
Abstract
Age-specific genetic and antigenic variations of influenza viruses have not been documented in tropical and subtropical regions. We implemented a systematic surveillance program in two tertiary hospitals in Hong Kong Island, to collect 112 A(H1N1)pdm09 and 254 A(H3N2) positive specimens from 2013 to 2014. Of these, 56 and 72 were identified as genetic variants of the WHO recommended vaccine composition strains, respectively. A subset of these genetic variants was selected for hemagglutination-inhibition (HI) tests, but none appeared to be antigenic variants of the vaccine composition strains. We also found that genetic and antigenicity variations were similar across sex and age groups of ≤18 yrs, 18 to 65 yrs, and ≥65 yrs. Our findings suggest that none of the age groups led other age groups in genetic evolution of influenza virus A strains. Future studies from different regions and longer study periods are needed to further investigate the age and sex heterogeneity of influenza viruses.
Similar content being viewed by others

Introduction
Influenza viruses undergo frequent antigenic drift, and cause winter epidemics in temperate regions and year-long circulation in tropical and subtropical regions. The World Health Organization (WHO) has established the Global Influenza Surveillance and Response System (GISRS) to track the antigenic change of influenza viruses worldwide, with the aim of guiding the selection of suitable influenza candidate vaccine viruses1. In Hong Kong, the influenza surveillance network managed by the Department of Health routinely selected potentially drifted specimens for genetic and antigenic characterization. However, temporal variations of genetic and antigenicity characteristics have never been studied, largely due to the relatively small number of specimens which were subjectively selected based on antigenic drifts and disease severity, rather than from a representative sample of strains currently circulating in the whole population.
Recent large-scale phylogenetic studies have demonstrated co-circulation of different influenza strains in tropical and subtropical regions of Southeast and East Asia, but it remains controversial whether novel viruses first originated from these regions2,3,4. Age discrepancy in susceptibility to different virus subtypes has also been reported5. For example, children were found more likely to be infected by A(H1N1)pdm09 as compared to adults, whereas A(H3N2) tended to affect more adults. However, age information has seldom been incorporated into genetic and antigenic surveillance, and to this date few studies have assessed the age difference in terms of genetic and antigenic variations.
In this study, we implemented a systematic surveillance program during 2013–2014, to randomly select a sample of positive specimens of children (aged below 18 years) and adults, from two tertiary hospitals in the Hong Kong Island each week. The systematically collected specimens were expected representative of concurrent circulating strains in the population6 and could also allow the investigation of temporal variations of genetic and antigenicity characteristics of concurrently circulating influenza viruses.
Results
Figure 1 shows the total number of specimens collected by the Queen Mary Hospital microbiology laboratory each week and also the number of specimens selected for sequencing in our study. The demographic characteristics of patients whose specimens were selected for sequencing and Hemagglutinin Inhibition (HI) tests were similar to those unselected patients, except that the percentage of children was slightly higher in the selected than in unselected group for A(H3N2) samples (Table 1). We speculate that this could be due to small numbers of positive specimens in this age group in some weeks.

Weekly numbers of collected samples (blue) and those selected for sequencing (red), 2013–2014.
Among 366 sequenced strains during 2013–2014, the proportion of amino acid mutations of the HA1 polypeptide for A(H1N1)pdm09 and A(H3N2) was 1.8–6.5% and 1.1–4.5%, respectively. During the study period, the maximum proportion of amino acid mutation per week remained at a relatively low level for subtype A(H1N1)pdm09, but a clear increasing trend was observed for A(H3N2) (Fig. 2).

Weekly maximum proportion of amino acid mutations for A(H1N1)pdm09 and A(H3N2), 2013–2014.
In the HI tests, over 99% of the isolates had HI titre over 320 for both A(H1N1)pdm09 and A(H3N2), suggesting that no obvious antigenic variants were detected in this study. There were no statistically significant differences in HI titres across age groups of ≤18 yrs, 18 to 65 yrs, and ≥65 yrs [A(H1N1)pdm09 p = 0.420; A(H3N2) p = 0.798]. No gender difference was found in HI titres [A(H1N1)pdm09 p = 0.304; A(H3N2) p = 0.294] (Table 2). For genetic variations, no statistically significant differences were found across different age or gender groups, in terms of amino acid mutations (Table 2).
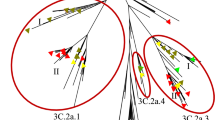
Two phylogenetic trees of the HA1 polypeptide sequences from all the A(H1N1)pdm09 and A(H3N2) sequences collected in this study, together with representative strains downloaded from the GISAID EpiFlu™ databank are shown in Supplementary Information. The phylogenetic tree of A(H1N1)pdm09 was separated into eight major genetic clades7. All of the A(H1N1)pdm09 strains that were isolated in this study belong to the clade 6 and 7 with a signature substitution of S220T in HA1 polypeptide. Mutations P100S, D114N, S220T, R240Q, and I338V were observed in over 90% of these A(H1N1)pdm09 isolates. Similarly, all the A(H3N2) isolates fell into the clade 3 and 4. There were 7 fixed amino acid mutations (H9Y, Q49R, N161S, Q172H, V202G, Y235S, and N294K) detected in nearly all the A(H3N2) strains (Table 3).
Discussion
Influenza A viruses are characterized by a high mutation rate2. Particularly, HA1 polypeptide exhibited higher sequence variations than HA2, which may be due to selection pressure from the immune system8,9,10. Previous studies had reported that 1% and 0.8% of amino acids in HA1 domain changed per year for H1 and H3, respectively11. In our study, although genetic variants were frequently detected, none of these genetic variants were identified as antigenic drift strains from the vaccine composition strains of A/California/07/2009 (H1N1) and A/Victoria/361/2011 (H3N2) recommended during the same season. These findings were consistent with those reported by USCDC that over 90% of A(H1N1)pdm09 and A(H3N2) strains isolated in the 2013–14 season were similar to the vaccine component strains12. Interestingly, an antigenic drift strain A/Switzerland/9715293/2013 (H3N2) appeared since January 2015 and caused a relatively severe winter epidemic13. Later WHO recommended this new strain as the vaccine composition strain for A(H3N2) in the 2015–2016 season for the Southern Hemisphere14. We further compared our sample strains with this new emerging strain and found that some of our sample strains clustered with the A/Switzerland/9715293/2013 (H3N2) into the sub-clade 3C.3 (see phylogenetic tree (b). in Supplementary Information). Our findings of an increasing trend in A(H3N2) mutation rates echo the findings of Shih and colleagues15. They also suggested that antigenic change of HA1 appears to be ongoing most of the time with occasional large changes. However, a mutation confers limited antigenic drift and its frequency increases only to a low level in the majority of cases, so that a linear increasing trend of mutations, possibly due to slow accumulation process, may be observed. These findings suggest that the influenza viruses evolve in a gradual process, and mutations at some critical sites that gain the ability of escape from the prior immunity could be crucial for emergence or migration of novel strains into Hong Kong. A future study with a longer study period using our systematic sampling approach could shed more light on whether influenza viruses in Hong Kong evolve through persistent local transmission or repeated introduction from the temperate regions.
Our findings further demonstrated the genetic variability of the influenza A viruses after the pandemic in 200916,17. Most A(H1N1)pdm09 isolates obtained during 2013 to 2014 belong to the clade 6 and 7 (see phylogenetic tree (a). in Supplementary Information). This echoes the previous findings of the co-circulation of these clades and high genetic diversity of influenza virus A(H1N1)pdm09 during the 2009–2013 seasons7,18,19. Mutations of P100S, D114N, S202T, S220T, and I338V had frequently been identified in temperate regions including China, France, and Canada, as well as in tropical region of Cuba and Thailand in the same period, whereas R240Q mutation was reported for the first time18,20,21,22,23. The A(H3N2) strains isolated in Hong Kong during 2013–2014 fell into the A/Victoria/361/2011 genetic clade, and particularly into the clade 3. 97.6% (248/254) of sequences were closely related to the sub-clade 3C and similar to A/Florida/21/2013 strain. The rest (6/254) were grouped into the sub-clade 3B, which were similar to A/Mahajanga/3628/2012 strain (see phylogenetic tree (b). in Supplementary Information). Mutations Q49R, N161S, Q172H, V202G, Y235S, and N294K were also reported in other countries20,21,22,24. To our surprise, although genetic changes of influenza viruses were continuously observed, none of them appeared antigenic variants from the vaccine composition strains. This could be due to the relatively short study period and antigenic variants emerging at the end of the period could have been missed. Future studies with intensive collection of sequence data over a longer period, together with more knowledge on the association of genetic and antigenic changes, could make possible the development of a forecast model on newly emerging antigenic variants. Several models have been adopted to predict influenza virus activity, such as the Seasonal Autoregressive Integrated Moving Average (SARIMA) model, Poisson model and SIR model, but most of these only consider environmental factors as the drivers for influenza seasonality25,26,27,28,29. Our previous study in Hong Kong also demonstrated that the switch of influenza seasonality from one-peak to two-peak patterns coincided with emergence of new A(H3N2) strains, suggesting a link between antigenic change and flu seasonality30. Therefore, one future direction of forecast model development could be incorporation of epidemiological and molecular data, similar to those collected in this study, in more sophisticated mathematical models.
Our study also investigated age and gender differences in antigenic and genetic variations. Previous studies demonstrated that the A(H1N1)pdm09 virus was associated with an age shift towards young people31,32. But inconsistent patterns of gender differences were found across different regions. A study conducted in Australia found more fatal cases were male33 while a Canadian study reported that the relative risk of influenza associated deaths in adult women is 1.5 times the risk in men34. Some studies observed relatively higher hospitalization risks associated with seasonal influenza A(H3N2) in older men than older women although the difference was not statistically significant35,36. Boys were found to have a weaker immune response against influenza infections than girls37, but this gender difference gradually vanished when they grow into adulthood38. In this study we did not find any significant differences across age and gender groups in genetic and antigenic variants of influenza A virus. We have previously analyzed age-specific sentinel surveillance data of laboratory confirmed influenza in Hong Kong, and found highly synchronized influenza seasonal epidemics across age groups in terms of the seasonal patterns, epidemic timing and durations5. Previous studies proposed that influenza seasonal variations could be driven by seasonal change of environmental factors (temperature, humidity, etc.), host immunity and antigenic change of viruses25,39. Hence this study provides more evidence to support the age synchrony in antigenic evolution of influenza viruses, which could partially explain the findings of our previous ecological study. Furthermore, our study has exemplified the plausibility of collecting individual demographical data during laboratory surveillance, and a better understanding could be achieved on age and gender synchrony of influenza infections when such data become available in a wider range.
There are several limitations in our study. First, the majority of samples were taken from inpatients admitted into two public hospitals. However, there is no strong evidence to suggest that influenza A viruses circulating in the community are different from those isolated from inpatients40,41. Second, we only sequenced the HA1 polypeptide of HA due to the limited budget. However, HA2 polypeptide of HA, neuraminidase (NA) and other proteins could also affect the genetic and antigenic characteristics of influenza A viruses, which have not been explored in this study. Third, HI tests are often criticized for its low sensitivity as compared to other tests such as micro-neutralization tests, which might lead to relatively low efficiency in detecting antigenic variants. Fourth, there are other available measurements for antigenic distance, such as antigenic maps which requires pairwise HI tests across different reference strains. However, given the high titres in HI tests, it is unlikely to observe obvious differences between these newly isolated strains if multiple reference strains are used. When potential antigenic variants appear in future studies, more sensitive tests and additional reference strains should be considered.
In conclusion, by using an age-stratified random selection strategy, we detected some genetic variants in Hong Kong, but no obvious antigenic variants from the WHO vaccine strains during the study period of 2013 to 2014. Further studies are warranted to integrate both antigenic profiles of locally circulating strains and other factors, allowing us to gain a better understanding on the mechanism of seasonal influenza epidemics.
Materials and Methods
Sample collection
The microbiology laboratory of Queen Mary Hospital routinely collects the nasopharyngeal aspirates (NPA) or nasopharyngeal swabs (NPS) from patients who are admitted into two tertiary hospitals (Queen Mary Hospital and Pamela Youde Nethersole Eastern Hospital) in Hong Kong Island, with influenza like symptoms (temperature >38 °C, cough and/or sore throat). These two public hospitals accept approximately 80% of all hospitalizations in Hong Kong Island which has a total population of one million42. Both NPA and NPS specimens were divided into two aliquots for direct immunofluorescence (IF) test and RT-PCR respectively. Direct IF D3 Ultra 8 DFA respiratory virus screening and identification kit (Diagnostic Hybrid, OH) was used to detect antigens of the specimens, and then viewed at a magnification of 400 under epifluorescent illumination of an Eurostar III plus (EUROIMMUN AG, Lübeck, Germany) fluorescence microscope43. Influenza A positive specimens were subsequently sub-typed into A(H3N2) and A(H1N1)pdm09 by RT-PCR44. Each week up to five influenza A positive specimens were selected for genetic sequencing between January 2013 to December 2014. If there were more than two positive isolates for each of two subtypes A(H1N1)pdm09 or A(H3N2), two were randomly selected from the subtype with less total number of positive specimens and three from the other. If one or two positive specimens were found for one subtype in that week, all of them were selected, and the rest were from the other subtype to get a total of five specimens. If there were five or less positive specimens in one week, all of them were selected. This selection procedure is shown in Fig. 3. In order to assess the age difference, positive specimens were randomly selected from different age groups of ≤18 yrs, 18 to 65 yrs, and ≥65 yrs, if any. For example, if there were three specimens to be selected from A(H3N2) positive specimens from three age groups, one would be randomly selected from each age group. If there were only two age groups, then one extra sample would be selected from the age group with more positive specimens. If there were five or less positive specimens in one week, all of them would be selected. Because in some weeks there were few or zero positive specimens isolated in some age groups, age distribution might be different between selected and unselected specimens due to the high chance of being selected in these age groups.

Flow chart of sample selection in this study.
The selected A(H1N1)pdm09 and A(H3N2) positive specimens were then isolated by cell culture on MDCK cells45, and subsequently sequenced using the Big Dye Terminator v3.1 Cycle Sequencing Kit. Only HA1 polypeptide was sequenced because mutations frequently occur in this polypeptide. It contains 329 amino acids, of which131 are located at or close to five antibody epitopes and are the critical sites determining the virus antigenicity10,46. Bioedit version 7.2.5 was used to assemble and edit sequences (http://www.mbio.ncsu.edu/bioedit/bioedit.html). We chose the representative isolates which had mutations identified from genetic sequencing, for further hemagglutinin-inhibition (HI) tests. The reference strains used in the HI tests were A/California/07/2009 (H1N1) and A/Victoria/361/2011 (H3N2), which were WHO recommended vaccine composition strains in the 2012/13 and 2013/14 seasons for the Northern Hemisphere. The HI tests started with 1:10 dilution of antiserum and the HI titre less than 40 was regarded as the threshold to define the antigenic variants47. Phylogenetic trees were produced by the G T R + G + Γ model of amino acid substitutions incorporated in the MrBayes v.3.2.5 software and visualized with the FigTree v.1.4.248. These gene sequences were compared with a sample of gene sequences that had been globally isolated in recent years, from the GISAID EpiFlu database (http://www.platform.gisaid.org) (Table S1). These reference strains selected from the database were representative of the major genetic clades defined by WHO7.
During the study period of January 1st, 2013 to December 31st, 2014, a total of 2,115 patients admitted to the two public hospitals in Hong Kong Island presented with influenza like symptoms (temperature >38 °C, cough and/or sore throat). Of these, 491 were confirmed infected with A(H1N1)pdm09 and 803 with A(H3N2). We randomly selected 366 positive specimens for sequencing [112 A(H1N1)pdm09 positive and 254A(H3N2) positive]. 56 of A(H1N1)pdm09 and 72 of A(H3N2) sequenced specimens with potential meaningful point mutations were further selected for HI test.
Statistical analysis
Genetic variation was measured by the proportion of amino acid mutations of the HA1 polypeptide relative to the reference strains, for A(H1N1)pdm09 and A(H3N2) subtypes, respectively21. The proportion of amino acid mutations was defined as

Antigenic variation was assessed by the HI tests against the reference strains. The Fisher exact tests were used to compare the rates across age and gender groups. In this study statistical significance was defined as p < 0.05. Statistical analyses were performed using R software version 3.0.049.
Ethical approval for this study was obtained from the Institutional Review Board of the University of Hong Kong/Hospital Authority Hong Kong West Cluster (Reference Number: UW 11-290). Informed consent was obtained from all subjects. All experiments were carried out in accordance with the approved guidelines.
Additional Information
How to cite this article: Cao, P. et al. Age-specific genetic and antigenic variations of influenza A viruses in Hong Kong, 2013–2014. Sci. Rep. 6, 30260; doi: 10.1038/srep30260 (2016).
References
Ampofo, W. K. et al. Improving influenza vaccine virus selection Report of a WHO informal consultation held at WHO headquarters, Geneva, Switzerland, 14–16 June 2010. Influenza Other Respir Viruses 6, 142–152, 10.1111/j.1750-2659.2011.00277.x (2012).
Rambaut, A. et al. The genomic and epidemiological dynamics of human influenza A virus. Nature 453, 615–619, 10.1038/nature06945 (2008).
Russell, C. A. et al. The global circulation of seasonal influenza A (H3N2) viruses. Science. 320, 340–346, 10.1126/science.1154137 (2008).
Bahl, J. et al. Temporally structured metapopulation dynamics and persistence of influenza A H3N2 virus in humans. Proc Natl Acad Sci USA 108, 19359–19364 (2011).
Yang, L. et al. Age-specific epidemic waves of influenza and respiratory syncytial virus in a subtropical city. Sci. Rep. 5, 10.1038/srep10390 (2015).
Van Kerkhove, M. D. et al. Studies needed to address public health challenges of the 2009 H1N1 influenza pandemic: insights from modeling. PLoS Med. 7, 711 (2010).
Klimov, A. I. et al. WHO recommendations for the viruses to be used in the 2012 Southern Hemisphere Influenza Vaccine: epidemiology, antigenic and genetic characteristics of influenza A (H1N1) pdm09, A (H3N2) and B influenza viruses collected from February to September 2011. Vaccine 30, 6461–6471 (2012).
Gupta, V., Earl, D. J. & Deem, M. W. Quantifying influenza vaccine efficacy and antigenic distance. Vaccine 24, 3881–3888, 10.1016/j.vaccine.2006.01.010 (2006).
Skehel, J. J. & Wiley, D. C. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem. 69, 531–569 (2000).
Wiley, D. C., Wilson, I. A. & Skehel, J. J. Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature 289, 373–378 (1981).
Wilson, I. A. & Cox, N. J. Structural basis of immune recognition of influenza virus hemagglutinin. Annu Rev Immunol. 8, 737–771, 10.1146/annurev.iy.08.040190.003513 (1990).
Centers for Disease Control and Prevention. Flu Activity During the 2013–2014 Season. (2014). Avaiable at: http://www.cdc.gov/flu/pastseasons/1314season.htm. (Accessed: 20th January (2015).
Ren, D. & Cheung, E. Deadly Hong Kong flu outbreak ‘won’t spread to mainland’. says disease expert. (2015). Avaiable at: http://www.scmp.com/news/china/article/1729166/china-top-respiratory-doctor-says-hong-kongs-deadly-h3n2-outbreak-could. (Accessed: 5th March (2015).
World Health Organization. Recommended composition of influenza virus vaccines for use in the 2015 southern hemisphere influenza season. (2015). Avaiable at: http://www.who.int/influenza/vaccines/virus/recommendations/2015_south/en/. (Accessed: 25th September, 2014).
Shih, A. C. C., Hsiao, T. C., Ho, M. S. & Li, W. H. Simultaneous amino acid substitutions at antigenic sites drive influenza A hemagglutinin evolution. Proc Natl Acad Sci USA 104, 6283–6288 (2007).
Fantoni, A. et al. Genetic drift of influenza A(H3N2) viruses during two consecutive seasons in 2011–2013 in Corsica, France. J Med Virol. 86, 585–591, 10.1002/jmv.23745 (2014).
Linderman, S. L. et al. Potential antigenic explanation for atypical H1N1 infections among middle-aged adults during the 2013–2014 influenza season. Proc Natl Acad Sci USA 111, 15798–15803 (2014).
Makkoch, J. et al. Whole genome characterization, phylogenetic and genome signature analysis of human pandemic H1N1 virus in Thailand, 2009–2012. PloS One. 7, e51275 (2012).
Arencibia, A. et al. New genetic variants of influenza A (H1N1) pdm09 detected in Cuba during 2011–2013. Infect Genet Evol. 32, 322–326 (2015).
Fantoni, A. et al. Genetic drift of influenza A (H3N2) viruses during two consecutive seasons in 2011–2013 in Corsica, France. J Med Virol. 86, 585–591, 10.1002/jmv.23745 (2014).
Fang, Q. et al. Molecular epidemiology and evolution of A (H1N1) pdm09 and H3N2 virus during winter 2012–2013 in Beijing, China. Infect Genet Evol. 26, 228–240 (2014).
Arencibia, A. et al. Genetic drift of hemagglutinin (HA) of influenza A (H3N2) viruses circulating in Cuba between 2011 and 2013. Infect Genet Evol. 28, 58–61 (2014).
Graham, M. et al. Nationwide molecular surveillance of pandemic H1N1 influenza A virus genomes: Canada, 2009. PLoS One. 6, e16087 (2011).
Bragstad, K. et al. Low vaccine effectiveness against influenza A (H3N2) virus among elderly people in Denmark in 2012/13–a rapid epidemiological and virological assessment. Euro Surveill 18, 11–17 (2013).
Lowen, A. C., Mubareka, S., Steel, J. & Palese, P. Influenza virus transmission is dependent on relative humidity and temperature. PLoS Pathog. 3, e151 (2007).
Shaman, J., Pitzer, V. E., Viboud, C., Grenfell, B. T. & Lipsitch, M. Absolute humidity and the seasonal onset of influenza in the continental United States. PLoS Biol. 8, e1000316, 10.1371/journal.pbio.1000316 (2010).
Shaman, J. & Kohn, M. Absolute humidity modulates influenza survival, transmission, and seasonality. Proc Natl Acad Sci USA 106, 3243–3248, 10.1073/pnas.0806852106 (2009).
Zuk, T., Rakowski, F. & Radomski, J. P. A model of influenza virus spread as a function of temperature and humidity. Comput Biol Chem. 33, 176–180 (2009).
Soebiyanto, R. P., Adimi, F. & Kiang, R. K. Modeling and predicting seasonal influenza transmission in warm regions using climatological parameters. PloS One. 5, e9450 (2010).
Yang, L. et al. Synchrony of clinical and laboratory surveillance for influenza in Hong Kong. PloS one 3, e1399 (2008).
Wong, J. Y. et al. Infection fatality risk of the pandemic A (H1N1) 2009 virus in Hong Kong. Am J Epidemiol. 177, 834–840 (2013).
Yang, L. et al. Hospitalisation associated with the 2009 H1N1 pandemic and seasonal influenza in Hong Kong, 2005 to 2010. Euro Surveill 17, 20317 (2012).
Cretikos, M. et al. Progression and impact of the first winter wave of the 2009 pandemic H1N1 influenza in New South Wales, Australia. Euro Surveill 14 (2009).
Campbell, A. et al. Risk of severe outcomes among patients admitted to hospital with pandemic (H1N1) influenza. CMAJ. 182, 349–355 (2010).
Crighton, E., Elliott, S., Moineddin, R., Kanaroglou, P. & Upshur, R. An exploratory spatial analysis of pneumonia and influenza hospitalizations in Ontario by age and gender. Epidemiol Infect. 135, 253–261 (2007).
Azziz-Baumgartner, E. et al. Incidence of influenza-associated mortality and hospitalizations in Argentina during 2002–2009. Influenza Other Respir Viruses 7, 710–717 (2013).
Engler, R. J. et al. Half-vs full-dose trivalent inactivated influenza vaccine (2004–2005): age, dose, and sex effects on immune responses. Arch Intern Med. 168, 2405–2414 (2008).
World Health Organization. In Addressing sex and gender in epidemic-prone infectious diseases (ed. Martha, Anker ) 12–13 (Geneva, 2007).
Tamerius, J. et al. Global influenza seasonality: reconciling patterns across temperate and tropical regions. Environ Health Perspect 119, 439 (2011).
Carville, K. S. et al. Understanding influenza vaccine protection in the community: An assessment of the 2013 influenza season in Victoria, Australia. Vaccine 33, 341–345 (2015).
Cowling, B. J. et al. Comparative epidemiology of pandemic and seasonal influenza A in households. N Engl J Med. 362, 2175–2184, 10.1056/NEJMoa0911530 (2010).
Hong Kong Census and Statistics Department. Hong Kong Statistics: 2006 Population Census. (2007). Avaiable at: http://www.census2011.gov.hk/en/census-result.html. (Accessed: 12th May, 2014).
Chan, K. H. et al. Assessment of Antigen and Molecular Tests with Serial Specimens from a Patient with Influenza A (H7N9) Infection. J Clin Microbiol. 52, 2272–2274 (2014).
Peiris, J. S. et al. Children with respiratory disease associated with metapneumovirus in Hong Kong. Emerg Infect Dis. 9, 628–633, 10.3201/eid0906.030009 (2003).
Lo, J. Y. et al. Respiratory infections during SARS outbreak, Hong Kong, 2003. Emerg Infect Dis. 11, 1738–1741, 10.3201/eid1111.050729 (2005).
Huang, J. W. & Yang, J. M. Changed epitopes drive the antigenic drift for influenza A (H3N2) viruses. BMC Bioinformatics. 12 Suppl 1, S31, 10.1186/1471-2105-12-s1-s31 (2011).
Nauta, J. J., Beyer, W. E. & Osterhaus, A. D. On the relationship between mean antibody level, seroprotection and clinical protection from influenza. Biologicals. 37, 216–221, 10.1016/j.biologicals.2009.02.002 (2009).
Ronquist, F. & Huelsenbeck, J. P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574 (2003).
R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. URL http://www.R-project.org/ (2013).
Acknowledgements
This study was supported by the Hong Kong Health and Medical Research Fund [12111232] and the Central Research Fund of the Hong Kong Polytechnic University. We thank Ms King Sau Chloe Wong and Mr Tsz Him Tsui of the University of Hong Kong for their services in performing the laboratory analysis. We also thank Dr Margaret O’Donoghue of the Hong Kong Polytechnic University for proofreading the manuscript.
Author information
Authors and Affiliations
Contributions
L.Y., C.-M.W., K.-H.C. and L.L.-M.P. designed the study; K.-H.C. collected the data; L.L.-M.P. did laboratory analysis; P.C., K.-P.C. and X.W. cleaned the data; P.C. conducted the data analysis; P.C., C.-M.W. and L.Y. drafted the manuscript; K.-H.C., J.S.M.P. and L.L.-M.P. revised the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Cao, P., Wong, CM., Chan, KH. et al. Age-specific genetic and antigenic variations of influenza A viruses in Hong Kong, 2013–2014. Sci Rep 6, 30260 (2016). https://doi.org/10.1038/srep30260
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep30260
This article is cited by
-
Individual immune selection pressure has limited impact on seasonal influenza virus evolution
Nature Ecology & Evolution (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
