
Abstract
BAC transgenic mammalian systems offer an important platform for recapitulating human gene expression and disease modeling. While the larger body mass, and greater genetic and physiologic similarity to humans render rats well suited for reproducing human immune diseases and evaluating therapeutic strategies, difficulties of generating BAC transgenic rats have hindered progress. Thus, an efficient method for BAC transgenesis in rats would be valuable. Immunodeficient mice carrying a human SIRPA transgene have previously been shown to support improved human cell hematopoiesis. Here, we have generated for the first time, human SIRPA BAC transgenic rats, for which the gene is faithfully expressed, functionally active, and germline transmissible. To do this, human SIRPA BAC was modified with elements to work in coordination with genome engineering technologies-piggyBac, CRISPR/Cas9 or TALEN. Our findings show that piggyBac transposition is a more efficient approach than the classical BAC transgenesis, resulting in complete BAC integration with predictable end sequences, thereby permitting precise assessment of the integration site. Neither CRISPR/Cas9 nor TALEN increased BAC transgenesis. Therefore, an efficient generation of human SIRPA transgenic rats using piggyBac opens opportunities for expansion of humanized transgenic rat models in the future to advance biomedical research and therapeutic applications.
Similar content being viewed by others

Introduction
Mammalian model systems provide an essential platform in biomedical research for deciphering the complexities underlying the pathogenesis of human disease, and for developing the applicative and translational potential of new therapies. Bacterial artificial chromosomes (BACs) have played an important role in these endeavors by providing the DNA source material with which transgenic animals are derived. BACs are E. coli based large insert DNA clones capable of carrying genomic fragments ranging in size between 150–300 kb1,2. Unlike transgenic animals created using small plasmids, the large insert size of BACs allows for the transgene to maintain stability and embody low chimerism3. Moreover, the inserts typically include enhancer and other regulatory elements, minimizing the undesirable consequences of position-effects, such as epigenetic silencing and unexpected splicing2,4,5. For these reasons, the past decade has witnessed a rapid growth of transgenic mice generated using BACs, rendering it the preferred method for creating animal models recapitulating human gene expression and disease modeling.
While the large genomic insert size of BACs is beneficial for creating animals with transgenes that are integration site independent and accurately expressed contingent on copy number in vivo, this quality also makes efficient integration of BACs into host genomes challenging. Furthermore, the BAC integration site is difficult to determine. In spite of these limitations, BACs continue to be widely used, mainly for the production of transgenic mice. In contrast, BAC transgenesis in rats has been less successful, due to inherent physiological differences between the two rodent species6, leading to barriers at the level of rat oocyte manipulation and ES cell derivation and expansion7. These obstacles have been unfortunate, as they hinder progress towards the applicative potential of the rat model system, which harbor distinct advantages over the mouse. For example, the rat offers the short generation time and cost effectiveness of the mouse model, yet, in contrast to their smaller counterparts, the rat is genetically and physiologically more similar to the human8, and the larger body mass is poised to yield more biological material per animal unit that can be harvested in the course of experimentation.
An area of research where BAC transgenic rat model system would be particularly useful is the humanization of the immune system. While immune-deficient mouse and rat strains devoid of key lymphoid components have played an important role in biomedical sciences by making feasible studies involving the engraftment of human cells, tissues and organs9,10,11,12, empirical evidence has shown that the absence of lymphoid cells alone is not sufficient for efficiently modulating the engraftment of human cells13. In 2007, Takenaka and colleagues14 reported for the first time that in the lymphocyte depleted non-obese diabetic (NOD) severe combined immunodefiency (SCID) mouse strain15, NOD strain specific polymorphisms in the signal-regulatory protein alpha (Sirpa) locus supported human hematopoiesis through enhanced binding of the SIRPα protein to the human CD47 ligand. Once bound, SIRPα acts as an inhibitory receptor for mouse macrophages, allowing human hematopoietic cells to escape host cell phagocytosis16,17,18,19. To confirm that the enhanced engraftment of human cells in NOD strains is dependent on polymorphic SIRPα, thereby ruling out the possibility of causation or influence by strain specific extraneous factors, Strowig et al.20 generated human SIRPA BAC transgenic mice on mixed 129/BALB/c background, and demonstrated that the expression of human SIRPA enhanced human cell engraftment and improved functionality of human adaptive immune system in vivo. For these reasons, an efficient method for generating human SIRPA BAC transgenic rats would allow for the building of a repository of humanized SIRPA rats on various immune-deficient rat strains21, for use as a tool for studying the engraftment potential of human cells and tissues, as well as for reproducing human immune diseases and evaluating therapeutic strategies.
Here, we aim to generate BAC transgenic rats faithfully expressing human SIRPα. To do this, we seek to develop strategies utilizing genome engineering technologies reported to be highly efficient for generating transgenic animal models. Specifically, we examined piggyBac transposon, CRISPR/Cas9 and TALEN mediated approaches, as they have emerged as powerful tools for manipulating the genome. The piggyBac transposon system is a genetic element capable of mobilizing a segment of DNA encased between terminal inverse repeat (TIR) elements in the presence of transposase proteins22,23,24. The mobilized DNA is then transpositioned into a TTAA site in a different location in the genome by the transposase, for which the insertion location can be precisely determined using PCR25,26,27. Taking advantage of the piggyBac system’s “cut and paste” mechanism, researchers have utilized the TIR elements to design strategies for carrying out high throughput insertional mutagenesis for cancer research28,29, cellular reprogramming of stem cells30,31, among a slew of other experimentations requiring genome engineering32,33. Of relevance to this study is a recent pioneer publication showing that BACs retrofitted with TIR elements can be efficiently transposed in mouse zygotes34.
In contrast to the piggyBac mediated approach, CRISPR/Cas9 and TALEN are a family of endonucleases capable of inducing double stranded break (DSB) at precise locations in the genome, initiating the stimulation of two different pathways of repair mechanism–non-homologous end-joining (NHEJ) and homology-directed repair (HDR)35,36,37,38,39,40. The ability to stimulate NHEJ and HDR by CRISPR/Cas9 and TALEN has ushered in an era in which precise editing of the genome with high efficiency has become possible. However, CRISPR/Cas9 has very recently been described in a manuscript, submitted concomitantly with our study, to support targeted integration of a single BAC41. TALENs have not yet been reported for targeted BAC integration in animals. In advancement of this field, we showed42 recently that in both mouse and rat models, CRISPR/Cas9 and TALEN were successfully used for targeting an eGFP expressing vector, approximately 4.5 kb in size, into the Rosa26 locus through HDR.
Encouraged by these findings, we seek to modify the human SIRPA carrying BAC to work in coordination with piggyBac transposition, CRISPR/Cas9 or TALEN mediated approaches for the generation of humanized BAC transgenic rats. Hence, the objective of this study is twofold: 1) to develop an efficient method for generating BAC transgenic rats by evaluating the piggyBac, CRISPR/Cas9 and TALEN mediated approaches in the zygote; and 2) to generate humanized SIRPA rats that are functionally viable by expressing the protein in leukocytes and actively interacting with human CD47 ligand.
Results
Conversion of a human SIRPA carrying BAC into a piggyBac transposon
The initial steps required for developing a piggyBac mediated BAC transposition system involves converting the large circular DNA into a transposon capable of being recognized and mobilized by the transposase proteins (Supp. Fig. 1). To do this, we selected a BAC clone (RP11-887J4) (Supp. Fig. 2) containing 176 kb of human genomic DNA segment covering the SIRPA transcript coding sequence, as well as the region spanning 52 kb bps up- and 78 kb down-stream of the transcript start and end points. The selected human SIRPA containing BAC was then retrofitted with a cassette containing the 5′ and 3′ piggyBac Terminal Inverted Repeat (TIR) elements flanking the spectinomycin resistance gene, by replacing the chloramphenicol resistance gene in the vector backbone (Fig. 1A) (from this point onward, the TIR retrofitted hSIRPA-BAC will be referred to as hSIRPA-BAC-TIRs DNA). The positioning of the TIR elements in the BAC vector backbone was designed to mimic the inherent ‘cut and paste’ mechanism of piggyBac-based gene transfer, for which DNA segment encapsulated between the two TIR elements is seamlessly excised out of the genome in the presence of the transposase proteins, and transpositioned into a different location in genomic sites containing the TTAA sequence (Supp. Fig. 1). In the case of the hSIRPA-BAC-TIRs DNA, the TIR sequences are oriented such that in the presence of the transposase proteins, the 5′ and 3′ TIR elements would be excised away from the spectinomycin resistance gene, allowing the mobilization of the entire BAC for transposition into one of the TTAA sequences in the rat genome (Fig. 1C). In this strategy, when the mRNA transcript of the piggyBac transposase is co-microinjected into the rat zygote with the hSIRPA-BAC-TIRs DNA, the transcribed transposase proteins bind to the inverted repeats in order to induce nicks at both ends of the transposon, exposing the 3′ hydroxyl group and TTAA tetranucleotide overhang in the opposite strands (Fig. 1C). The 3′ hydroxyl end launches a hydrophilic attack on the TTAA overhang, creating a hairpin formation, which initiates the release of the hSIRPA-BAC-TIRs DNA from the spectinomycin resistance gene. Once released, the hairpin structure is resolved by leaving TTAA overhang at the 5′ ends of the transposon, and 3′ hydroxyl group exposed at the opposite strands. During this process, the transposase proteins identify other TTAA sequences in the genome, and likewise, induce nicks and hydrophilic 3′ hydroxyl attacks on the TTAA tetranucleotide sequences. The resolution of the hairpin formation in the genome involves target joining of the 3′ hydroxyl groups at the transposon ends, with the 5′ staggered TTAA overhanging sequence in the genomic DNA. Through this process, the hSIRPA-BAC-TIRs DNA transposon gets inserted into the genome, by duplicating the TTAA sequence, and positioning itself such that each tetranucleotide sequence ends up residing at opposite ends of the transpositioned hSIRPA-BAC-TIRs.

(A) Diagram illustrating the strategy used for retrofitting hSIRPA-BAC DNA (RP11-887J4) with piggyBac TIR elements. 5′ TIR (green) and 3′ TIR (orange) elements were sub-cloned into pUC19 vector backbone with spectinomycin resistance gene (purple), and 50 bp homology arm sequences (red) used for replacing the chloramphenicol resistance gene in the BAC vector backbone via recombineering technology. The diagram also indicates that the genomic DNA insert in the RP11-887J4 BAC is 176,233 bps, covering the SIRPA genic region, on chromosome 20 between 1,842,086-2,018,318. (B) The green arrows indicate the primer pairs used to verify hSIRPA-BAC retrofitting after the recombineering process. (C) A schematic diagram describing the transpositioning strategy of hSIRPA-BAC retrofitted with TIR elements mediated by piggyBac transposase. Illustration (i) shows the retrofitted BAC DNA. Illustrations (ii) and (iii) show the process by which the piggyBac transposase proteins bind to the TIR sequences, initiating nicking of the DNA strands, allowing 3′ hydroxyl group at both ends of the transposon to hydrophilic attack the flanking TTAA sequence and freeing the BAC from the spectinomycin resistance gene by forming hairpin structure at the TIR ends. Once the BAC DNA is released from spectinomycin resistance gene, illustration (iv) shows repairing of the linearized BAC DNA by ligating into the complementary TTAA overhangs in the genomic DNA through the mediation of the piggyBac transposase proteins.
Generation of piggyBac mediated humanized SIRPA rats via zygote injection
The implementation of the piggyBac mediated hSIRPA-BAC-TIRs DNA transposition strategy was carried out by co-injecting 2 or 3 ng/ul of BAC DNA with 25, 50 or 100 ng/ul of hyperactive transposase (hyPBase) mRNA into the pronucleus of rat zygotes (Table 1). Co-injections of 25 ng/ul of hyPBase and 2 ng/ul of BAC yielded 25 pups (13.8% relative to the number of embryos transferred), 50 ng/ul of hyPBase and 3 ng/ul of BAC yielded 7 pups (13.1% relative to the number of embryos transferred), and 100 ng/ul of hyPBase and 3 ng/ul of BAC yielded 13 pups (36.8% relative to the number of embryos transferred). To evaluate the efficiency of BAC insertion in these pups, tissue biopsies were performed on the tails for genomic DNA isolation, which was used to genotype for the presence of the human SIRPA gene (Supp. Table 1). In both conditions with either 50 ng/ul or 100 ng/ul of hyPBase, 1 pup was found to be PCR positive (14.3% and 7.7%, respectively, relative to the total number of pups), while 6 pups were found to be PCR positive in the 25 ng/ul of hyPBase condition (24.0% relative to the total number of pups) (Table 1 and Fig. 2A). As a control experiment, we replaced the hyPBase mRNA for the Cas9 mRNA. Cas9 in the absence of sgRNA will not cleave the DNA39 and thus would essentially be a bystander mRNA to be co-injected with the hSIRPA-BAC-TIRs. Because of the higher percentage of PCR positive pups relative to the total number of pups in the condition in which 2 ng/ul of BAC and 25 ng/ul of hyPBase mRNA transcripts were used, negative controls were set up using 2 ng/ul of hSIRPA-BAC-TIRs DNA and 25 ng/ul of Cas9 mRNA transcripts (Fig. 2C,D and Table 1). In the negative control experiment, 25 embryos (E15) were genotyped (32.1% relative to the number of embryos transferred), of which 1 embryo was found to be PCR positive (1.3% relative to the total number of embryos transferred) (Table 1). These findings suggest that: 1) co-injecting hSIRPA-BAC-TIRs DNA with hyPBase piggyBac increases the number of pups that are PCR positive for the presence of hSIRPA; and 2) while increasing the amount of hyPBase co-injected with 100 ng may give rise to higher number of pups relative to the number of embryos injected, the genotyping result indicates a higher percentage of PCR positive pups relative to the total number of pups when the amount of hyPBase injected is decreased to 25 ng.

(A) Genotyping result showing PCR positive pups for a 201 bp regions in the human SIRPA gene. The diagram shows pup numbers 1.6, 4.2, 5.4, 5.5, 5.6, 5.7, 8.1, 9.2 and 9.4 to be positive for PCR. In the upper panel, BAC refers to RP11-887J4 used as a positive control, gDNA refers to human genomic DNA as a positive control, and Negative as a negative control. In the lower panel, 5.7 (Control) refers to genomic DNA from pup number 5.7 used as a positive control. (B and C) Show PCR result of rat zygotes injected with Cas9 mRNA transcript, instead of piggyBac transposase, as a negative control. BAC refers to RP11-887J4 used as a positive control. To determine transposition, splinkerette PCR was performed in order to sequence the region adjacent to the TIR ends. (D) Shows the sequencing result, indicating that six pups (1.6, 5.4, 5.7, 8.1, 9.2 and 9.4) carry transpositioned hSIRPA-BACs. The first column shows the pup identities, the second and third columns show the location of the transposition sites, the fourth column lists the TTAA site of transposition, and sequences ten base up and downstream. The last column indicates whether the transposition occurred in genic or intergenic region.
Identification of transposition sites in the genome
Having shown that co-injecting the piggyBac TIR retrofitted hSIRPA BAC with hyPBase mRNA transcripts increases the number BAC insertions relative to the negative control, we sought to determine whether these insertions are transposition events mediated by piggyBac transposase. To do this, splinkerette PCR was utilized to: 1) identify the precise location of the insertion sites; and 2) determine whether the BAC transposon ends are flanked by TTAA sequence, since the hallmark feature of piggyBac mediated transposition is the mobilization of the transposon from one TTAA region into another, by duplicating the tetranucleotide sequence, and inserting itself in between the duplicated sequences (Supp. Figs 1 and 3). Among the 8 pups genotyped to be positive for hSIRPA BAC insertions (1.6, 4.2, 5.4, 5.5, 5.7, 8.1, 9.2, 9.4), 6 were found to have duplicated TTAA sequences, each tetranucleotide residing immediately adjacent to the 5′ and 3′ TIR elements (1.6, 5.4, 5.7, 8.1, 9.2, 9.4) (Fig. 2D). Detailed analysis of the integration sites show that pups 1.6, 8.1, 9.2 and 9.4 have hSIRPA-BACs transpositioned at intergenic regions, whereas, pups 5.4 and 5.7 have BAC transpositioned at genic regions, Ppp2r5e and Lasp1, respectively (Fig. 2D and Supp. Fig. 4a–f).
Validation of transpositioned BAC integrity
To determine whether the transpositioned hSIRPA-BAC-TIRs DNA are fully intact, a series of primer pairs were designed at approximately 20 kb intervals, along the BAC insert (Fig. 3A, Supp. Table 1). Polymerase chain reactions showed that all the transpositioned BACs in pups 1.6, 5.4, 5.7, 8.1, 9.2, 9.4 are fully intact (Fig. 3B). Analysis of the two pups (4.2 and 5.5) that were genotyped to be PCR positive for a section of the human SIRPA gene, but negative for transposition, showed that: 1) pup 4.2 was PCR positive only at primer pair 5, suggesting random integration of a fragmented BAC consistent with the absence of TIRs; whereas 2) pup 5.5 showed random insertion of a full BAC DNA without transposition (Fig. 3C). However, the results for pup 5.5 do not establish how the circular BAC was disrupted during the random integration, and it remains to be established if the hSIRPA locus is present as one contiguous segment. Our findings indicate that converting the circular BAC DNA into a large piggyBac transposon is an efficient and robust approach for generating transgenic rats, for which the transposition site can be precisely determined, and the likelihood of a complete insert is increased.

(A) Graphic illustration showing the genomic DNA insert in RP11-887J4. The chromosomal locations of start and end points are indicated. Exon in the SIRPA gene are blocked in bold. Green arrows indicate the primer pairs (10) spanning along entire BAC at approximately 20 kb intervals, used to verify the presence of the full BAC in pups confirmed to have hSIRPA-BAC transpositioned. (B) PCR result showing that six pups (1.6, 5.4, 5.7, 8.1, 9.2 and 9.4) with transpositioned hSIRPA-BACs are confirmed to be positive for all ten primer pairs. Genomic DNAs from pups 2.3, 3.2 and 6.2 were used as negative controls (Fig. 2A). (C) PCR result two pups (4.2 and 5.5) that were found to be positive for the presence of the human SIRPA gene during the genotyping screening, but negative for piggyBac mediated transposition. The results show that pup 4.2 is positive only for primer pair 5, suggesting random insertion of a fragmented BAC DNA. Pup 5.5 is found to be positive for all ten primer pairs, suggesting random insertion of hSIRPA-BAC DNA. (D) Bar graph showing RT-qPCR result indicating the copy number of human SIRPA BAC in piggyBac transpositioned founders (1.6, 5.4, 5.7, 8.1, 9.2, 9.4), random insertion founder (5.5), two negative controls (#1, #2). RT-qPCR results were analyzed relative to the rat GAPDH, for which there are two copies in the genome.
Analysis of copy number integration
Unlike transgenic rodents generated using small plasmids where multiple copies of transgene can be integrated leading to abnormally high expression of the transgene, BAC transgenics tend to have a single copy integration, thereby more accurately reflecting normal physiological level of transgene expression. To evaluate whether piggyBac mediation effects copy number integration of BAC DNA, we further characterized the transposition positive founders (1.6, 5.4, 5.7, 8.1, 9.2, 9.4), as well as the pup with random hSIRPA-BAC-TIRs DNA insertion (5.5), using qPCR on genomic DNA. When normalized to the Ct values derived from the endogenous rat Gapdh gene, for which there are two copies, we found that all of the founders carry a single copy of BAC integration (Fig. 3D).
Analysis of germline transmission
Having shown that the transgenic founder pups generated by piggyBac transposition events carry a single copy of the full length hSIRPA-BAC-TIRs DNA, we sought to determine the germline transmission efficiency. To do this, hSIRPA-BAC-TIRs DNA founders (with the exception of 8.1, which died prior to mating) were mated with wild-type animals to obtain F1 offspring, and analyzed for the rate of BAC transmission (Table 2). PCR genotyping of offspring revealed that founders 1.6 and 9.4 transmitted the BAC to the F1 offspring in a Mendelian manner, whereas founder 5.7 transmitted in a low proportion of the offspring. Founder 4.2 with a fragmented BAC insertion transmitted to the offspring as well. Founders 5.4, 5.5 and 9.2 on the other hand, did not transmit the transgene to their offspring. These analyses indicate that 3 out of 5 of viable hSIRPA-BAC-TIRs DNA transgenic founders derived from piggyBac transposition events transmitted the BAC transgene to their F1 offspring, whereas 2 out of 5 may likely be mosaics resulting in failure of transmission.
Functional analysis of hSIRPA expression in BAC transgenic rats
In order to evaluate the functionality of hSIRPA in the transpositioned BAC DNA, thirteen F1 offspring derived from founder 1.6 were analyzed first for the presence of the transgene (Fig. 4A) and then for the expression of human SIRPα on CD11b+ monocytes, using a human specific CD47-Fc fusion molecule, which binds to human, but not rat Sirpα. All F1 animals positive for the transgene bound human CD47-Fc, but none did among F1 animals negative for the transgene, as shown for two representative animals from each group (Fig. 4B and data not shown). The rationale for evaluating CD11b+ cells among rat leukocyte populations stems from the fact that among human17 and rat43 peripheral blood mononuclear cells, SIRPα expression is mainly restricted to monocytes (essentially all CD11b+) (data not shown). Furthermore, in mice transgenic for human SIRPα expression, human SIRPα detection was restricted to mouse monocytes20, and mononuclear phagocytes are the main cells involved in clearing human cells in immune humanized mice17.

(A) The PCR analysis shows human SIRPA BAC detection in 9 out of 13 offspring from the founder 1.6. Genomic DNA from founder 5.7 was used as a positive control. (B) Animals 1.6 F1.3 and 1.6 F1.4 are F1 offspring animals representative of PCR SIRPA-BAC positive or negative animals, respectively. Peripheral blood mononuclear cells (PBMCs) were first gated by morphology (dot plot SSC-FSC), then CD11b+ cells (mostly expressed by monocytes) were labelled with a rat anti-CD11b antibody (dot plot CD11b-FSC) and finally, cells were stained with combinations of human and rat anti-SIRPα antibodies (left dot-plots) or human anti-SIRPA antibodies and human CD47-Fc (right dot-plots). The large majority of rat monocytes from BAC-SIRPA-TIR+ transgenic rats expressed human SIRPα, as detected using both anti-human SIRPα antibodies and human CD47-Fc whereas all monocytes from BAC-SIRPA-TIR− transgenic rats were negative for both labels.
We also evaluated the expression of hSIRPα in rat monocytes by staining with anti-human and rat specific SIRPα monoclonal antibodies. The results confirmed that genotyped positive offspring (1.6F1.3) faithfully expressed both human and rat SIRPα protein on CD11b+ cells, while the genotyped negative offspring (1.6F1.4) showed expression of only the rat SIRPα protein (Fig. 4B). Additional analysis using SE5A5 monoclonal antibody confirmed our findings (data not shown). Interestingly, all monocytes expressing human SIRPα also expressed rat SIRPα (Fig. 4B), suggesting a common set of underlying molecular mechanisms governing the expression of both human and rat SIRPA gene, and supporting the future use of human SIRPα transgenic and deficient for Rag121 and Il2rg (unpublished) genes as recipients for immune humanization experiments. These findings indicate that piggyBac mediated hSIRPA-BAC-TIRs transpositioned rats give rise to progeny that faithfully express functionally viable human SIRPα protein in rat monocytes.
Generation of CRISPR/Cas9 and TALEN mediated hSIRPA-BAC transgenic rats
We have thus far shown the efficiency with which functionally viable humanized SIRPA transgenic rats can be generated when mediated by piggyBac transposition. While the BAC insertion sites can be readily determined using splinkerette PCR, one of the major limitations of the piggyBac mediated approach is that the BAC can be inserted anywhere in the genome with a TTAA tetranucleotide sequence. From this vantage point, both CRISPR/Cas9 and TALEN endonucleases offer the potential benefit of targeting the BAC into a specific location in the genome. To test this possibility, we designed a strategy for applying the same CRISPR/Cas9 and TALENs, that we previously used42 for inducing DSB in the rat Rosa26 locus, to stimulate HDR44. To take advantage of the HDR repair mechanism, we modified the hSIRPA BAC by retrofitting a cassette containing 900 base pairs of Rosa26 homology arm with sequences flanking the endonuclease cut site. In our laboratories, we have shown that 900 base pair homology arm length is sufficient for targeting circular KOMP vectors (approximately 9 kb) in the mouse genome at high efficiency (data not shown), and that 4.5 kb linearized constructs with similar homology arms can be targeted using CRISPRs and TALENs to Rosa26 and Hprt in mouse and rat zygotes42. Two different cassettes were designed such that one contains the same rat Rosa26 sgRNA or TALEN recognition sequence separating the 5′ and 3′ homology arms, and the other cassette that lacks the recognition sequence. By designing cassettes with or without the sgRNA and TALEN recognition sequences, the aim was to simultaneously linearize or keep the BAC circular in the injected embryo when the endonuclease cleaved the genomic DNA at the targeted Rosa26 locus. The hSIRPA-BAC that was retrofitted with the linearization site will be referred to as hSIRPA-BAC-Rosa26+, and the BAC without the linearization site will be referred to as hSIRPA-BAC-Rosa26−.
The modified BACs were microinjected with the Rosa26 sgRNA and Cas9 protein because we previously demonstrated that Cas9 protein was more efficient than mRNA to obtain genome insertion of donor DNA42. Microinjection with the hSIRPA-BAC-Rosa26+ did not show toxic effects on embryo survival immediately before microinjection (76.3% viable), yielded 60 day-15 fetuses (40.5% relative to the number of zygotes transferred), resulted in 1 founder PCR positive for the BAC (1.7% of fetuses) and 22 founders (36.7% of fetuses) with mutations around the cleavage site in the Rosa26 locus (Table 3). Microinjection with the hSIRPA-BAC-Rosa26− yielded roughly similar results, no toxic effects on embryo survival immediately before microinjection (69.6% viable), yielded 28 day-15 fetuses (32.2% relative to the number of zygotes transferred), resulted in 1 founder PCR positive for the BAC (3.6% from fetuses) and in 12 founders (42.8% from the fetuses) with mutations around the cleavage site of the Rosa26 locus (Table 3). The founder obtained with hSIRPA-BAC-Rosa26+ did not show insertion in the Rosa26 locus as depicted by negative junction PCRs (data not shown). Therefore, despite that sgRNAs for the Rosa26 locus were highly active, as evidenced by high rates of mutations, indicating that the hSIRPA-BAC-Rosa26+ was likely linearized, the frequency of transgenic BAC rats was not increased as compared to the one observed using the circular hSIRPA-BAC-Rosa26−.
We also tested the microinjection of the hSIRPA-BAC-Rosa26 with TALE nuclease mRNA which we previously showed to efficiently and reproducibly target plasmid donor DNA integration by HDR in the Rosa26 locus38. Microinjection with the hSIRPA-BAC-Rosa26+ did not show toxic effects on embryo survival immediately before microinjection (68.9% viable), yielded 39 day-15 fetuses (32% relative to the number of zygotes transferred), resulted in no founders PCR positive for the BAC (0% of fetuses), and in 9 founders (23.1% of fetuses) with NHEJ mutations around the cleavage site of the Rosa26 locus (Table 3). Microinjection with the hSIRPA-BAC-Rosa26− yielded roughly similar results, no toxic effects on embryo survival immediately before microinjection (66% viable), yielded 13 day-15 fetuses (19.7% relative to the number of zygotes transferred), resulted in 0 founder PCR BAC positive (0% from fetuses) and in 4 founders (30.8% from the fetuses) with mutations around the cleavage site of the Rosa26 locus (Table 3). Thus, the TALENs also did not result in integration of the BAC in the targeted locus.
Discussion
In this study, we have generated for the first time: 1) highly efficient piggyBac mediated BAC transgenic rats that are germline transmissible; and 2) rats faithfully expressing functionally viable human SIRPα. While the procedures involved in the production of transgenic rats and mice are similar, the efficiency of creating transgenic rats has remained more difficult than mice6,45. This difficulty is attributed to inherent physiological differences between the two rodent species, giving rise to lower transformation rate of microinjected pronuclear stage zygotes in rats relative to mice. Moreover, unlike in mice, the derivation of germline competent rat embryonic stem (ES) cells could not be achieved until only recently with the discovery of MEK, GSK-3 and FGFR inhibiting strategy46,47,48. Therefore, while improved methodologies for manipulating rat zygotes and ES cells are facilitating the production of transgenic rats, the associated challenges have been immense, leading many to overlook rats as a potential model system for biomedical and translational science, and instead, relying on transgenic mice as the primary source of animal research model.
As methods for generating transgenic rats improve over time, the last decade has also witnessed a rapid advancement in genome engineering technologies. Among them, tools based on transposable element systems such as piggyBac and Sleeping Beauty, and engineered endonucleases such as ZFNs, TALENs and CRISPRs bursted onto the scene allowing manipulating and editing the genome with efficiency and precision never before possible. Because producing transgenic mice have traditionally been less difficult than rats, a large body of literature has accumulated demonstrating the ease with which the genomes of mice can be manipulated using these tools to, for example, reprogram somatic cells in into iPS cells, induce random mutagenesis, conduct precise genome editing through oligo replacement, and knock out gene function via indels33,37,39,49,50. Although much smaller in number relative to mouse models, transgenic rats have been generated using these tools as well42,51,52,53,54,55.
In spite of the great potential engendered in these newly emerging genome engineering tools, BAC transgenesis continues to be a method of choice for many. The advantages imbued in BAC-based transgenics stem from the large genomic insert size, encompassing one or more genes with essential regulatory elements for proper expression of the transgene, and insulating elements that curb epigenetic silencing due to position effect variegation56. Moreover, the time and cost effectiveness of modifying BACs have allowed for high throughput production projects such as the Gene Expression and Nervous System AtLas (GENSAT)57,58 and Knock Out Mouse Project (KOMP)59 which continue to play a pivotal role for enhancing our understanding of gene function and regulation. These are some of the examples delineating the importance and benefits attributed to generating model organisms using BAC transgenesis. Perhaps more importantly, for many research studies involving human therapy and disease modeling, BACs provide the most effective method of recapitulation60,61,62,63,64, as demonstrated by humanized SIRPA mice.
The major limitations of using BACs have been the cumbersome handling procedure and low insertion efficiency in the genome. A major breakthrough came in 2011 when Li and colleagues65 demonstrated for the first time that piggyBac TIR elements can be used to mobilize a fraction of BAC as large as 100 kb into the genome of mouse ES cells, and Suster and colleagues also showed in the same year BAC transposition in zebrafish using Tol2 transposable elements66. In 2013, Rostovskaya et al. reported that BACs modified with piggyBac TIRs can be injected directly into mouse zygotes for efficient generation of founders carrying the transgene67. In rats however, the mediation by piggyBac, nor any other genome engineering tools, has yet to be reported for generating BAC transgenics.
Here, we aimed to examine whether piggyBac, CRISPR/Cas9 and TALEN mediated approaches may enhance hSIRPA-BAC transgenesis in the rat zygote. To do this, we strategically modified the BAC with elements compatible for recognition by these tools. Through this approach, we discovered that piggyBac provided an efficient approach for generating hSIRPA-BAC transgenic rats that are germline transmissible, and faithfully express functionally viable human SIRPα protein in the monocytes of F1 offspring. We have also demonstrated that monocytes from BAC transgenic rats express human SIRPα, as observed in human monocytes, indicating that regulation of expression by hSIRPA-BAC is conserved. Since rat monocytes from BAC transgenic rats express human SIRPα which interacts with human CD47, it is likely that the transgenic rat monocytes will receive phagocytosis inhibitory signals from human SIRPα. Immune humanization of immunodeficient rats has been reported to be undetectable68, and backcrossing with human SIRPA transgenic rats will likely increase efficiency.
By laying out a blueprint for generating piggyBac mediated hSIRPA-BAC transgenesis in rats, we provide an efficient tool in the hands of the research community, for expanding the existing platform for generating hSIRPA-BAC transgenic rats in various immune compromised strains21, as has been done for the mouse immune humanized models13,20. Humanized SIRPA transgenic rats will likely be useful in other biomedical and translational research arena as well, for example, a growing body of literature suggests the relevance of SIRPA in inflammation69, stem cells70, cancer71 and cell cycle regulation72.
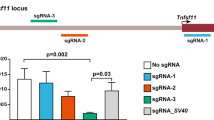
Concomitantly with the submission of our manuscript, the genomic insertion of a BAC using the CRISPR/Cas9 approach has been described in the rat41. The authors describe the use of ssODNs with short homology sequences bridging BAC sequences (human SIRPA) and the genomic region into which the BAC was targeted (rat Sirpa). The efficiency of BAC transgenesis by this approach was comparable to that of the same BAC using classical transgenesis (13.3 and 9.2% of born animals, respectively). In contrast, the piggyBac-mediated approach increased the efficacy of BAC transgenesis compared to that of classical transgenesis (24 vs. 6.7%, respectively). BAC insertion using ssODNS and guide RNAs occurred in the targeted locus whereas piggyBac-mediated BAC transgenesis resulted in random insertions in TTAA sites. Nevertheless, the insertion site of the piggyBac-mediated BAC transgene can be easily determined by splinkerette PCR, and thus the risk of insertional mutations can be evaluated. It is surprising that Yoshimi et al.’s CRISPR/Cas9 approach using ssODNs resulted in BAC insertion into the targeted locus, whereas the use of longer homology arms in our study did not allow for targeted integration. The different outcome may be attributed to the differences in mechanisms underlying insertions using ssODNs versus homology arms. In the case of ssODNs, it is likely that the insertion involved ssODN-mediated nonhomologous end-joining73,74, whereas the homology arms may have required homologous recombination. These two repair pathways not only use completely different protein complexes but also occur at different times of the cell cycle (G0/G1 for NHEJ and G2/S for homologous recombination). Furthermore, it is conceivable that different loci (rat Sirpa vs. rat Rosa26) may be differentially susceptible to insertion of BACs by the two repair pathways. Importantly, researchers now have the choice of performing BAC transgenesis either using Cas9/ssODNs- or piggyBac-mediated integration, with each system having advantages and disadvantages. The CRISPR/Cas9 approach may be particularly attractive once optimal parameters for this approach have been established, as it conceivably allows for precise editing of the genome including the creation of models where the human gene replaces the mouse or rat homolog.
The report by Rostovskaya et al.34 was the first to demonstrate piggyBac mediated BAC transposition in mouse zygotes. Our manuscript shows that piggyBac transposition of BACs is also an efficient method in rats, resulting in increased transgenesis with typically a single copy insertion faithfully expressing the transgene. Moreover, the insertion sites are easily identifiable using simple PCR. Our results support the possibility that this approach may also be useful for generating transgenic animals in other mammalian species of biomedical or agricultural significance. We envision that in the near future, rapidly advancing technologies will allow for an efficient targeting of BACs into specific loci in the genome for the production of rat transgenics. Likewise, the emergence of newer, more advanced, technologies may no longer benefit from BAC-based transgenics for simple procedures such as knocking out genes or knocking in reporters. However, it is premature to disregard BACs as “obsolete” as recently claimed75. BACs have served an important function in science thus far, and we foresee continued relevance. Therefore, improving the method for generating BAC transgenic animals, such as the rat, using rapidly advancing genome engineering technologies can only benefit research, and lead us closer to translation and therapeutic applications.
Material and Methods
Conversion of hSIRPA BAC into a piggyBac transposon
Human BAC, RP11-887J4, carrying 176,233 bps of genomic DNA segment covering the SIRPA transcript coding sequence, as well as the region spanning 52,081 bps up- and 78,424 bps down-stream of the transcript start and end points, was selected for retrofitting with the piggyBac TIR elements. The RP11-887J4 BAC clone was acquired from BAC/PAC Resources (bacpac.chori.org), and retrofitted using the recombineering technology. The recombineering cassette carrying the TIR elements was generated by subcloning the 5′ TIR and 3′ TIR elements derived from pPB-CAG-EBNXN (Wellcome Trust Sanger Institute) into a pUC19 vector backbone with the spectinomycin resistance genes in between the TIR elements. The recombineered BAC clone was PCR and sequence verified.
Modification of hSIRPA BAC with rat Rosa26 homology arms flanking Rosa26 or AAVS1 sgRNA recognition sequences
The RP11-887J4 BAC clone was retrofitted with a recombineering cassette carrying 1049 and 1038 bp length rat Rosa26 homology arms flandking the Rosa26 sgRNA recognition sequence. The cassette was generated by PCR amplifying the 5′ and 3′ homology arms from CHORI230-418P10, for which one of the primers carried the Rosa26 sgRNA recognition sequence. The PCR amplified fragments were subcloned into a pUC19 vector backbone with neomycin resistance gene driven by PGK and EM7 as a selection cassette using the Gibson Assembly kit (NEB).
HyPBase mRNA transcript preparation
To prepare the hyPBase mRNA transcript, pCMV-hyPBase (generous gift from the Wellcome Trust Sanger Institute) was digested with SalI restriction endonuclease, and cleaned up using the PCR Cleanup Kit (Macherey Nagel). 500 ng of the linearized plasmid was used as a template to transcribed in vitro using the mMESSAGE mMACHINE Kit (Ambion) according to the manufacturer’s instructions. The transcribed mRNA was purified using the MEGAclear Kit (Ambion).
Zygote injections
The rats used in this work were from the Sprague-Dawley (SD/Crl) strain (Charles River, L’Arbresle, France). The study was approved by the Ethics Committee on Animal Experimentation of the Pays de la Loire Region, France, in accordance with the guidelines from the French National Research Council for the Care and Use of Laboratory Animals (Permit Numbers: APAFIS#692), as such, the methods themselves were conducted in “accordance” with the approved guidelines. The BAC constructs and hyperactive transposase were injected at different concentrations indicated in the results section (Tables 1 and 3). The BAC was diluted in a buffer containing 10 mM Tris-HCl, pH 7.5, 0, 1 mM EDTA, 30 uM spermine, 70 uM spermidine, 100 mM NaCl76. The BAC were handled gently and kept on ice during microinjection. Fertilized 1-cell stage embryo collection and sequential microinjection into the male pronucleous and into the cytoplasm using different BAC and hyPBase concentrations, Cas9 protein and sgRNA for rat Rosa26 (sgRNA and Cas9 protein incubated before microinjection), as well as TALENs mRNA for rat Rosa26 and donor DNA as previously been described in detail42. We demonstrated that Cas9 protein was more efficient than its mRNA to generate knockin by homologous recombination42. Microinjected embryos were maintained in 5% CO2 at 37 °C for at least 1 h. Surviving embryos were then implanted on the same day of microinjection in the oviduct of pseudo-pregnant females (0.5 days post coitum) and allowed to develop until embryonic day 15 or until normal delivery. Offspring were obtained by crossing BAC positive founders with wild-type rats.
Genotyping analysis
Embryos or pups were identified by PCR using the following primers: 5′-CTCTACGCGCTTTCTTGTCC-3′ and 5′- AACGTCAGCCTCCAGGTATG-3′. The standard amplification profile consisted of 5 mn at 95 °C, 2 mn at 62 °C followed by 35 cycles of 30 s at 72 °C, 10 s at 95 °C, 10 s at 60 °C and 3 min at 72 °C. Amplicons were analyzed using microfluidic capillary electrophoresis77.
Splinkerette PCR for sequencing of transposon integration sites
Genomic DNA was isolated and digested with Sau3AI. The digested genomic DNA was ligated to 50 nM of splinkerette adaptors using T4 ligase and ligation buffer from NEB. Nested PCR was used to amplify the genomic DNA sequence adjacent to the 5′ and 3′ TIR ends. The PCR products were sequenced to determine the precise transposition sites.
Realtime Quantitative PCR (qPCR)
Using 20 ng of isolated genomic DNA, qRT-PCR reactions were performed with SYBR Green PCR Master Mix (Applied Biosystems) according to manufacturer’s instructions. 200 nM of primer pair used for genotyping the presence of the human SIRPA gene was used for qRT-PCR. The Ct values were calculated using Applied Biosystems’ SDS2.4 software. To determine the copy number of the transpositioned BACs, the Ct values derived from the primer pair used for amplifying the human SIRPA gene was normalized to the GAPDH gene in the rat genome.
Flow cytometry
Rat peripheral blood leukocytes were isolated from heparinized blood by red blood cell lysis. hSIRPA was detected using an anti-human SIRPA monoclonal antibodies (clones REA144, Miltenyi or SE5A5, BD Biosciences, both IgG1). Recombinant human CD47-Fc (hCD47-Fc, R&D Systems) was also used to detect human SIRPA expression since it does not bind to rat SIRPA. Rat SIRPA was detected using anti-rat SIRPA-FITC monoclonal antibody (OX41, European Collection of Cell Culture). Several isotype negative controls were used: APC-mouse IgG1, FITC-mouse IgG2a, PE-mouse IgG1 (BD Pharmingen). All monoclonal antibodies, hCD47-Fc and human IgGs were used at 10 ug/ml. Fluorescence was analyzed with a FACSVerse flow cytometer (BD Biosciences), and FlowJo software was used to analyze data.
Additional Information
How to cite this article: Jung, C. J. et al. Comparative Analysis of piggyBac, CRISPR/Cas9 and TALEN Mediated BAC Transgenesis in the Zygote for the Generation of Humanized SIRPA Rats. Sci. Rep. 6, 31455; doi: 10.1038/srep31455 (2016).
References
Shizuya, H. et al. Cloning and stable maintenance of 300-kilobase-pair fragments of human DNA in Escherichia coli using an F-factor-based vector. Proc Natl Acad Sci USA 89, 8794–8797 (1992).
Osoegawa, K. et al. Bacterial artificial chromosome libraries for mouse sequencing and functional analysis. Genome Res 10, 116–128 (2000).
Marra, M. A. et al. High throughput fingerprint analysis of large-insert clones. Genome Res 7, 1072–1084 (1997).
Yang, X. W., Model, P. & Heintz, N. Homologous recombination based modification in Escherichia coli and germline transmission in transgenic mice of a bacterial artificial chromosome. Nat Biotechnol 15, 859–865, 10.1038/nbt0997-859 (1997).
Heintz, N. BAC to the future: the use of bac transgenic mice for neuroscience research. Nat Rev Neurosci 2, 861–870, 10.1038/35104049 (2001).
Charreau, B., Tesson, L., Soulillou, J. P., Pourcel, C. & Anegon, I. Transgenesis in rats: technical aspects and models. Transgenic Res 5, 223–234 (1996).
Pinkert, C. A. Transgenic animal technology: a laboratory handbook. 2nd edn, (Academic Press, 2002).
Abbott, A. Laboratory animals: the Renaissance rat. Nature 428, 464–466, 10.1038/428464a (2004).
Mosier, D. E., Gulizia, R. J., Baird, S. M. & Wilson, D. B. Transfer of a functional human immune system to mice with severe combined immunodeficiency. Nature 335, 256–259, 10.1038/335256a0 (1988).
Bosma, G. C., Custer, R. P. & Bosma, M. J. A severe combined immunodeficiency mutation in the mouse. Nature 301, 527–530 (1983).
Ito, R., Takahashi, T., Katano, I. & Ito, M. Current advances in humanized mouse models. Cell Mol Immunol 9, 208–214, 10.1038/cmi.2012.2 (2012).
Shultz, L. D., Ishikawa, F. & Greiner, D. L. Humanized mice in translational biomedical research. Nat Rev Immunol 7, 118–130, 10.1038/nri2017 (2007).
Yamauchi, T. et al. Polymorphic Sirpa is the genetic determinant for NOD-based mouse lines to achieve efficient human cell engraftment. Blood 121, 1316–1325, 10.1182/blood-2012-06-440354 (2013).
Takenaka, K. et al. Polymorphism in Sirpa modulates engraftment of human hematopoietic stem cells. Nat Immunol 8, 1313–1323, 10.1038/ni1527 (2007).
Shultz, L. D. et al. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J Immunol 154, 180–191 (1995).
Barclay, A. N. Signal regulatory protein alpha (SIRPalpha)/CD47 interaction and function. Curr Opin Immunol 21, 47–52, 10.1016/j.coi.2009.01.008 (2009).
Barclay, A. N. & Van den Berg, T. K. The interaction between signal regulatory protein alpha (SIRPalpha) and CD47: structure, function, and therapeutic target. Annu Rev Immunol 32, 25–50, 10.1146/annurev-immunol-032713-120142 (2014).
Oldenborg, P. A. et al. Role of CD47 as a marker of self on red blood cells. Science 288, 2051–2054 (2000).
Kharitonenkov, A. et al. A family of proteins that inhibit signalling through tyrosine kinase receptors. Nature 386, 181–186, 10.1038/386181a0 (1997).
Strowig, T. et al. Transgenic expression of human signal regulatory protein alpha in Rag2-/-gamma(c)-/- mice improves engraftment of human hematopoietic cells in humanized mice. Proc Natl Acad Sci USA 108, 13218–13223, 10.1073/pnas.1109769108 (2011).
Menoret, S. et al. Generation of Rag1-knockout immunodeficient rats and mice using engineered meganucleases. FASEB J 27, 703–711, 10.1096/fj.12-219907 (2013).
Fraser, M. J., Ciszczon, T., Elick, T. & Bauser, C. Precise excision of TTAA-specific lepidopteran transposons piggyBac (IFP2) and tagalong (TFP3) from the baculovirus genome in cell lines from two species of Lepidoptera. Insect Mol Biol 5, 141–151 (1996).
Mitra, R., Fain-Thornton, J. & Craig, N. L. piggyBac can bypass DNA synthesis during cut and paste transposition. EMBO J 27, 1097–1109, 10.1038/emboj.2008.41 (2008).
Ding, S. et al. Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell 122, 473–483, 10.1016/j.cell.2005.07.013 (2005).
Horn, C. et al. Splinkerette PCR for more efficient characterization of gene trap events. Nat Genet 39, 933–934, 10.1038/ng0807-933 (2007).
Potter, C. J. & Luo, L. Splinkerette PCR for mapping transposable elements in Drosophila. PLoS One 5, e10168, 10.1371/journal.pone.0010168 (2010).
Uren, A. G. et al. A high-throughput splinkerette-PCR method for the isolation and sequencing of retroviral insertion sites. Nat Protoc 4, 789–798, 10.1038/nprot.2009.64 (2009).
Rad, R. et al. PiggyBac transposon mutagenesis: a tool for cancer gene discovery in mice. Science 330, 1104–1107, 10.1126/science.1193004 (2010).
Rad, R. et al. A conditional piggyBac transposition system for genetic screening in mice identifies oncogenic networks in pancreatic cancer. Nat Genet 47, 47–56, 10.1038/ng.3164 (2015).
Lacoste, A., Berenshteyn, F. & Brivanlou, A. H. An efficient and reversible transposable system for gene delivery and lineage-specific differentiation in human embryonic stem cells. Cell Stem Cell 5, 332–342, 10.1016/j.stem.2009.07.011 (2009).
Woltjen, K. et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature 458, 766–770, 10.1038/nature07863 (2009).
Woodard, L. E. & Wilson, M. H. piggyBac-ing models and new therapeutic strategies. Trends Biotechnol 33, 525–533, 10.1016/j.tibtech.2015.06.009 (2015).
Yusa, K. piggyBac Transposon. Microbiol Spectr 3, MDNA3-0028-2014, 10.1128/microbiolspec.MDNA3-0028-2014 (2015).
Rostovskaya, M. et al. Transposon mediated BAC transgenesis via pronuclear injection of mouse zygotes. Genesis 51, 135–141, 10.1002/dvg.22362 (2013).
Chu, V. T. et al. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat Biotechnol 33, 543–548, 10.1038/nbt.3198 (2015).
Maruyama, T. et al. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat Biotechnol 33, 538–542, 10.1038/nbt.3190 (2015).
Sander, J. D. & Joung, J. K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol 32, 347–355, 10.1038/nbt.2842 (2014).
Ran, F. A. et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308, 10.1038/nprot.2013.143 (2013).
Gaj, T., Gersbach, C. A. & Barbas, C. F. 3rd . ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31, 397–405, 10.1016/j.tibtech.2013.04.004 (2013).
Voytas, D. F. & Joung, J. K. Plant science. DNA binding made easy. Science 326, 1491–1492, 10.1126/science.1183604 (2009).
Yoshimi, K. et al. ssODN-mediated knock-in with CRISPR-Cas for large genomic regions in zygotes. Nat Commun 7, 10431, 10.1038/ncomms10431 (2016).
Menoret, S. et al. Homology-directed repair in rodent zygotes using Cas9 and TALEN engineered proteins. Sci Rep 5, 14410, 10.1038/srep14410 (2015).
Adams, S. et al. Signal-regulatory protein is selectively expressed by myeloid and neuronal cells. J Immunol 161, 1853–1859 (1998).
Remy, S. et al. Efficient gene targeting by homology-directed repair in rat zygotes using TALE nucleases. Genome Res 24, 1371–1383, 10.1101/gr.171538.113 (2014).
Mullins, L. J. & Mullins, J. J. Transgenesis in the rat and larger mammals. J Clin Invest 97, 1557–1560, 10.1172/JCI118579 (1996).
Buehr, M. et al. Capture of authentic embryonic stem cells from rat blastocysts. Cell 135, 1287–1298, 10.1016/j.cell.2008.12.007 (2008).
Li, P. et al. Germline competent embryonic stem cells derived from rat blastocysts. Cell 135, 1299–1310, 10.1016/j.cell.2008.12.006 (2008).
Meek, S. et al. Efficient gene targeting by homologous recombination in rat embryonic stem cells. PLoS One 5, e14225, 10.1371/journal.pone.0014225 (2010).
Copeland, N. G. & Jenkins, N. A. Harnessing transposons for cancer gene discovery. Nat Rev Cancer 10, 696–706, 10.1038/nrc2916 (2010).
Yang, H., Wang, H. & Jaenisch, R. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nat Protoc 9, 1956–1968, 10.1038/nprot.2014.134 (2014).
Furushima, K. et al. Insertional mutagenesis by a hybrid piggyBac and sleeping beauty transposon in the rat. Genetics 192, 1235–1248, 10.1534/genetics.112.140855 (2012).
Li, W. et al. Genetic modification and screening in rat using haploid embryonic stem cells. Cell Stem Cell 14, 404–414, 10.1016/j.stem.2013.11.016 (2014).
Tesson, L. et al. Knockout rats generated by embryo microinjection of TALENs. Nat Biotechnol 29, 695–696, 10.1038/nbt.1940 (2011).
Geurts, A. M. et al. Knockout rats via embryo microinjection of zinc-finger nucleases. Science 325, 433, 10.1126/science.1172447 (2009).
Larcher, T. et al. Characterization of dystrophin deficient rats: a new model for Duchenne muscular dystrophy. PLoS One 9, e110371, 10.1371/journal.pone.0110371 (2014).
Bian, Q. & Belmont, A. S. BAC TG-EMBED: one-step method for high-level, copy-number-dependent, position-independent transgene expression. Nucleic Acids Res 38, e127, 10.1093/nar/gkq178 (2010).
Heintz, N. Gene expression nervous system atlas (GENSAT). Nat Neurosci 7, 483, 10.1038/nn0504-483 (2004).
Gong, S. et al. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature 425, 917–925, 10.1038/nature02033 (2003).
Skarnes, W. C. et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474, 337–342, 10.1038/nature10163 (2011).
Blesa, J. & Przedborski, S. Parkinson’s disease: animal models and dopaminergic cell vulnerability. Front Neuroanat 8, 155, 10.3389/fnana.2014.00155 (2014).
Justice, M. J., Siracusa, L. D. & Stewart, A. F. Technical approaches for mouse models of human disease. Dis Model Mech 4, 305–310, 10.1242/dmm.000901 (2011).
Doyle, A., McGarry, M. P., Lee, N. A. & Lee, J. J. The construction of transgenic and gene knockout/knockin mouse models of human disease. Transgenic Res 21, 327–349, 10.1007/s11248-011-9537-3 (2012).
Song, H., Chung, S. K. & Xu, Y. Modeling disease in human ESCs using an efficient BAC-based homologous recombination system. Cell Stem Cell 6, 80–89, 10.1016/j.stem.2009.11.016 (2010).
Peters, O. M. et al. Human C9ORF72 Hexanucleotide Expansion Reproduces RNA Foci and Dipeptide Repeat Proteins but Not Neurodegeneration in BAC Transgenic Mice. Neuron 88, 902–909, 10.1016/j.neuron.2015.11.018 (2015).
Li, M. A. et al. Mobilization of giant piggyBac transposons in the mouse genome. Nucleic Acids Res 39, e148, 10.1093/nar/gkr764 (2011).
Suster, M. L., Abe, G., Schouw, A. & Kawakami, K. Transposon-mediated BAC transgenesis in zebrafish. Nat Protoc 6, 1998–2021, 10.1038/nprot.2011.416 (2011).
Rostovskaya, M. et al. Transposon-mediated BAC transgenesis in human ES cells. Nucleic Acids Res 40, e150, 10.1093/nar/gks643 (2012).
Mashimo, T. et al. Generation and characterization of severe combined immunodeficiency rats. Cell Rep 2, 685–694, 10.1016/j.celrep.2012.08.009 (2012).
Zen, K. et al. Inflammation-induced proteolytic processing of the SIRPalpha cytoplasmic ITIM in neutrophils propagates a proinflammatory state. Nat Commun 4, 2436, 10.1038/ncomms3436 (2013).
Dubois, N. C. et al. SIRPA is a specific cell-surface marker for isolating cardiomyocytes derived from human pluripotent stem cells. Nat Biotechnol 29, 1011–1018, 10.1038/nbt.2005 (2011).
Theocharides, A. P. et al. Disruption of SIRPalpha signaling in macrophages eliminates human acute myeloid leukemia stem cells in xenografts. J Exp Med 209, 1883–1899, 10.1084/jem.20120502 (2012).
Poon, I. K., Lucas, C. D., Rossi, A. G. & Ravichandran, K. S. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol 14, 166–180, 10.1038/nri3607 (2014).
Maresca, M., Lin, V. G., Guo, N. & Yang, Y. Obligate ligation-gated recombination (ObLiGaRe): custom-designed nuclease-mediated targeted integration through nonhomologous end joining. Genome Res 23, 539–546, 10.1101/gr.145441.112 (2013).
Renaud, J. B. et al. Improved Genome Editing Efficiency and Flexibility Using Modified Oligonucleotides with TALEN and CRISPR-Cas9 Nucleases. Cell Rep 14, 2263–2272, 10.1016/j.celrep.2016.02.018 (2016).
Beil, J., Fairbairn, L., Pelczar, P. & Buch, T. Is BAC transgenesis obsolete? State of the art in the era of designer nucleases. J Biomed Biotechnol 2012, 308414, 10.1155/2012/308414 (2012).
Montoliu, L., Bock, C. T., Schutz, G. & Zentgraf, H. Visualization of large DNA molecules by electron microscopy with polyamines: application to the analysis of yeast endogenous and artificial chromosomes. J Mol Biol 246, 486–492 (1995).
Chenouard, V. et al. A rapid and cost-effective method for genotyping genome-edited animals: a heteroduplex mobility assay using microfluidic capillary electrophoresis. J. Genetics and Genomics 43, 341–348, 10.1016/j.jgg.2016.04.005 (2016).
Acknowledgements
This work was financially supported by the “TEFOR” project funded by the ≪Investissements d’Avenir≫ French Government program, managed by the French National Research Agency (ANR) (ANRII-INSB-0014). (ANR II-INSB-0014) and by Institut National of Cancer (INCA) (project MDScan). This work was realized in the context of the Labex IGO project (no. ANR-11-LABX-0016-01) and the IHU-Cesti project (ANR-10-IBHU-005) which both are part of the ≪Investissements d’Avenir≫ French Government program managed by the ANR. The IHU-Cesti project is also supported by Nantes Métropole and Région Pays de la Loire. Work at CHORI was funded by BACPAC Resources Center.
Author information
Authors and Affiliations
Contributions
Conceptual framework: C.J.J., S.M., P.J.d.J. and I.A. Designed experiments: C.J.J., S.M., P.J.d.J. and I.A. Analyzed data: C.J.J., S.M., P.J.d.J. and I.A. Performed Experiments: C.J.J., S.M., L.B., L.T., C.U., V.C., S.R., L.-H.O., N.P. and B.V. Wrote the manuscript: C.J.J., S.M., P.J.d.J. and I.A.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Jung, C., Ménoret, S., Brusselle, L. et al. Comparative Analysis of piggyBac, CRISPR/Cas9 and TALEN Mediated BAC Transgenesis in the Zygote for the Generation of Humanized SIRPA Rats. Sci Rep 6, 31455 (2016). https://doi.org/10.1038/srep31455
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep31455
This article is cited by
-
Revolutionizing Agriculture: Harnessing CRISPR/Cas9 for Crop Enhancement
Indian Journal of Microbiology (2024)
-
Electroporation of mice zygotes with dual guide RNA/Cas9 complexes for simple and efficient cloning-free genome editing
Scientific Reports (2018)
-
A versatile genetic tool: haploid cells
Stem Cell Research & Therapy (2017)
-
Advances in transgenic animal models and techniques
Transgenic Research (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
