
Abstract
Resistance exercise (RE) activates signalling by the mammalian target of rapamycin (mTOR) and it has been suggested that rapamycin-sensitive mTOR signalling controls RE-induced changes in protein synthesis, ribosome biogenesis, autophagy and the expression of peroxisome proliferator gamma coactivator 1 alpha (PGC-1α). However, direct evidence to support the aforementioned relationships is lacking. Therefore, in this study, we investigated the role of rapamycin-sensitive mTOR in the RE-induced activation of muscle protein synthesis, ribosome biogenesis, PGC-1α expression and hypertrophy. The results indicated that the inhibition of rapamycin-sensitive mTOR could prevent the induction of ribosome biogenesis by RE, but it only partially inhibited the activation of muscle protein synthesis. Likewise, the inhibition of rapamycin-sensitive mTOR only partially blocked the hypertrophic effects of chronic RE. Furthermore, both acute and chronic RE promoted an increase in PGC-1α expression and these alterations were not affected by the inhibition of rapamycin-sensitive mTOR. Combined, the results from this study not only establish that rapamycin-sensitive mTOR plays an important role in the RE-induced activation of protein synthesis and the induction of hypertrophy, but they also demonstrate that additional (rapamycin-sensitive mTOR-independent) mechanisms contribute to these fundamentally important events.
Similar content being viewed by others


Introduction
Protein metabolism plays a critical role in the regulation of skeletal muscle mass and recent studies have demonstrated that signalling by the mechanistic/mammalian target of rapamycin (mTOR) plays a central role in the regulation of both protein synthesis and protein degradation1,2. It is known that mTOR can be found in at least two multi-protein complexes called mTORC1 and mTORC2. The defining component of mTORC1 is a protein called Raptor and it has been shown that a subset of mTORC1-dependent, but not mTORC2-dependent, signalling events are highly sensitive to inhibition by rapamycin3,4,5. For this reason, it has been widely assumed that mTORC1 is responsible for the rapamycin-sensitive and mTOR-dependent signalling events that regulate protein metabolism, yet recent studies have shown that this assumption may not be entirely correct6. Nonetheless, it is clear that rapamycin-sensitive mTOR signalling can regulate protein synthesis and autophagy and these effects are mediated, at least in part, through changes in the phosphorylation of downstream molecules such as p70S6K, 4E-BP1 and ULK1 1,2.
Consistent with its critical role in the regulation of protein metabolism, previous studies have shown that rapamycin-sensitive mTOR signalling is necessary for the hypertrophy that occurs in response to chronic mechanical overload7,8. Moreover, numerous studies have reported that rapamycin-sensitive mTOR is robustly activated for a relatively long duration (e.g. >24 h after exercise) after a single bout of RE9,10,11,12. Thus, it has been widely suggested that rapamycin-sensitive mTOR is the key signalling node through which RE induces hypertrophy. However, this hypothesis has not been fully tested in a model that mimics human RE. This is a critical point because the mode and pattern of muscle contraction and the time course and the extent of adaptations that occur in response to chronic mechanical overload (via synergist ablation, SA) are quite different from RE. For example, SA increases muscle mass more than 50% within 2 weeks while RE takes a few months to increase muscle mass by 10%. Furthermore, a recent study which used a rodent model of RE reported that the inhibition of rapamycin-sensitive mTOR signalling only partially inhibits the increase in protein synthesis that is observed after RE13, indicating that, in contrast to SA, RE might induce muscle hypertrophy via both rapamycin-sensitive mTOR-dependent and -independent mechanisms.
In addition to its role in the regulation of protein metabolism, rapamycin-sensitive mTOR has also been shown to regulate mitochondria biogenesis via control of peroxisome proliferator gamma coactivator 1 alpha (PGC-1α) expression14. Increased PGC-1α expression occurs after endurance exercise via robust AMP-activated protein kinase (AMPK) activation15 and RE is known to increase PGC-1α expression16. Moreover, recent studies reported that the addition of RE or protein diet supplementation to endurance exercise results in amplified PGC-1α expression as compared with endurance exercise alone17,18. It has been proposed that these effects are at least partly due to rapamycin-sensitive mTOR activation; however, the role of rapamycin-sensitive mTOR in this process has not been directly addressed.
As described above, it has been widely suggested that rapamycin-sensitive mTOR signalling plays a key role in several of the adaptive responses that are observed following RE: however, the validity of these claims has not been fully investigated. Therefore, the purpose of this study was to define the role that rapamycin-sensitive mTOR plays in the RE-induced activation of muscle protein synthesis, ribosome biogenesis, PGC-1α expression and hypertrophy using a rodent model of resistance exercise.
Results
The effect of rapamycin on RE-induced Akt-rapamycin-sensitive mTOR signalling
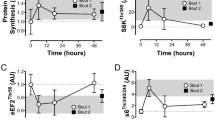
Akt is known as effector of insulin/IGF-1 signalling and it can induce muscle hypertrophy through a pathway involving rapamycin-sensitive mTOR19. As shown in Fig. 1, the phosphorylation of Akt at Ser473 increased 1 h after RE independent of rapamycin administration while no significant differences were observed at 6 and 24 h post RE (Fig. 1). In contrast to the changes in Akt phosphorylation, rapamycin completely inhibited the RE-induced increase in p70S6K Thr389 phosphorylation at all time points. This is important because p70S6K Thr389 phosphorylation is a validated marker of rapamycin-sensitive mTOR signalling7 and thus, these results establish that the rapamycin treatment was fully effective at inhibiting rapamycin-sensitive mTOR signalling.

The effect of acute RE and rapamycin administration on the phosphorylation and total protein content of Akt, p70S6K, 4E-BP1 and rpS6.
RE, resistance exercise; RAPA, rapamycin; PLA, placebo. Values in the graphs are means + SE. *P < 0.05 vs. no exercise control muscle in the same group; #P < 0.05 vs. corresponding muscle in the placebo group.
We next investigated other commonly used markers of rapamycin-sensitive mTOR signalling including 4E-BP1 and rpS6. Phosphorylation of 4E-BP1 at Thr37/46, which is known to be a direct target of rapamycin-sensitive mTOR20, increased only 6 h after RE and was rapamycin-sensitive. The active isoform ratio of 4E-BP1 (active γ-form / total α+β+γ), which is a marker of 4E-BP1 activity21,22, also increased 1 and 6 h after exercise and this effect was also completely inhibited by rapamycin. Moreover, RE also induced a significant increase in rpS6 Ser240/244 phosphorylation at all time points and consistent with other studies23,24, this effect was not entirely blocked by rapamycin. Interestingly, RE also induced a small but significant increase total rpS6 at all time points and this effect was rapamycin-sensitive.
ULK1 can activate autophagy and rapamycin-sensitive mTOR has been reported to inhibit ULK1 via phosphorylation of the Ser757 residue25. As shown in Fig. 2, the phosphorylation of ULK at Ser757 increased 1 h after RE independent of rapamycin administration, while no significant effect of RE was observed at 6 and 24 h post RE. In addition, no changes in LC3-I and -II proteins (autophagosome marker) were observed after RE and rapamycin administration. Combined, these results indicate that RE can increase ULK1 phosphorylation through an rapamycin-sensitive mTOR-independent process, but this event is not sufficient to alter LC3-based markers of autophagosome formation.

The effect of acute RE and rapamycin administration on the phosphorylation and total protein content of ULK1 and LC3.
RE, resistance exercise; RAPA, rapamycin; PLA, placebo. Values in the graphs are means + SE. *P < 0.05 vs. no exercise control muscle in the same group; #P < 0.05 vs. corresponding muscle in the placebo group.
We further investigated MAPK signalling molecules, ERK1/2 and p38 MAPK, to determine whether there are off-target rapamycin effects. As shown in Fig. S2, the phosphorylation of ERK1/2 at Thr202/Tyr204 and p38 MAPK at Thr180/Tyr182 did not change after acute RE or rapamycin administration. Similarly, RE or rapamycin did not alter total ERK1/2 and p38 MAPK proteins.
The effect of chronic rapamycin administration and RE on Akt-rapamycin-sensitive mTOR signalling
Chronic rapamycin administration (8 mg/kg rapamycin every other day for 4 weeks) has been reported to decrease basal phosphorylation of p70S6K, rpS6 and 4E-BP1 (increase in inactive α-form/total 4E-BP1) in skeletal muscle26 and similar results were observed in the present study (Fig. 3). Moreover and in contrast to acute rapamycin administration, we observed that chronic rapamycin administration led to a decrease in basal Akt Ser473 phosphorylation (a marker of mTORC2 signalling). The decrease in Akt Ser473 phosphorylation is consistent with a previous study that administered 2 mg/kg/day of rapamycin for 4 weeks27 and therefore, lends support to the hypothesis that chronic rapamycin administration can inhibit both mTORC1 and mTORC2 in skeletal muscle27,28.

The effect of chronic RE and rapamycin administration on the phosphorylation and total protein content of molecules associated with RSmTOR signalling.
RE, resistance exercise; RAPA, rapamycin; PLA, placebo. Values in the graphs are means + SE. *P < 0.05 vs. no exercise control muscle in the same group; #P < 0.05 vs. corresponding muscle in the placebo group.
In contrast to downstream signalling molecules of rapamycin-sensitive mTOR that regulate protein synthesis, chronic rapamycin administration unexpectedly increased the phosphorylation and total content of ULK1 (Fig. 3), while the ratio of phosphorylated to total ULK1 was not changed. These results suggest that, at least in skeletal muscle, rapamycin-sensitive mTOR does not phosphorylate ULK1, but it may negatively control ULK1 expression and therefore the capacity of ULK1 signalling that leads to autophagy. Similarly, chronic rapamycin administration significantly increased LC3-I as well as LC3-II to an even greater extent (Fig. 3). It is known that immediately after translation, proLC3 is cleaved to inactive form, LC3-I 29. During autophagy, LC3-I is processed to the autophagosomal membrane-associated form, LC3-II and the amount of LC3-II is correlated with the extent of autophagosome formation29. Thus, our results suggest that chronic inhibition of rapamycin-sensitive mTOR controls LC3 expression, but not necessarily autophagosome formation.
Basal phosphorylation of ERK1/2 at Thr202/Tyr204 and p38 MAPK at Thr180/Tyr182 were not changed after chronic rapamycin administration (Fig. S2). In contrast, chronic RE decreased basal phosphorylation of ERK1/2 at Thr202/Tyr204 but not p38 MAPK at Thr180/Tyr182 independent of rapamycin administration.
The role of rapamycin-sensitive mTOR signalling in the regulation of RE-induced PGC-1α expression
Rapamycin-sensitive mTOR has also been reported to regulate PGC-1α expression, which is a key regulator of mitochondria biogenesis14 and the expression of PGC-1α has been implicated in the control of skeletal muscle mass30,31. We found that RE potently increased the mRNA abundance of PGC-1α independent of rapamycin administration at 6 h after RE (Fig. 4A). However, no significant changes in PGC-1α protein levels were detected at any time point after acute RE. On the other hand, PGC-1α protein levels were significantly elevated after chronic RE, but rapamycin did not alter this response RE (Fig. 4B). Taken together, these results indicate that chronic RE induces an increase in PGC-1α and that this effect is mediated through an rapamycin-sensitive mTOR-independent mechanism.

The effect of RE and rapamycin administration on PGC-1α mRNA (A) and protein (B). CON, control; RE, resistance exercise; PLA, placebo; RAPA, rapamycin. Values in the graphs are means + SE. *P < 0.05 vs. no exercise control muscle in the same group.
The role of rapamycin-sensitive mTOR signalling in the regulation of RE-induced ribosome biogenesis
Rapamycin-sensitive mTOR is known to stimulate not only translation initiation but also ribosome biogenesis32,33. Ribosomes act as the translation machinery and thus their volume reflects the translational capacity of the cells. While enhanced translational efficiency, mainly via an increase in translation initiation, is considered to be important in enhancing muscle protein synthesis, ribosome biogenesis can also play a major regulatory role32. In the present study, RE did not alter rRNA content at 1 and 6 h after exercise, but significant increases in rRNA were observed 24 h after acute RE and chronic RE. These alterations were largely abolished by rapamycin (Fig. 5A). Thus, it can be concluded that both acture and chronic RE induce ribosome biogenesis through an rapamycin-sensitive mTOR-dependent pathway.

The effect of RE and rapamycin administration on rRNA (A), c-myc and UBF proteins (B) and c-myc mRNA (C). CON, control; RE, resistance exercise; PLA, placebo; RAPA, rapamycin. Values in graphs are means + SE. *P < 0.05 vs. no exercise control muscle in the same group; #P < 0.05 vs. corresponding muscle in the placebo group.
To examine possible mediators, we first looked at UBF because it is a transcription factor that induces rDNA transcription and it is recognized as a downstream regulator of rapamycin-sensitive mTOR34. However, neither acute nor chronic RE significantly altered UBF expression (Fig. 5B). Next, we looked at c-myc because ribosome biogenesis is known to be regulated by c-myc and RE has been reported to promote an increase in c-myc expression16,35,36. Moreover, it has been suggested that c-myc expression might be regulated, at least in part, by rapamycin-sensitive mTOR37. Similar to previous studies, we found that both acute and chronic RE increased c-myc expression (Fig. 5B,C). Interestingly, rapamycin inhibited the increase in c-myc with chronic RE but not acute RE. Taken together, these data indicate that c-myc is not part of the rapamycin-sensitive mTOR pathway that drives acute RE-induced changes in ribosome biogenesis, however, c-myc might play role in the rapamycin-sensitive mTOR pathway that mediates ribosome biogenesis during chronic RE.
The role of rapamycin-sensitive mTOR in RE-induced protein synthesis and muscle hypertrophy
Numerous studies have shown that RE induces an increase in muscle protein synthesis38,39 and it is thought that the increase in protein synthesis plays a central role in the concomitant hypertrophic response40. In accordance with previous studies38,39, we found that RE induced an increase in muscle protein synthesis at all of the time points that were analyzed (Fig. 6). Interestingly, however, rapamycin only partially inhibited the RE-induced increase in protein synthesis. Our observations are very similar to those recently reported by West et al.13 and therefore, provide further evidence in support the conclusion that both rapamycin-sensitive mTOR-dependent and -independent mechanisms contribute to the increase in protein synthesis that occurs after an acute bout of RE.

The effect of acute RE and rapamycin administration on muscle protein synthesis (puromycin-labeled peptides).
CON, control; RE, resistance exercise; PLA, placebo; RAPA, rapamycin. Values are expressed relative to the no exercise placebo control group and presented as the means + SE. *P < 0.05 vs. no exercise control muscle in the same group; #P < 0.05 vs. corresponding muscle in the placebo group.
Our observation that an rapamycin-sensitive mTOR-independent mechanism contributes to the RE-induced increase in protein synthesis led us to the hypothesis that an rapamycin-sensitive mTOR-independent mechanism would also contribute to the hypertrophy that occurs in response to chronic RE. Thus, we next investigated the role of rapamycin-sensitive mTOR in chronic RE-induced muscle hypertrophy. The results show that chronic RE increased muscle wet weight (MWW) and fiber CSA in both placebo and rapamycin groups although the magnitude of increase in MWW and fiber CSA was significantly greater in the placebo group (Fig. 7).

The effect of chronic RE and rapamycin administration on muscle wet weight, MWW (A,B) and fiber cross-sectional area, CSA (C,D). Representative images of PLA-CON (E), PLA-RE (F), PARA-CON (G) and RAPA-RE (H). CON, control; RE, resistance exercise; PLA, placebo; RAPA, rapamycin. Values are expressed relative to the no exercise placebo control group and presented as the means + SE. *P < 0.05 vs. no exercise control muscle in the same group; #P < 0.05 vs. corresponding muscle in the placebo group; +P < 0.05 between groups.
Discussion
RE is known to activate rapamycin-sensitive mTOR signalling and it has been widely assumed that that the activation of rapamycin-sensitive mTOR plays a critical role in the concomitant hypertrophic response41. However, very little is known about how, or if, RE-induced rapamycin-sensitive mTOR signalling regulates downstream process such as protein synthesis, ribosome biogenesis and mitochondria biogenesis. More importantly, whether rapamycin-sensitive mTOR signalling is necessary for chronic RE-induced hypertrophy has never been investigated. Here we have demonstrated that inhibition of rapamycin-sensitive mTOR signalling via rapamycin injection completely inhibited the induction of ribosome biogenesis by acute RE, but it did not fully prevent acute RE from inducing an increase in c-myc expression or muscle protein synthesis. More importantly, we discovered that rapamycin only partially inhibits the hypertrophy that occurs in response to chronic RE. This is a fundamentally important observation because it indicates that rapamycin-sensitive mTOR-independent mechanisms contribute not only to RE-induced changes in protein synthesis, but also the induction of hypertrophy.
To the best of our knowledge, only three studies have investigated the effects of rapamycin on acute RE-induced changes in protein synthesis. Two of these studies have reported that rapamycin completely inhibited the RE-induced increase in protein synthesis42,43. However, one study conducted in humans did not demonstrate an effective inhibition of p70S6K phosphorylation42; probably due to difficulties in administering rapamycin to humans; moreover, the other study employed a rat squat model that does not induce muscle hypertrophy44. In the current study, we used an electrical stimulation-induced RE model, which is known to induce muscle adaptations that mimic those observed with RE in humans12,45. Our results demonstrate that RE induces an increase muscle protein synthesis and rapamycin only partially blocked this response. Our results are in accordance with those of another recent study in rodents that used electrical stimulation-induced eccentric contractions13. In addition, another recent study has reported increases in muscle protein synthesis after endurance exercise in the absence of rapamycin-sensitive mTOR signalling46, indicating that both rapamycin-sensitive mTOR-independent and rapamycin-sensitive mTOR-dependent processes regulate muscle protein synthesis following an acute bout of endurance exercise or RE.
A lot of previous studies have identified rapamycin-sensitive mTOR as mTORC1 42,47. However, similar to previous studies27,28, we observed a decrease in the phosphorylation of Akt Ser473, a marker of mTORC2 signalling, after chronic rapamycin administration. Thus, our observations are consistent with previous studies which have concluded that prolonged rapamycin administration can disrupt signaling by both mTORC1 and mTORC2. Therefore, the rapamycin-sensitive components of RE-induced hypertrophy might be not fully mediated by mTORC1. Moreover, previous studies have never excluded the possibility of unknown forms of rapamycin-sensitive mTOR. Therefore, it is important to consider that the effects of rapamycin may be mediated by more than just the inhibition of mTORC1.
Ribosome biogenesis is currently thought to regulate both muscle protein synthesis and hypertrophy and it can be regulated by both rapamycin-sensitive mTOR-dependent and rapamycin-sensitive mTOR-independent mechanisms32. Previous studies have shown that acute RE increase ribosome biogenesis in both human and rodent skeletal muscles13,36,48. Moreover, chronic RE in human has also shown to induce ribosome biogenesis49. In this study, consistent with these studies, we found that acute RE induced ribosome biogenesis and, to our knowledge, we observed for the first time that chronic RE in rodents also induced ribosome biogenesis as well as in human skeletal muscle. Moreover, we determined that rapamycin completely inhibited this effect. Combined, our results indicate that RE induces ribosome biogenesis through an rapamycin-sensitive mTOR-dependent mechanism and also suggest that ribosome biogenesis is part of the rapamycin-sensitive mTOR-dependent components through which chronic RE induces hypertrophy.
It is known that c-myc regulates protein synthesis and this effect is due, at least in part, to its ability to control the expression genes that regulate ribosome biogenesis32,50,51,52. Previous study has shown that mechanical overloading by SA increased association of c-myc at the rDNA promoter53. Moreover, recent studies have reported that c-myc-induced ribosomal biogenesis and protein synthesis are, in part, mediated by mTOR signalling51,52. Previous studies have also shown that RE increases c-myc expression in both human and rodent skeletal muscles13,16,35,36,48. Similarly, in the present study, we found that c-myc expression was increased after acute RE and this effect was rapamycin-insensitive, which is also in accordance with another study13. Therefore, it is possible that the RE induced increase in protein synthesis and subsequent hypertrophy are partially mediated through the rapamycin-sensitive mTOR-independent increase in c-myc expression. However, our results also indicate that the increase in c-myc is not sufficient to induce ribosome biogenesis when rapamycin-sensitive mTOR is inhibited. Thus, the exact role that c-myc plays in RE-induced hypertrophy remains to be defined and this will likely be a fruitful area for future studies.
The mechanisms that regulate c-myc expression in skeletal muscle are relatively unknown. However, similar to Akt phosphorylation (a marker of mTORC2 signalling), c-myc protein expression decreased after chronic rapamycin administration. Interestingly, previous studies have shown that mTORC2 can function as an upstream regulator of c-myc54,55, suggesting that c-myc expression in skeletal muscles might be regulated by mTORC2. Compared to mTORC1 or rapamycin-sensitive mTOR, the roles of mTORC2 in skeletal muscle are less understood. Nonetheless, previous studies have concluded that mTORC2 can regulate cell growth through a c-myc-dependent pathway54,56. Combined, these points suggest that the rapamycin-sensitive mTOR-independent increase in protein synthesis by acute RE and the rapamycin-sensitive mTOR-dependent inhibition of hypertrophy by chronic RE, might be mediated by changes in mTORC2/c-myc signalling.
In summary, one of the most important conclusions that can be drawn from this study is that rapamycin-sensitive mTOR-independent signalling events contribute to the hypertrophic effects of RE. This point raises questions about the possible rapamycin-sensitive mTOR-independent events that might be involved in this process. Obviously these mechanisms remain to be defined, but if future studies are able to define these mechanisms, then they will expose a previously unappreciated and major component, of the pathway through which RE promotes an increase in muscle mass.
Methods
Animal experimental procedures
All experimental procedures in this study were performed in accordance with the guidelines for the Care and Use of Laboratory Animals of Ritsumeikan University and approved by the Ethics Committee for Animal Experiments at Ritsumeikan University. Male Sprague–Dawley rats (~330 g, 11 weeks old) purchased from CLEA Japan (Tokyo, Japan) were housed for 1 week in an environment maintained at 22–24 °C with a 12 h light/dark cycle, with food and water ad libitum. After an overnight fast, under isoflurane anesthesia, the right gastrocnemius muscle of the animals was isometrically contracted (ten 3-s stimulation, with a 7-s interval between contraction for 5 sets with 3 min rest intervals) via percutaneous electrical stimulation as previously described12 and the left gastrocnemius muscle served as a control. The mTOR inhibitor rapamycin (1.5 mg/kg, 0.25 mg/ml in PBS containing 0.5%DMSO) or placebo (PBS containing 0.5%DMSO) was injected intraperitoneally 1 h before exercise. Muscle samples were obtained 1, 6 and 24 h after a bout of RE and 48 h after chronic RE (3 times a week for 4 weeks) (n = 5 in each time point). Tissues were rapidly frozen in liquid nitrogen and stored at −80 °C until use.
Western blotting
Frozen muscle samples were powdered using a beads crusher (μT-12, TAITEC, Saitama, Japan) and 20 mg of powdered samples were homogenized in 10 volumes of homogenization buffer containing 20 mM Tris-HCl (pH 7.5), 1% NP40, 1% sodium deoxycholate, 1 mM EDTA, 1 mM EGTA, 150 mM NaCl and HaltTM protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific, Waltham, MA). The homogenates were centrifuged at 10,000 × g for 10 min at 4 °C. The supernatant was collected and the protein concentration of each sample was determined using a Protein Assay Rapid kit (WAKO, Osaka, Japan). The samples were diluted in 3 × sample buffer (Cell Signaling Technology, Danvers, MA) and boiled at 95 °C for 5 min. Then, using 5–20% or 10–20% sodium dodecyl sulfate-polyacrylamide gradient gels, equal amounts of protein (20 or 50 μg) were separated using electrophoresis and subsequently transferred to ClearTrans® SP polyvinylidene difluoride membranes (WAKO). After transfer, the membranes were washed in Tris-buffered saline containing 0.1% Tween-20 (TBST) and blocked with RAPIDBLOCKTM SOLUTION (AMRESCO, Solon, OH) for 5 min at room temperature. Membranes were washed and incubated overnight at 4 °C with primary antibody. Antibodies against phospho-Akt (Ser473, cat#9271), total-Akt (cat#9272), phospho-p70S6K (Thr389, cat#9205), total-p70S6K (cat#2708), phospho-rpS6 (Ser240/244, cat#2215), total-rpS6 (cat#2217), phospho-4E-BP1 (Thr37/46, cat#9459), total-4E-BP1 (cat#9452), phospho-ERK1/2 (Thr202/Tyr204, cat#9101), total-ERK1/2 (cat#9102), phospho-p38 MAPK (Thr180/Tyr182, cat#9211), total p38 MAPK (cat#9212), phospho-ULK1 (Ser757, cat#14202), total-ULK1 (cat#8054), LC3 (cat#2775) and c-myc (cat#9402) were obtained from Cell Signaling Technology (Danvers, MA). The UBF (cat#sc-13125) and PGC-1α (cat#516557) antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA) and Millipore (Billerica, MA), respectively. The membranes were then washed again in TBST and incubated for 1 h at room temperature with the appropriate secondary antibody. The membranes were visualized using chemiluminescent reagents and the bands were detected using the C-DiGit Blot Scanner (LI-COR, Lincoln, NE). The membranes were then stained with Coomassie blue to verify equal loading in all lanes. Band intensities were quantified using Image Studio (LI-COR, Lincoln, NE).
Muscle protein synthesis
Muscle protein synthesis was measured by the in vivo SUnSET method57. Under the anaesthesia, 0.04 μmol puromycin/g body wt (Wako, Tokyo, Japan) diluted in a 0.02 M PBS stock solution was injected intraperitoneally and the gastrocnemius muscle was removed exactly 15 min after puromycin administration. Following homogenization as described above and centrifugation at 2,000 g for 3 min at 4 °C, the supernatant was collected and processed for western blotting. A mouse monoclonal anti-puromycin antibody (cat#MABE343, Millipore, Billerica, MA) was used to detect puromycin incorporation, which was evaluated as the sum of intensity of all protein bands in the western blot.
Real-time PCR
Total RNA was extracted from the powdered muscle sample using ISOGEN II (Nippon Gene, Tokyo, Japan) according to the manufacturer’s instructions. Total RNA concentrations were measured using a Synergy HT (BioTek, Winooski, VT) and 500 ng of total RNA was reverse transcribed into cDNA using a High Capacity cDNA RT kit (Applied Biosystems, Foster City, CA).
Real-time PCR was performed using a TaqMan® Fast Universal PCR Master Mix (Applied Biosystems) and a Mini Optiocon Real-Time PCR System (Bio-Rad, Hercules, CA). TaqMan® Gene Expression Assays (Applied Biosystems) were used to measure the expression of PGC-1α (Rn00580241_m1), c-myc (Rn00561507_m1), β-actin (Rn00667869_m1), GAPDH (Rn01775763_g1) and β2-microgloblin (Rn00560865_m1). In a rodent model of resistance exercise used in this study, GAPDH and β2-microgloblin did not change after RE and rapamycin administration while β-actin significantly increased after RE (Fig. S1). Furthermore, GAPDH was more stable than β2-microgloblin. Therefore, GAPDH was used as the reference gene in this study.
Ribosomal RNA content analysis
Five microliters of total RNA solution (25 μg muscle/μl TE) was mixed with 1 μl GR Red Loading Buffer (Biocraft, Tokyo, Japan) and 1 μl 100% glycerol and then loaded on a 1% agarose gel in TBE buffer. The band intensities of 18S and 28S ribosomal RNA (rRNA) were measured using ImageJ software (NIH, Bethesda, MD).
Myofiber CSA analysis
Mid-belly cross-sections (5 μm thick) were fixed with 10% neutral buffered formalin for 30 min at room temperature and then sections were stained with hematoxylin and eosin to measure the myofiber cross-sectional area (CSA). The CSA of approximately 300 randomly selected myofibers per muscle were measured by a blinded investigator with ImageJ software (NIH, Bethesda, MD).
Statistical analyses
Two-way ANOVA (rapamycin × exercise) was used to evaluate changes in the phosphorylated protein, total protein, mRNA, rRNA, muscle wet weight and muscle fiber CSA. Post-hoc analyses were performed using t-tests, with a Benjamini and Hochberg false discovery rate correction for multiple comparisons when significant main effects or interactions were found. Relative changes were compared between groups by paired t-test. The level of significance was set at P < 0.05.
Additional Information
How to cite this article: Ogasawara, R. et al. The role of mTOR signalling in the regulation of skeletal muscle mass in a rodent model of resistance exercise. Sci. Rep. 6, 31142; doi: 10.1038/srep31142 (2016).
References
Laplante, M. & Sabatini, D. M. mTOR signaling in growth control and disease. Cell 149, 274–293 (2012).
Sengupta, S., Peterson, T. R. & Sabatini, D. M. Regulation of the mTOR complex 1 pathway by nutrients, growth factors and stress. Mol Cell 40, 310–322 (2010).
Benjamin, D., Colombi, M., Moroni, C. & Hall, M. N. Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat Rev Drug Discov 10, 868–880 (2011).
Kang, S. A. et al. mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science 341, 1236566 (2013).
Zhou, H. & Huang, S. The complexes of mammalian target of rapamycin. Curr Protein Pept Sci 11, 409–424 (2010).
Patursky-Polischuk, I. et al. Reassessment of the role of TSC, mTORC1 and microRNAs in amino acids-meditated translational control of TOP mRNAs. PLoS One 9, e109410 (2014).
Goodman, C. A. et al. The role of skeletal muscle mTOR in the regulation of mechanical load-induced growth. J Physiol 589, 5485–5501 (2011).
Bodine, S. C. et al. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol 3, 1014–1019 (2001).
Ogasawara, R., Sato, K., Higashida, K., Nakazato, K. & Fujita, S. Ursolic acid stimulates mTORC1 signaling after resistance exercise in rat skeletal muscle. American journal of physiology. Endocrinology and metabolism 305, E760–E765 (2013).
Wilkinson, S. B. et al. Differential effects of resistance and endurance exercise in the fed state on signalling molecule phosphorylation and protein synthesis in human muscle. J Physiol 586, 3701–3717 (2008).
Dreyer, H. C. et al. Resistance exercise increases AMPK activity and reduces 4E-BP1 phosphorylation and protein synthesis in human skeletal muscle. J Physiol 576, 613–624 (2006).
Ogasawara, R., Sato, K., Matsutani, K., Nakazato, K. & Fujita, S. The order of concurrent endurance and resistance exercise modifies mTOR signaling and protein synthesis in rat skeletal muscle. American journal of physiology. Endocrinology and metabolism 306, E1155–E1162 (2014).
West, D. W. et al. Acute resistance exercise activates rapamycin-sensitive and -insensitive mechanisms that control translational activity and capacity in skeletal muscle. J Physiol 594, 453–468 (2016).
Cunningham, J. T. et al. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 450, 736–740 (2007).
Atherton, P. J. et al. Selective activation of AMPK-PGC-1alpha or PKB-TSC2-mTOR signaling can explain specific adaptive responses to endurance or resistance training-like electrical muscle stimulation. FASEB J 19, 786–788 (2005).
Apro, W., Wang, L., Ponten, M., Blomstrand, E. & Sahlin, K. Resistance exercise induced mTORC1 signaling is not impaired by subsequent endurance exercise in human skeletal muscle. American journal of physiology. Endocrinology and metabolism 305, E22–E32 (2013).
Hill, K. M., Stathis, C. G., Grinfeld, E., Hayes, A. & McAinch, A. J. Co-ingestion of carbohydrate and whey protein isolates enhance PGC-1alpha mRNA expression: a randomised, single blind, cross over study. J Int Soc Sports Nutr 10, 8 (2013).
Wang, L., Mascher, H., Psilander, N., Blomstrand, E. & Sahlin, K. Resistance exercise enhances the molecular signaling of mitochondrial biogenesis induced by endurance exercise in human skeletal muscle. J Appl Physiol (1985) 111, 1335–1344 (2011).
Rommel, C. et al. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat Cell Biol 3, 1009–1013 (2001).
Morley, S. J., Coldwell, M. J. & Clemens, M. J. Initiation factor modifications in the preapoptotic phase. Cell Death Differ 12, 571–584 (2005).
Ferguson, G., Mothe-Satney, I. & Lawrence, J. C., Jr. Ser-64 and Ser-111 in PHAS-I are dispensable for insulin-stimulated dissociation from eIF4E. J Biol Chem 278, 47459–47465 (2003).
Mothe-Satney, I., Yang, D., Fadden, P., Haystead, T. A. & Lawrence, J. C., Jr. Multiple mechanisms control phosphorylation of PHAS-I in five (S/T)P sites that govern translational repression. Mol Cell Biol 20, 3558–3567 (2000).
Miyazaki, M., McCarthy, J. J., Fedele, M. J. & Esser, K. A. Early activation of mTORC1 signalling in response to mechanical overload is independent of phosphoinositide 3-kinase/Akt signalling. J Physiol 589, 1831–1846 (2011).
You, J. S., Frey, J. W. & Hornberger, T. A. Mechanical stimulation induces mTOR signaling via an ERK-independent mechanism: implications for a direct activation of mTOR by phosphatidic acid. PLoS One 7, e47258 (2012).
Kim, J., Kundu, M., Viollet, B. & Guan, K. L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13, 132–141 (2011).
Garelick, M. G. et al. Chronic rapamycin treatment or lack of S6K1 does not reduce ribosome activity in vivo. Cell Cycle 12, 2493–2504 (2013).
Lamming, D. W. et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 335, 1638–1643 (2012).
Sarbassov, D. D. et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 22, 159–168 (2006).
Kabeya, Y. et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19, 5720–5728 (2000).
Kang, C., Goodman, C. A., Hornberger, T. A. & Ji, L. L. PGC-1alpha overexpression by in vivo transfection attenuates mitochondrial deterioration of skeletal muscle caused by immobilization. FASEB J 29, 4092–4106 (2015).
Sandri, M. et al. PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc Natl Acad Sci USA 103, 16260–16265 (2006).
Chaillou, T., Kirby, T. J. & McCarthy, J. J. Ribosome biogenesis: emerging evidence for a central role in the regulation of skeletal muscle mass. J Cell Physiol 229, 1584–1594 (2014).
Iadevaia, V., Huo, Y., Zhang, Z., Foster, L. J. & Proud, C. G. Roles of the mammalian target of rapamycin, mTOR, in controlling ribosome biogenesis and protein synthesis. Biochem Soc Trans 40, 168–172 (2012).
Hannan, K. M. et al. mTOR-dependent regulation of ribosomal gene transcription requires S6K1 and is mediated by phosphorylation of the carboxy-terminal activation domain of the nucleolar transcription factor UBF. Mol Cell Biol 23, 8862–8877 (2003).
Drummond, M. J. et al. Expression of growth-related genes in young and older human skeletal muscle following an acute stimulation of protein synthesis. J Appl Physiol (1985) 106, 1403–1411 (2009).
Figueiredo, V. C. et al. Impact of resistance exercise on ribosome biogenesis is acutely regulated by post-exercise recovery strategies. Physiol Rep 4, e12670 (2016).
Teleman, A. A., Hietakangas, V., Sayadian, A. C. & Cohen, S. M. Nutritional control of protein biosynthetic capacity by insulin via Myc in Drosophila. Cell Metab 7, 21–32 (2008).
Phillips, S. M., Tipton, K. D., Aarsland, A., Wolf, S. E. & Wolfe, R. R. Mixed muscle protein synthesis and breakdown after resistance exercise in humans. Am J Physiol 273, E99–107 (1997).
Fry, C. S. et al. Aging impairs contraction-induced human skeletal muscle mTORC1 signaling and protein synthesis. Skelet Muscle 1, 11 (2011).
Brook, M. S. et al. Skeletal muscle hypertrophy adaptations predominate in the early stages of resistance exercise training, matching deuterium oxide-derived measures of muscle protein synthesis and mechanistic target of rapamycin complex 1 signaling. FASEB J 29, 4485–4496 (2015).
Egan, B. & Zierath, J. R. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab 17, 162–184 (2013).
Drummond, M. J. et al. Rapamycin administration in humans blocks the contraction-induced increase in skeletal muscle protein synthesis. J Physiol 587, 1535–1546 (2009).
Kubica, N., Bolster, D. R., Farrell, P. A., Kimball, S. R. & Jefferson, L. S. Resistance exercise increases muscle protein synthesis and translation of eukaryotic initiation factor 2Bepsilon mRNA in a mammalian target of rapamycin-dependent manner. J Biol Chem 280, 7570–7580 (2005).
Krisan, A. D. et al. Resistance training enhances components of the insulin signaling cascade in normal and high-fat-fed rodent skeletal muscle. J Appl Physiol (1985) 96, 1691–1700 (2004).
Ogasawara, R. et al. mTOR signaling response to resistance exercise is altered by chronic resistance training and detraining in skeletal muscle. J Appl Physiol (1985) 114, 934–940 (2013).
Philp, A. et al. Rapamycin does not prevent increases in myofibrillar or mitochondrial protein synthesis following endurance exercise. J Physiol 593, 4275–4284 (2015).
Ramos, F. J. et al. Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function and extends survival. Sci Transl Med 4, 144ra103 (2012).
Mobley, C. B. et al. Comparative effects of whey protein versus L-leucine on skeletal muscle protein synthesis and markers of ribosome biogenesis following resistance exercise. Amino Acids 48, 733–750 (2016).
Figueiredo, V. C. et al. Ribosome biogenesis adaptation in resistance training-induced human skeletal muscle hypertrophy. American journal of physiology. Endocrinology and metabolism 309, E72–E83 (2015).
Dang, C. V. M. Y. C., metabolism, cell growth and tumorigenesis. Cold Spring Harb Perspect Med 3, a014217 (2013).
Elkon, R. et al. Myc coordinates transcription and translation to enhance transformation and suppress invasiveness. EMBO Rep 16, 1723–1736 (2015).
Pourdehnad, M. et al. Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. Proc Natl Acad Sci USA 110, 11988–11993 (2013).
von Walden, F., Casagrande, V., Ostlund Farrants, A. K. & Nader, G. A. Mechanical loading induces the expression of a Pol I regulon at the onset of skeletal muscle hypertrophy. Am J Physiol Cell Physiol 302, C1523–C1530 (2012).
Kuo, Y., Huang, H., Cai, T. & Wang, T. Target of Rapamycin Complex 2 regulates cell growth via Myc in Drosophila. Sci Rep 5, 10339 (2015).
Masui, K. et al. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab 18, 726–739 (2013).
Carayol, N. et al. Critical roles for mTORC2- and rapamycin-insensitive mTORC1-complexes in growth and survival of BCR-ABL-expressing leukemic cells. Proc Natl Acad Sci USA 107, 12469–12474 (2010).
Goodman, C. A. et al. Novel insights into the regulation of skeletal muscle protein synthesis as revealed by a new nonradioactive in vivo technique. FASEB J 25, 1028–1039 (2011).
Acknowledgements
This work was supported by JSPS KAKENHI Grant No. 26702028 (to R. Ogasawara) and No. 15H03078 (to N. Ishii) as well as the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Number AR057347 (to T. Hornberger). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
Conception and design of the experiments: R.O., S.F., T.A.H., K.N. and I.N. Collection, analysis and interpretation of data: R.O., Y.K. and Y.M. Drafting the article or revising it critically for important intellectual content: R.O., S.F. and T.A.H. All authors approved the final version of the manuscript and listed qualify for authorship.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Ogasawara, R., Fujita, S., Hornberger, T. et al. The role of mTOR signalling in the regulation of skeletal muscle mass in a rodent model of resistance exercise. Sci Rep 6, 31142 (2016). https://doi.org/10.1038/srep31142
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep31142
This article is cited by
-
Low-frequency electrical stimulation of bilateral hind legs by belt electrodes is effective for preventing denervation-induced atrophies in multiple skeletal muscle groups in rats
Scientific Reports (2022)
-
β2-adrenergic receptor agonist counteracts skeletal muscle atrophy and oxidative stress in uremic mice
Scientific Reports (2021)
-
Changes in UPR-PERK pathway and muscle hypertrophy following resistance training and creatine supplementation in rats
Journal of Physiology and Biochemistry (2021)
-
The distribution of eukaryotic initiation factor 4E after bouts of resistance exercise is altered by shortening of recovery periods
The Journal of Physiological Sciences (2020)
-
HS-1793 protects C2C12 cells from oxidative stress via mitochondrial function regulation
Molecular & Cellular Toxicology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
