier is reported if the number of uniquely matching k-mers in that contig is above a threshold, which is adaptively determined based on the contig length. We set this threshold in the range of (0.001~0.02)*Contig_Length. This easily tuneable threshold parameter allows the user to find a balance between sensitivity and specificity. With low coverage datasets, this parameter is set lower to achieve better sensitivity; otherwise it is set to higher to enforce better accuracy. In all our experiments, the contigs are generated with our previous SparseAssembler5.
After this procedure, each read is converted into an ordered list of contig identifiers. An example of such a list is {Contig_a, Contig_b}, where Contig_a and Contig_b are identifiers of two different contigs. We also record the orientations of these contigs in the mapping. This compact representation is a lossy compression of the original long reads. We term the converted reads as compressed reads in this work. A compressed read is considered to be equivalent to its reverse complement and the same compressed reads are then collapsed. Since a de Bruijn graph can efficiently partition the genome into chunks of bases as contigs, this lossy compression leads to orders of magnitude reduction in data size. Moreover, the compact representation can span through small regions with low or even no NGS coverage; these important gap regions in NGS assembly can be covered by 3GS data. Likewise, small 3GS sequencing gaps may be covered by NGS contigs. These sequencing gaps will be bridged in the final stage. Similarity detection between these compressed reads becomes a simpler bookkeeping problem with the identifiers and can be done quickly with low memory. To demonstrate the effectiveness of this strategy, we ran it over five datasets including genomes of different sizes and different sequencing technologies (Table 1, resources can be found in the Supplementary Materials). The compression usually leads to three factors of reduction in read length with 3GS.
Ultra-fast Pair-wise Alignments
Most existing algorithms rely on sensitive algorithms27,28 to align reads to other reads or assemblies. In our approach, since the compressed reads are usually much shorter than the original reads, alignments of these compressed reads can be calculated far more efficiently. We adopt a simple bookkeeping strategy and use the contig identifiers to build an inverted-index. Each identifier points to a set of compressed reads that contain this identifier. This inverted-index helps us to quickly select the potentially overlapping reads based on shared contig identifiers. Alignments are calculated only with these candidate compressed reads. The alignment score is calculated using the Smith-Waterman algorithm29; the contig identifiers that can be matched are positively scored while the mismatched contig identifiers are penalized. Scores for match/mismatch are calculated based on the involved contig lengths or the number of matching k-mers in the previous step. With the compressed reads, our algorithm can finish pair-wise alignments in a small amount of time.
As discussed previously, state-of-the-art assembly pipelines usually resort to costly base-level error correction algorithms to correct each individual read2,7,8,10,13, which they then feed into an existing assembler. However, an important finding of this work is that per-base accuracy may not be a major roadblock for assembly contiguity. Rather, the chimeras or structural sequencing errors are the major “hot spots” worth putting major effort into. Without cleaning these chimeras, the overlap relations include many falsely generated reads and will lead to a tangled overlap graph. To resolve this issue, we compute multiple sequence alignments (MSA) by aligning each compressed read with all other candidate compressed reads. With MSA we can detect the chimeric reads and the spurious contig identifiers in each read (Fig. 2). Both of these errors are cleaned up. The major side effect of this correction is a slightly increased requirement of the 3GS data coverage so that each compressed read can be confirmed by at least another one. The remaining minor errors (mostly false negatives) in the cleaned compressed reads will be tolerated by the alignment algorithm. In our experiments, we noticed that this algorithm is accurate enough to find high quality overlaps and can be used for constructing draft genomes as assembly backbones.
Read Overlap Graph
Compared with most hybrid approaches that used long reads to link together the short read contigs, our approach takes the unorthodox way–we use the short read contigs to help link together the long reads. We construct a best overlap graph21 using the above-described alignment algorithm with the compressed reads. In the best overlap graph, each node represents a compressed read. For each node, the best overlapped nodes (one before and the other after) are found based on the overlap score and the links between these nodes are recorded. The overlap graph is calculated in two rounds (Fig. 1B). In round 1, all the contained nodes (with respect to other nodes) are filtered off. For example, {Contig_a, Contig_b} is removed if {Contig_a, Contig_b, Contig_c} is present. With this strategy, alignments with repeating and contained nodes are avoided. In round 2, all suffix-prefix overlaps among the remaining nodes are detected with the alignment algorithm. Nodes are chained one to another in both directions and in the best overlapped fashion. Graph simplification is applied to remove tiny tips and merge bubbles in the best overlap graph. Truly unresolvable repeats result in branches in the graph21 and will be kept as the assembly breakpoints.
Note that constructing the overlap graph with the compressed reads offers us several major benefits. (1) Long read information is sufficiently utilized. (2) The costly long read alignments are accelerated with the easily available NGS contigs. (3) The expensive graph search algorithms (with exponential complexity to the search depth) often used for graph resolving in many existing genome assembly programs are no long needed in our software.
Consensus
It is noteworthy that only in this final stage that the compressed reads are converted back to the raw nucleotide reads for polishing purpose. Linear unbranched regions of the best overlap graph encode the unambiguously assembled sequences. Uncompressed long reads that lie in these regions are laid out in the best-overlapped fashion and patched one after another (Fig. 1C). NGS contigs are included when there is a gap in the 3GS data. Reads that are related to each backbone are collected based on the contig identifiers. A consensus module is finally called to align these reads to each backbone and calculate the polished assembly (Fig. 1D). To polish the 3GS assembly backbone, we use an efficient consensus module Sparc30. Sparc builds an efficient sparse k-mer graph structure5 using a collection of sequences from the same genomic region. The heaviest path approximates the most likely genome sequence (consensus) and is found in a sparsity-induced reweighted graph.
Results
We conducted a comprehensive comparison on a small yeast genome (12 Mbp) dataset to provide a scope of the performance of each software program we compared in this study. Since most other programs do not scale linearly with the data scale and require thousands of hours per-run on genomes larger than 100 Mbp, the readers are encouraged to read through their original publications for the performance results of those programs.
As a side note, the advent of 3GS long reads has raised the bar to a higher level compared with previous sequencing techniques: existing reference genomes usually contain a large number of structural errors and/or variations that can surpass the number of assembly errors using the long reads. In most cases we select assemblies by other assemblers with more coverage (~100x) as references. If high quality reference genome is available, thorough evaluations of our algorithm show that DBG2OLC can provide high quality results with fewer structural errors and comparable per-base accuracy. This has been recently demonstrated in the case study of D. melanogaster genome by Chakraborty et al.31 who compared our pre-released software with other premier programs for 3GS data. In this paper, we demonstrate results on some other well-studied species and use existing high coverage assemblies as quality checks. On medium to large genomes, DBG2OLC can produce comparably good results with one to two orders of magnitude less time and memory usages than most existing pipelines. A draft assembly (without polishing) of a 3 Gbp H. sapiens can be finished in 3 CPU days with our pipeline, utilizing 30 × 3GS and 50x NGS data. This computational time is roughly comparable to many existing NGS assemblers. The time consumption of each step running different genomes can be found in Table 2.
We compared our algorithm results with Celera Assembler (CA, version 8.3rc2), PacBioToCA (in CA8.3rc2)2, ECTools9, MHAP (in CA8.3rc2)7, HGAP (in SMRT Analysis v2.3.0)13 and Falcon assembler (v0.3.0), which are well recognized as the best-performed genome assemblers for 3GS technologies. Data from PacBio SMRT RS-II (the currently leading platform of 3GS technology) was used to perform the comparative experiments (50x Illumina MiSeq reads were additionally used for PacBioToCA and DBG2OLC, the two hybrid methods). The experiments are run on a server with eight Intel Xeon E7-8857 v2 CPUs (each has 12 cores) and 2 TB memory. For all DBG2OLC experiments in this paper we used SparseAssembler (Ye et al.5) to preassemble 50x Illumina short reads into contigs and then to compress the 3GS reads. Similarly, Celera Assembler was used to assemble the same short reads into contigs for ECTools. Unassembled short reads were fed into PacBioToCA according to its specification. At the time of this work, 50x Illumina reads cost less and also can be obtained more easily, than 1 × 3GS reads. Celera Assembler could be run with uncorrected reads on small datasets, so we run it as a baseline.
It is noteworthy that in our current implementation, most of the computation time (~90%) is spent on the consensus step, in which BLASR28 is called to align all raw reads to the assembly backbone. Since the alignments are multi-threaded, the wall time can be reduced depending on the available threads. The consensus step is relatively independent in genome assembly and is open to any future improvements and accelerations. The overall computational time of the whole pipeline scales near linearly to the data size, which is a highly valuable property to large-scale genome assembly problems. Using 10x–20x coverage of PacBio sequence data, we obtained assembly N50s that are significantly (>10x) better than Illumina data alone (Table 1). The datasets, commands and parameters can be found in the Supplementary Materials. We used QUAST 3.032 in its default setting to evaluate the assembly results; these are reported as the NGA50, per-base identity rates and misassembly errors. In analyzing 3GS assembly results, the NGA50 is a measure of the average length of high quality region before reaching a poor quality region in the assembly. The identity rates were calculated by summarizing the single base mismatches and insertion/deletion mismatches. Relocations, inversions and translocations are regarded as misassembly errors32. The alignment dot plots can be found in the Supplementary Materials. The nearly perfect diagonal dot plots indicate that DBG2OLC can produce structurally correct assemblies from as low as 10x long read data.
For the yeast dataset we picked an assembly from 454 data (NCBI Accession No.: GCA_000292815.1) and another assembly generated using MHAP with high coverage data as references. DBG2OLC takes advantage of different sequencing types and obtains the most contiguous results using 10x–40x data with comparable levels of accuracy (Table 3). Some non-hybrid assemblers are not able to fully assemble the yeast genome with 10x–20x PacBio data. It is also worth mentioning a caveat in many current hybrid error correction approaches. These pipelines use NGS contigs to correct the 3GS reads, which seem to have improved the accuracy of each individual 3GS read. However, the errors in NGS contigs may have corrupted the originally correct 3GS reads and lead to consensus errors in the final assembly. For example, we notice the identity rates of the ECTools assembly are higher when aligned to the 454 reference, contrary to all other pipelines. With high enough coverage (and significantly increased sequencing cost), the 3GS self-correction based assembly methods produce better assembly results. Since our pipeline has a major advantage in low coverage data and efficiency, it is expected to scale well to large genomes where low coverage data and computational time becomes major concerns.
We tested DBG2OLC on other medium to large genomes from PacBio sequencers (Table 4). On the A. thaliana genome (120 Mbp), the computations with DBG2OLC finish in one hour, with an additional hour spent constructing the initial NGS contigs. The consensus module takes another 10–20 CPU hours to get the final assembly. The peak memory usage is 6 GB. In comparison, existing pipelines can take over one thousand CPU hours with problems of this scale. On a large 54x human (H. sapiens) dataset, DBG2OLC is able to produce an assembly with high contiguity starting from 10x PacBio data (NG50 433 kbp) and DBG contigs generated from 50x Illumina reads (Table S1 in Supplementary Materials). To produce a better assembly, the longest 30x of the reads in this dataset (mean length 14.5 kbp) are selected (Table S2 in Supplementary Materials). DBG2OLC occupies 70 GB memory to store the 17-mer index and takes 37 CPU hours to compress and align the 30x longest PacBio reads. The pair-wise alignment takes only 3 hours and takes less than 6 hours on the full 54x dataset. The final consensus takes roughly 2000 CPU hours. In an initial report by PacBio scientists, the overlapping process took 405,000 CPU hours4. Our final assembly quality (N50 = 6 Mbp) is comparable to the state-of-the-art results obtained using orders of magnitude more resources. When evaluating this assembly, QUAST 3.0 can take weeks to finish the full evaluation even on our best workstation. We therefore only align our assembly to the longest 500 Mbp assembly generated by the Pacific Biosciences and report the NGA50 and identity rate in this portion.
DBG2OLC was also tested on an Oxford Nanopore MinION sequencing dataset (Table 4). According to initial studies, this type of data has higher (up to ~40%) error rates3 compared to PacBio SMRT sequencing. However, we find DBG2OLC still successfully assembled the E. coli into one single contig. The polished assembly has an error rate of 0.23%. The dot plot of the alignment of the assembly to the reference can be found in Fig. 13 of the Supplementary Materials.
Compared with the state-of-the-art assemblers for 3GS technologies, our proposed method produces assemblies with high contiguity using lower sequencing coverage and memory and is orders of magnitude faster on large genomes. Its combination of different data types leads to both computation and cost efficiency. These advantages are gained from three general and basic design principles: (i) Compact representation of the long reads leads to efficient alignments. (ii) Base-level errors can be skipped, but structural errors need to be detected and cleaned. (iii) Structurally correct 3GS reads are assembled and polished. DBG2OLC is a specific and simple realization of these principles. Interestingly, this implementation builds a nice connection between the two major assembly frameworks and even though DBG2OLC is majorly developed for 3GS data, this strategy of compression and converting a de Bruijn graph to an overlap graph is general and can be used for popular NGS data. A preliminary showcase on a purely NGS dataset can be found in the Supplementary Materials. The strategy of compressing long reads and performing the most computationally expensive tasks in the compressed domain strikes a balance between the DBG and OLC frameworks.
Summary and Discussion
In summary, we have built and validated a new de novo assembly pipeline that significantly reduces the computational and sequencing requirements of 3GS assembly. We demonstrate that the erroneous long reads can be directly assembled and can lead to significantly improved assembly without base-level error correction. This strategy, first publicly demonstrated in our pre-released pipeline in 2014, has paved the road for several subsequent development attempts on efficient utilization of 3GS data and promises even more efficient 3GS assemblers. Another major finding in developing DBG2OLC is that 3GS technologies generate chimeric reads and the problem seems to be severer with the PacBio platform. These structural errors lead to tangles in the assembly graph and greatly hamper the assembly contiguity. The most straightforward way to clean up the chimeric reads resorts to multiple sequence alignment, as implemented in DBG2OLC, which leads to a slightly increased coverage requirement. This limitation will serve as the starting point for future development. We conjecture that near perfect assemblies can be reached with even lower coverage if the chimeras/structural errors can be removed.
Additional Information
How to cite this article: Ye, C. et al. DBG2OLC: Efficient Assembly of Large Genomes Using Long Erroneous Reads of the Third Generation Sequencing Technologies. Sci. Rep. 6, 31900; doi: 10.1038/srep31900 (2016).
References
Venter, J. C. et al. The sequence of the human genome. Science 291, 1304–1351, doi: 10.1126/science.1058040 (2001).
Koren, S. et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nature biotechnology 30, 693–700, doi: 10.1038/nbt.2280 (2012).
Laver, T. et al. Assessing the performance of the Oxford Nanopore Technologies MinION. Biomolecular Detection and Quantification 3, 1–8 (2015).
Pacific Biosciences of California, I. Data Release: ~54x Long-Read Coverage for PacBio-only De Novo Human Genome Assembly, http://www.pacb.com/blog/data-release-54x-long-read-coverage-for (Published: 2014, Date of access: 17/03/2016).
Ye, C., Ma, Z. S., Cannon, C. H., Pop, M. & Yu, D. W. Exploiting sparseness in de novo genome assembly. BMC Bioinformatics 13 Suppl 6, S1, doi: 10.1186/1471-2105-13-S6-S1 (2012).
Koren, S. & Phillippy, A. M. One chromosome, one contig: complete microbial genomes from long-read sequencing and assembly. Current Opinion in Microbiology 23, 110–120, doi: 10.1016/j.mib.2014.11.014 (2015).
Berlin, K. et al. Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nature biotechnology 33, 623–630 (2015).
Salmela, L. & Rivals, E. LoRDEC: accurate and efficient long read error correction. Bioinformatics doi: 10.1093/bioinformatics/btu538 (2014).
Lee, H. et al. Error correction and assembly complexity of single molecule sequencing reads. BioRxiv. 006395, doi: 10.1101/006395 (2014).
Hackl, T., Hedrich, R., Schultz, J. & Forster, F. proovread: large-scale high-accuracy PacBio correction through iterative short read consensus. Bioinformatics doi: 10.1093/bioinformatics/btu392 (2014).
Boetzer, M. & Pirovano, W. SSPACE-LongRead: scaffolding bacterial draft genomes using long read sequence information. BMC Bioinformatics 15, 211, doi: 10.1186/1471-2105-15-211 (2014).
Deshpande, V., Fung, E. K., Pham, S. & Bafna, V. In Algorithms in Bioinformatics Vol. 8126 Lecture Notes in Computer Science (eds Aaron Darling & Jens Stoye ) Ch. 27, 349–363 (Springer: Berlin Heidelberg,, 2013).
Chin, C. S. et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nature methods 10, 563–569, doi: 10.1038/nmeth.2474 (2013).
Ribeiro, F. J. et al. Finished bacterial genomes from shotgun sequence data. Genome research 22, 2270–2277, doi: 10.1101/gr.141515.112 (2012).
English, A. C. et al. Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PloS one 7, e47768, doi: 10.1371/journal.pone.0047768 (2012).
Bashir, A. et al. A hybrid approach for the automated finishing of bacterial genomes. Nature biotechnology 30, 701–707, doi: 10.1038/nbt.2288 (2012).
Au, K. F., Underwood, J. G., Lee, L. & Wong, W. H. Improving PacBio long read accuracy by short read alignment. PloS one 7, e46679, doi: 10.1371/journal.pone.0046679 (2012).
Nagarajan, N. & Pop, M. Sequence assembly demystified. Nature reviews. Genetics 14, 157–167, doi: 10.1038/nrg3367 (2013).
Myers, E. W. The fragment assembly string graph. Bioinformatics 21 Suppl 2, ii79–85, doi: 10.1093/bioinformatics/bti1114 (2005).
Pevzner, P. A., Tang, H. & Waterman, M. S. An Eulerian path approach to DNA fragment assembly. Proceedings of the National Academy of Sciences 98, 9748–9753, doi: 10.1073/pnas.171285098 (2001).
Miller, J. R. et al. Aggressive assembly of pyrosequencing reads with mates. Bioinformatics 24, 2818–2824, doi: 10.1093/bioinformatics/btn548 (2008).
Simpson, J. T. & Durbin, R. Efficient de novo assembly of large genomes using compressed data structures. Genome research 22, 549–556, doi: 10.1101/gr.126953.111 (2012).
Treangen, T. J., Sommer, D. D., Angly, F. E., Koren, S. & Pop, M. Next generation sequence assembly with AMOS. Current Protocols in Bioinformatics, doi: 10.1002/0471250953.bi1108s33 (2011).
Batzoglou, S. et al. ARACHNE: a whole-genome shotgun assembler. Genome research 12, 177–189, doi: 10.1101/gr.208902 (2002).
Myers, G. Efficient local alignment discovery amongst noisy long reads. Algorithms in Bioinformatics 52–67, doi: 10.1007/978-3-662-44753-6 (2014).
Chaisson, M. J., Brinza, D. & Pevzner, P. A. De novo fragment assembly with short mate-paired reads: Does the read length matter? Genome research 19, 336–346, doi: 10.1101/gr.079053.108 (2009).
Kurtz, S. et al. Versatile and open software for comparing large genomes. Genome Biology 5, R12 (2004).
Chaisson, M. & Tesler, G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics 13, 238 (2012).
Smith, T. F. & Waterman, M. S. Identification of Common Molecular Subsequences. J Mol Biol 147, 195–197, doi: 10.1016/0022-2836(81)90087-5 (1981).
Ye, C. & Ma, Z. S. Sparc: a sparsity-based consensus algorithm for long erroneous sequencing reads. PeerJ 4, e2016 (2016).
Chakraborty, M., Baldwin-Brown, J. G., Long, A. D. & Emerson, J. J. A practical guide to de novo genome assembly using long reads. bioRxiv. 029306 (2015).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075, doi: 10.1093/bioinformatics/btt086 (2013).
Acknowledgements
We appreciate Prof. Mihai Pop, Prof. James Yorke, Dr. Aleksey Zimin and their groups at the University of Maryland for supports and helpful discussions. We thank Dr. Sergey Koren and Daniel Liang for helping us to improve our manuscript. This research received funding from the following sources: National Science Foundation of China (Grants No. 61175071, 71473243), the Exceptional Scientists Program and Top Oversea Scholars Program of Yunnan Province and Yunling Industrial Innovation Grant.
Author information
Authors and Affiliations
Contributions
C.Y. and Z.M. conceived and designed the study; C.Y. and C.M.H. wrote and tested the software; C.Y. and Z.M. wrote the paper; C.M.H. and J.R. participated in performance evaluation and discussion; S.W. helped in running test cases.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Ye, C., Hill, C., Wu, S. et al. DBG2OLC: Efficient Assembly of Large Genomes Using Long Erroneous Reads of the Third Generation Sequencing Technologies. Sci Rep 6, 31900 (2016). https://doi.org/10.1038/srep31900
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep31900
This article is cited by
-
Whole genome sequencing of a novel sea anemone (Actinostola sp.) from a deep-sea hydrothermal vent
Scientific Data (2024)
-
Genomic resources for the Yellowfin tuna Thunnus albacares
Molecular Biology Reports (2024)
-
Conserved chromatin and repetitive patterns reveal slow genome evolution in frogs
Nature Communications (2024)
-
Chromosome-level genome assembly of the deep-sea snail Phymorhynchus buccinoides provides insights into the adaptation to the cold seep habitat
BMC Genomics (2023)
-
A review of the pangenome: how it affects our understanding of genomic variation, selection and breeding in domestic animals?
Journal of Animal Science and Biotechnology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
