
Abstract
Current therapies for metastatic clear cell renal cell carcinoma (ccRCC) show limited efficacy. Drug efficacy, typically investigated in preclinical cell line models during drug development, is influenced by pharmacogenes involved in targeting and disposition of drugs. Here we show through genome-wide DNA methylation profiling, that methylation patterns are concordant between primary ccRCC and macro-metastases irrespective of metastatic sites (rs ≥ 0.92). However, 195,038 (41%) of all investigated CpG sites, including sites within pharmacogenes, were differentially methylated (adjusted P < 0.05) in five established RCC cell lines compared to primary tumors, resulting in altered transcriptional expression. Exemplarily, gene-specific analyses of DNA methylation, mRNA and protein expression demonstrate lack of expression of the clinically important drug transporter OCT2 (encoded by SLC22A2) in cell lines due to hypermethylation compared to tumors or metastases. Our findings provide evidence that RCC cell lines are of limited benefit for prediction of drug effects due to epigenetic alterations. Similar epigenetic landscape of ccRCC-metastases and tumors opens new avenue for future therapeutic strategies.
Similar content being viewed by others


Introduction
Renal cell carcinoma (RCC) affects approximately 60,000 new patients annually in the United States and 84,400 in the European Union. Histologically, three major subtypes of RCC exist, namely clear cell RCC (ccRCC), chromophobe RCC, and papillary RCC. Among those, ccRCC is the most frequent subtype, accounting for approximately 80% of all RCCs1,2,3. Currently used first line treatment strategy for ccRCC is partial or radical nephrectomy, but patients are at risk of recurrence and development of metastases in different organs after surgery. Thus, several prognostic scores, including DNA methylation-based risk scores, have been developed to identify patients at risk of worse outcome4,5,6. Despite implementation of targeted therapies, using mTOR inhibitors or the multitarget tyrosine kinase inhibitors sunitinib or pazopanib, the survival rate for metastatic ccRCC is only about 20% after 5-year follow-up1,2,3.
Generally, ccRCC is mainly considered as a metabolic disease7,8 with consequences on treatment strategies. However, the efficacy and toxicity of therapeutic strategies will strongly depend not only on the expression of the drug targets (e.g. VEGF receptor) in primary tumors or their metastases, but also on pharmacogenes involved in drug absorption, distribution, metabolism and excretion (ADME). For instance, genetic polymorphisms in ADME genes like the efflux transporter ABCB1 are associated with survival and toxicity in patients with metastatic RCC treated with sunitinib9,10,11.
The importance of pharmacogenes for the drug discovery process is increasingly recognized, since a substantial proportion of drugs are withdrawn during preclinical stage or during clinical studies not only due to toxicity, but also due to lack of efficacy related to ADME processes like poor drug absorption in target cells12,13.
In the initial stages of the drug discovery process of novel drugs, cell lines or cell line derived xenograft mouse models play an important role14. For instance, the National Cancer Institute introduced an in vitro screening platform based on 60 human tumor cell lines (NCI60), including kidney cancers and a recent report demonstrated that use of a large cell-line collection may improve preclinical stratification for anticancer agents15. Cell based model systems also served to study molecular mechanisms of drug effects or multidrug resistance. The investigation of pharmacogenes, e.g. using preclinical cell models, is also recommended by the FDA, and for instance preclinical study of membrane drug transporters is part of the submission and approval process of new drugs16,17,18.
Based on cell line experiments, the benefit of DNA demethylating agents as novel therapeutic strategy in RCC has been proposed19. This is of importance since epigenetic regulation through DNA methylation is crucial for cancer progression and metastases. Regarding cell line models, there is increasing evidence from several tumor entities other than renal tumors that cell lines might show different DNA methylation or expression patterns compared to primary tumors with consequences on their use as model systems20,21. For instance, Nestor et al. demonstrated that rapid epigenetic re-programming occurred in cell culture of CD4+ T-cells22. However, whether pharmacogenes, particularly involved in the absorption, distribution, and metabolism of drugs, display aberrant methylation patterns in RCC cell lines compared to primary tumors or metastases has not been comprehensively investigated, but will be essential for their value as model systems. In general, epigenetic regulation is of importance for the transcriptional expression of ADME genes23,24 and we could previously demonstrate that drug transporters involved in ADME processes are regulated through DNA methylation, e.g. in primary RCC or hepatocellular carcinoma25,26.
Moreover, in contrast to other tumor entities27,28,29, it is also unclear whether global DNA methylation patterns and specifically those of pharmacogenes are shared between metastases and primary renal tumors with consequences on gene expression. The identification of particularly metastasis-associated markers may serve as novel targets for therapy in the metastatic setting.
Thus, the aim of our study is to comprehensively investigate genome-wide DNA methylation patterns in ccRCC and distant metastases in different organs to identify epigenetic regulation of pharmacogenes with potential as drug targets or for prediction of drug response. DNA methylation and gene expression profiles are compared to those of RCC cell lines in order to elucidate the predictive value of the established RCC cell lines for the study of pharmacogene-related processes. Validation experiments including gene expression on RNA and protein level corroborate major differences for epigenetic regulation between RCC cell lines and primary renal tumors, while ccRCCs and metastases show similar patterns.
Results
Genome-wide DNA methylation in primary ccRCC and metastases
Genome-wide DNA methylation analyses of tissue samples of our cohort (Table 1) were performed using the Illumina HumanMethylation 450 BeadChip. In total, 20 metastases in different organs (Table 1) and 34 primary ccRCC tumors were investigated, including tumor and metastasis tissue that were derived from the same patient (n = 6) as well as metastases occurring in different organs of the same patients (n = 3). Prior to all our analyses, methylation sites located on the X- and Y-chromosome as well as control probes were excluded, resulting in 473,864 CpG sites. As shown in Fig. 1A, all metastases clustered with the primary ccRCC tumor samples, indicating that global DNA methylation profiles in metastases of ccRCC show a high similarity to DNA methylation patterns observed in primary ccRCC samples. Of note, methylation patterns of samples derived from the same patient showed not only a high correlation (rS > 0.96) (Fig. 1B), but also small absolute differences in DNA methylation levels (Supplementary Fig. S1A,B; mean absolute differences in β-values < 0.060), except for one case (marked in red) which revealed a slight deviation in β-values (mean absolute differences in β-values = 0.092) and lower correlation (rS = 0.92) (Fig. 1A,B). Moreover, the local recurrence samples clustered with the primary tumor and distant metastasis samples (Supplementary Fig. S1C). In addition, we analyzed intra-tumor heterogeneity of DNA methylation in three ccRCC tumors from which genome-wide DNA methylation data were obtained from two regions of each tumor. As shown in Fig. 1A, samples derived from the same tumor (marked in identical grey shades) clustered together and DNA methylation patterns of these samples derived from the same tumor were not only significantly correlated (rS > 0.97; Fig. 1B,C), but also showed similar methylation levels (mean absolute differences in β-values 0.026, 0.038, and 0.047, respectively; Fig. S1B). Of note, their correlation coefficients and differences in methylation levels were comparable to those of technical replicates (rS = 0.99 and mean absolute differences in β-values = 0.020; Supplementary Fig. S2).

(A) Hierarchical cluster analysis of ccRCC (n = 34) and metastases (n = 20) samples shows similarity between tumor and metastases samples, as well as similarities between samples derived from the same patients (samples from the same patient are marked by identical colors). (B) Heatmap showing Spearman’s correlation coefficients for patients (n = 11) for which multiple samples were available. Samples from the same patient are marked as in Fig. 1A by identical colors in the vertical and horizontal side bar. Calculation of correlation coefficients is based on batch-corrected β-values of 473,864 CpG sites (including cross-reactive probes and probes that are impaired by the presence of SNPs). Correlation coefficients are color-coded as indicated. (C) Scatter plots with smoothed densities color representation for three different ccRCC tumor tissues, each showing DNA methylation data (β-values) of two regions per tumor. (D) Heatmap and cluster dendrogram of primary ccRCC (n = 34), metastases of ccRCC (n = 20), and the three TCGA RCC subtypes (Illumina 450K data on primary tumors: KIRC (ccRCC, n = 319), KICH (chromophobe RCC, n = 66), and KIRP (papillary RCC, n = 226)). The analysis is based on 41,322 CpG sites differentially methylated between the three RCC subtypes. (E) Violin plots depicting differences between median β-values of ccRCCs (n = 34) and metastases (n = 20) across various genomic regions. Here, all 46,691 CpG sites with an unadjusted P-value ≤ 0.05 between ccRCC and metastases in multivariate analysis were considered. Black bars represent 25% and 75% quantiles; white dots mark the medians, and the gray shape represents the density. (F) Volcano plot showing results of multivariate linear mixed model analysis for individual CpG sites, comparing metastases (n = 20) and primary ccRCC tumors (n = 34). (G) Manhattan plot showing results of multivariate linear mixed model analysis for individual CpG sites, comparing ccRCC (n = 34) and metastases (n = 20) samples. Analyses are based on 473,864 CpG sites (including cross-reactive probes and probes that are impaired by the presence of SNPs). Significantly different CpG sites that are hypomethylated in metastasis compared to primary ccRCC are marked in orange and hypermethylated ones are marked in brown.
Genome-wide DNA methylation in RCC subtypes and metastases
We next compared the DNA methylation data of our cohort to those of three RCC subtypes collected by TCGA: ccRCC (KIRC; n = 319), chromophobe RCC (KICH; n = 66), and papillary RCC (KIRP; n = 226). For this purpose, cluster analysis was performed based on 41,322 CpG sites which differed significantly and relevantly (Benjamini-Hochberg –adjusted [BH-adj.] P-value ≤ 0.05, absolute difference in median β-value ≥ 0.20) between the three RCC subtypes of TCGA. As shown in Fig. 1D, all RCC samples were clustered into three subgroups separating mainly ccRCC, chromophobe and papillary RCC subtypes. Interestingly, 15 ccRCC samples of the KIRC TCGA cohort revealed DNA methylation profiles similar to the majority of chromophobe RCC signatures. Moreover, ten ccRCC samples of the KIRC TCGA cohort displayed a papillary-like pattern. Re-evaluation of the histological diagnosis of these cases by two independent pathologists with expertise in renal tumor pathology was performed using the publicly available TCGA diagnostic and histological slide images. Results from this re-evaluation indicated that the 15 chromophobe-like ccRCC cases include chromophobe RCC, oncocytoma and other rare variants of RCC and the 10 papillary-like ccRCC cases also include non-ccRCC samples, thereby confirming our results from the cluster analysis (Supplementary Table S1). Almost all metastases samples of our cohort, irrespective of the affected organ, as well as the primary ccRCC samples of our cohort, clustered with the ccRCC samples (KIRC) of the TCGA cohort. Only one metastasis sample, derived from the pancreas, showed a papillary-like pattern and one of our primary ccRCC samples displayed a chromophobe-like signature. Additionally, one tumor sample was not consistently clustered within the ccRCC cohort, indicating a clear cell RCC phenotype with papillary features.
Concordant DNA methylation in primary ccRCC and metastases
Further analyses to identify individual CpG sites or gene regions that are differentially methylated between metastases and primary tumor tissue of our cohort were performed using linear mixed model analyses. Here, we corrected for age, sex, array batch and also considered that samples were in part derived from the same patient (five patients with paired RCC and metastasis, one patient had one RCC and two metastases, and two patients with two metastases each; regarding the three ccRCCs for which two regions of each tumor were measured, DNA methylation levels were averaged prior to all linear mixed model analyses; see Supplementary Methods). We first investigated whether alterations occur depending on the genomic location of CpG sites. As depicted in the Violin plots (Fig. 1E), no considerable differences in promoter regions, CpG islands or in the gene-bodies were found. Notably, only a few significantly differentially methylated CpG sites were identified after adjustment for multiple testing (Fig. 1F,G; Supplementary Table S2). Interestingly, moderately increased methylation levels in metastases compared to primary tumors were observed for a CpG site in the IGDCC4 (immunoglobulin superfamily, DCC subclass, member 4) gene region (Supplementary Fig. S3). Of note, no significantly differentially methylated regions (DMR) were observed when at least three CpG sites with BH-adj. P ≤ 0.05 per region were required. Significant DMR, using a relaxed false discovery rate (FDR) threshold of 15% for at least two CpG sites per region, are shown in Supplementary Table S3 and Supplementary Fig. S4. Here, DNA methylation in WHSC2 was reduced in metastases compared to ccRCC tumors, whereas GALR1 methylation was increased.
Next, DNA methylation profiles were compared between metastases occurring in lymph nodes and those in other organs to investigate whether DNA methylation is influenced by the organ in which the ccRCC metastasis occurred. No differentially methylated gene regions or CpG sites could be identified after adjustment for multiple testing (Supplementary Fig. S5A). Additionally, DNA methylation patterns between synchronous and metachronous metastasis were compared and no statistically significant differences were found after adjustment for multiple testing (Supplementary Fig. S5B), even when excluding the local recurrence samples (data not shown). The same held true when CpG sites that are influenced by SNPs and probes with potential cross-hybridization were excluded (data not shown). Additionally, restriction of analyses to patients without metastatic ccRCC (M0 and N0), or comparing metastases with either primary RCCs for which no metastasis occurred during follow-up, or primary RCCs for which one or more metastases occurred during follow-up did not reveal additional significantly different CpG sites (Supplementary Fig. S5C–F).
Exemplarily, DNA methylation patterns of candidate genes TINAGL1, ESYT3, ITGA5, FKBP1030, and CYTIP31 recently identified to be involved in epithelial-to mesenchymal transition (EMT) are depicted in Supplementary Fig. S6A,B. None of these regions were significantly differentially methylated in metastasis samples compared to primary tumor samples of our cohort after adjustment for multiple testing. Moreover, no significant differences between methylation levels in tumor and metastases were observed in the VHL promoter region (Supplementary Fig. S6C), which is known to be differentially methylated in about 7%7 of ccRCC compared to non-tumor tissue.
Genome-wide alterations of DNA methylation in RCC cell lines
Furthermore, genome-wide DNA methylation profiles of five RCC cell lines (Caki-1, Caki-2, A-498, ACHN, and 786-O) were compared with 20 metastases and 34 primary ccRCC tumors of our cohort. As shown in Fig. 2A, cell lines did not cluster with metastases or tumor samples. Detailed analyses revealed that genome-wide 195,038 individual CpG sites (41.2%) differed significantly between RCC cell lines and primary tumors (Fig. 2B), and 97,370 sites (20.5%) differed between RCC cell lines and metastases samples, respectively. Among those CpG sites, 41,682 and 8,194 CpG sites with an adjusted P < 1E-05 were found comparing cell lines and primary ccRCC (Fig. 2B) or metastases, respectively. Further analyses considering genomic locations showed that differences occurred in all investigated regions. However, predominantly hypermethylation in CpG islands and related shores was observed in cell lines compared to primary tumors (Fig. 2C). Furthermore, CpG sites throughout the genome were altered in cell lines compared to primary ccRCC (Fig. 2D). 73.5% of CpG sites were significantly hypermethylated in the five investigated cell lines compared to primary ccRCC (Fig. 2E,F). The same trend was observed for the comparison of cell lines and metastases (Fig. 2F).

(A) Hierarchical cluster analysis of five RCC cell lines, ccRCC (n = 34) and metastases (n = 20) samples (samples from the same patient are marked by identical colors). (B) Volcano plot showing results of multivariate linear mixed model analysis for individual CpG sites, comparing RCC cell lines and primary tumors. (C) Violin plots depicting differences between median β-values of ccRCCs (n = 34) and RCC cell lines (n = 5) across various genomic regions. Here, 195,038 CpG sites significantly differing between ccRCC and RCC cell lines in multivariate analysis were considered. (D) Manhattan plot showing results of linear mixed model analysis of differentially methylated CpG sites, comparing ccRCC (n = 34) and five RCC cell lines. Analyses are based on 473,864 CpG sites (including cross-reactive probes and probes that are impaired by the presence of SNPs). (E) Scatter plots with smoothed densities color representation of median DNA methylation levels (β-values) in primary ccRCC (n = 34) and RCC cell lines (n = 5), indicating hypermethylation of RCC cell lines compared to primary tumors. (F) Venn diagram depicting the number of significantly hyper-and hypomethylated CpG sites in pairwise comparisons of primary tumors, metastases and RCC cell lines.
Since RCC cell lines are frequently used to study the effects of drugs and for identification of drug targets, we asked whether pharmacogenes particularly involved in ADME processes like drug uptake or drug targeting are differentially methylated in RCC cell lines compared to primary RCC. A list of ADME related genes, which is based on the PharmaADME initiative (www.pharmaadme.org), as well as drug targets (based on www.broadinstitute.org/cancer/cga/target) was assembled (see Supplementary Table S4). The list contains 298 genes involved in e.g., drug metabolism or drug transport and 135 drug target genes, such as EGFR or BRAF. In summary, 1981 (41.7%) CpG sites within ADME genes and 1688 (39.3%) individual CpG sites within target gene regions were significantly (adjusted P < 0.05) differentially methylated between cell lines and primary ccRCC (see Volcano plot Fig. 3A). Moreover, 399 (8.4%) of those CpG sites within ADME genes and 366 (8.5%) individual CpG sites within target gene regions were differentially methylated between cell lines and primary ccRCC with an adjusted P < 1E-05 (see Volcano plot Fig. 3A). Most of these significant sites in ADME (78.0%) and drug target genes (82.5%) were hypermethylated in cell lines compared to primary ccRCC tumors, which is comparable to the methylation alterations observed for other genes.

(A) Volcano plot showing differentially methylated CpG sites in ADME and drug target genes between RCC cell lines and primary tumors. (B,C) Cluster analyses showing significantly differentially methylated gene regions (DMR) within (B) drug target genes or (C) ADME genes and particularly membrane transport proteins of the solute carrier (SLC) family.
In addition to individual CpG sites, also differentially methylated gene regions (DMR) were identified within drug target genes (n = 53; e.g. JAK3; Fig. 3B) or ADME genes (n = 74; Fig. 3C) with P < 0.006. We next focused on drug transporters which play an important role in the uptake and efflux of drugs in cancer cells, thereby contributing to drug resistance and non-response. Notably, several important uptake transport proteins are hypermethylated in RCC cell lines (e.g. SLC22A2, SLC22A8; Fig. 3C, right panel). In contrast, DNA methylation levels of ADME genes or drug targets were similar between metastases and primary ccRCC samples, as shown in Fig. 3B,C.
Epigenetic regulation of the expression of pharmacogenes
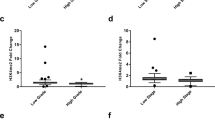
Since aberrant methylation of pharmacogenes and drug targets might result in altered gene expression of the respective genes, we next investigated their transcriptional expression in cell lines as well as primary ccRCC and metastases samples using Affymetrix Human Transcriptome 2.0 microarrays. Gene expression levels of 134 ADME and 93 drug target genes were significantly altered in cell lines compared to primary ccRCC tumors (Fig. 4A,B). Notably, about 7% of ADME genes (e.g., organic cation transporter OCT2) normally expressed in ccRCC tumors and metastases were expressed at very low levels or even not expressed at all in RCC cell lines. The same was true for drug target genes (e.g., type III receptor tyrosine kinase KDR, known as VEGFR). We further performed correlation analyses of DNA methylation levels and gene expression of ADME genes and drug targets. Therefore, only CpG sites significantly and relevantly (absolute difference in β-values > 0.1) differentially methylated as well as genes significantly and relevantly (absolute log2 fold change > 1, mean log2 signal intensity > 6) differentially expressed between primary tumors and cell lines were considered. For each gene, Spearman’s correlation coefficients were calculated between the mRNA expression and the DNA methylation levels of the CpG sites within the gene. As shown in Table 2, significant correlations between DNA methylation and gene expression were detected for 18 ADME as well as 8 drug target genes (results for each individual CpG site are given in Supplementary Table S5). Focusing on DNA methylation in promoter regions (TSS gene regions), which might directly influence gene expression levels, revealed significant correlations for e. g., the organic cation transporter OCT2/SLC22A2 and the drug target KDR (Supplementary Table S5).

Heatmaps showing significantly differentially expressed ADME genes (A) or drug target genes (B).
The organic cation transporter 2 (encoded by the SLC22A2 gene) is well characterized as uptake drug transporter for platinum drugs as well as other anti-cancer agents and the current FDA Guidance32 on “Drug Interaction Studies” recommends OCT2 as an important ADME target to be considered in the preclinical/clinical drug development process. Therefore, we further investigated its DNA methylation and expression on mRNA and protein level. Since the Illumina HumanMethylation 450 BeadChip does not cover every single CpG site in the human genome, we first validated DNA methylation data through more comprehensive MALDI-TOF mass spectrometry analyses. The MALDI-TOF MS assay is located in the SLC22A2 promoter region which was previously identified to be important for regulation of OCT2/SLC22A2 expression24,33. As shown in Fig. 5A, DNA methylation levels were significantly increased in RCC cell lines. These results were consistent in three replicates. In addition, no significant differences between DNA methylation in metastasis and tumor tissue were observed (Fig. 5A). Next, we analysed SLC22A2/OCT2 expression on mRNA and protein level. The mRNA levels were similar between primary tumor and metastasis tissue (Fig. 5B), but in RCC cell lines the OCT2 expression was below the limit of quantification. Investigation of OCT2 protein expression through Western blotting revealed that OCT2 protein was not detectable in these cell lines (Fig. 5C). To further assess a causal association between DNA methylation and expression, we examined the effect of AZA (5-Aza-2′-deoxycytidine, decitabine), which is a well-established DNA methylation inhibitor, on OCT2 expression in Caki-2 cells. To verify the effect of AZA treatment, global DNA methylation status was determined using an established LC-MS-MS method34. The proportion of 5-methylcytosine residues in genomic DNA of Caki-2 cells decreased from 4.50% to 2.20% after treatment with 1 μM AZA, clearly demonstrating the demethylating effect (Fig. 5D). Subsequently, mRNA expression was analyzed using real-time PCR technology. As shown in Fig. 5E, treatment of Caki-2 cells led to an increase of expression of OCT2. Since especially SLC transporters (Fig. 3C) displayed alterations in DNA methylation, we next assessed the expression of 55 SLC transporters in Caki-2 cells treated with decitabine using real-time PCR technology. Quantification of their mRNA expression levels indicated that several transporters are upregulated on mRNA level through demethylation (see Supplementary Fig. S7). Since organic cation transporters mediate cell sensitivity to platinum drugs like cisplatin35, we tested the effects of 5-Aza-2′-deoxycytidine (decitabine) pretreatment on cisplatin sensitivity in Caki-2 cells. The percentage of dead cells was quantified by FACS analysis using Annexin V staining36. As shown in Supplementary Fig. S8, the combination of both drugs showed significantly greater induction of apoptosis than either drug alone.

(A) DNA methylation at the SLC22A2 promoter in metastases, ccRCCs and five RCC cell lines was quantified using MALDI-TOF MS. No significant differences between metastases and ccRCCs were observed, but DNA methylation in all five RCC cell lines is significantly increased compared to tumor and metastases tissue. (B) SLC22A2/OCT2 mRNA expression in tumor and metastases tissue samples. mRNA expression was determined by quantitative real-time PCR and normalized to β-actin expression. (C) Protein expression of OCT2 in five RCC cell lines investigated through Western blotting revealed that OCT2 is not expressed in RCC cell lines on protein level. OCT2 transfected HEK-cells served as positive controls. β-actin expression was used as loading control. (D) Effect of treatment of Caki-2 cells with 5-Aza-2´-deoxycytidine (AZA) on global DNA methylation. Cells were either untreated or treated with 1 μM AZA and the amount of 5-methylcytosine was quantified using LC-MS-MS to verify the effect of AZA treatment on global DNA methylation. Results represent mean of 2 experiments ± SE. (E) Effect of AZA treatment on mRNA expression. Cells were cultured with 1 μM AZA and mRNA levels (normalized to β-actin) were determined using TaqMan technology. Fold increase in expression compared to untreated cells was calculated. (F) Immunohistochemical staining of OCT2 exemplarily in normal kidney tissue, ccRCC tissue samples and metastases of ccRCC. Protein expression was investigated in TMAs by semiquantitative immunohistochemistry (right panel), indicating that OCT2 is expressed both in ccRCC and metastases.
In addition, OCT2 protein expression in metastatic and primary tumor tissue was investigated by immunohistochemical staining of tissue microarrays. Representative staining of tissue cores is shown in Fig. 5F. In all metastasis samples, irrespective of the involved organ, a strong membranous staining of OCT2 was observed (Fig. 5F, left panel). Semiquantitative analyses of protein expression levels indicated that expression levels in metastases were within the range of tumor tissue (Fig. 5F, right panel). Thus, macro-metastases of ccRCC not only retain their SLC22A2 DNA methylation profile, but as a consequence also display the same high OCT2 expression as primary ccRCC tumors, irrespective of the affected organ. In RCC cell lines, OCT2 protein expression is downregulated due to hypermethylation in the SLC22A2 promoter region compared to primary tumors or metastases.
Discussion
Increasing evidence suggests that epigenetic mechanisms play an important role in the dynamic process of metastasis. Therefore, detailed knowledge not only of the epigenetic alterations present in primary tumor tissue, but also in metastatic tissue is of great interest especially for the development of novel treatment strategies for patients with metastatic disease. However, the efficacy of all therapeutic strategies will depend on the uptake, metabolism or excretion of the drugs in the tumor tissue as well as in metastases. For drug discovery and cancer research, cancer cell lines are still essential and extensively used, despite knowledge about their limitations.
In-depth molecular profiling on primary ccRCC tumors and non-tumor kidney tissue has been performed e.g. by The Cancer Genome Atlas (TCGA). However, corresponding metastases of ccRCC were not studied in the TCGA initiative. Since therapeutic response to anticancer agents might differ between primary tumors and metastases, we performed for the first time a comprehensive analysis of genome-wide DNA methylation in ccRCC metastases including local recurrence and metastasis samples derived from different organs (e.g. lymph node, liver, lung or adrenal gland). Moreover, paired tumor and metastases tissue as well as different metastases of the same patient were investigated.
Cluster analyses including all samples from our cohort as well as samples from TCGA of different RCC entities (clear cell, chromophobe, and papillary RCC) confirmed the ccRCC subtype in all our metastatic tissues, except for one metastasis. Interestingly, some ccRCC samples of the TCGA KIRC cohort did not cluster within the ccRCC cluster. Histopathological re-evaluation confirmed a non-ccRCC morphology and phenotype in most of these samples (Supplementary Table S1). This is in agreement with our previous findings using gene expression levels4 and results of the TCGA consortium, that in some cases the tumors were not conventional ccRCCs (see Supplementary Information in ref. 7). Furthermore, the present findings indicate that subtypes of RCC can be identified based on DNA methylation levels, in line with recent findings from Malouf et al.37.
In general, our data provide novel evidence that DNA methylation profiles are shared between primary ccRCC tumors and corresponding macro-metastases in various distant organs. Moreover, DNA methylation levels of CpG sites in gene regions previously identified to be important in epithelial(-to-)mesenchymal transition (EMT) in renal cell models are not altered in macro-metastasis. For instance, DNA methylation patterns of four candidate gene regions (TINAGL1, ESYT3, ITGA5, FKBP10), recently identified to be involved in EMT in a canine kidney cell model and confirmed in breast cancer30, were not significantly differentially methylated in metastasis samples compared to primary tumor samples of our cohort. The same result was found for the cytohesin interacting protein CYTIP, previously identified to be important in the process of EMT in ccRCC31.
Since we investigated metastases of ccRCC in nine different organs (Table 1), we had the possibility to examine the DNA methylome in its organ microenvironment, which cannot be restored using cell lines or cell line-derived xenografts. Our findings indicate that alterations of DNA methylation in metastasis generally seem not to be majorly impaired by the microenvironment of distant organs. Based on the hypothesis of two recent publications that clusters of cells that escape from the primary tumor have a higher ability to form distant metastasis compared to circulating single cells38,39, our results indicate that DNA methylation features influenced by the ccRCC microenvironment might be preserved in these cell clusters during the process of macro-metastases formation.
Notably, also DNA methylation and expression patterns of pharmacogenes, such as drug target genes and ADME-related genes, were shared between primary tumors and metastases. Thus, ADME processes, e.g. drug uptake processes and subsequent drug effects are comparable between tumors and metastases. However, all five studied RCC cell lines displayed not only genome-wide hypermethylation compared to primary tumors or metastases, but also altered DNA methylation of drug targets and important ADME genes was observed. Our data indicating that the investigated renal cancer cell lines generally are hypermethylated compared to primary renal tumors, are in line with previous data of other tumor entities (e.g. breast, prostate, colon) demonstrating CpG island hypermethylation in cancer cell lines compared to primary tumors40. The effect of cell culture on epigenetic alterations was also investigated for other tumor entities through genome-wide analyses of cell lines and primary tumors by Varley et al.20, though the underlying mechanisms have not been completely elucidated. Antequera et al.41 proposed that CpG island hypermethylation in cell culture is associated with the loss of DNA demethylase activity and recently, Nestor et al.22 provided first evidence that loss of TET-mediated demethylase activity is responsible for cell culture-induced hypermethylation.
In our study, we could further demonstrate that alterations in DNA methylation lead to expression differences in pharmacogenes involved in ADME processes. For instance, important drug transporters (e.g. the organic cation transporter OCT2/SLC22A2) were expressed at high levels in primary tumors and metastases, but seem to be downregulated in any of the investigated RCC cell lines due to hypermethylation. Interestingly, these cell lines differ in their basal phenotypes and genomes, such as the mutation status in the von Hippel-Lindau (VHL) gene. Although DNA methylation analyses through Illumina HumanMethylation 450 BeadChip is highly reliable, not every single CpG site present in the human genome is covered by the array. Moreover, some DNA methylation values are impaired by cross-reactive probes present on the microarray. Thus, additional experiments using independent methods e.g., MALDI-TOF MS or pyrosequencing, might still be necessary not only for validation, but also for fine-mapping of DNA methylation in the promoter region to identify single, relevant CpG sites not covered by the Illumina HumanMethylation 450 BeadChip, as we recently investigated for the monocarboxylate transporter MCT425.
Notably, in the present study the results of genome-wide methylation analyses of SLC22A2 were validated by gene-specific analyses, using an independent MALDI-TOF MS assay covering the CpG site previously identified to regulate OCT2 expression. Further analyses of the expression data from 533 tumors of the TCGA ccRCC cohort indicated that indeed 88% of all ccRCC tumors showed an OCT2 expression, which was higher than the median gene expression of all genes expressed in ccRCC (Supplementary Fig. S9A). Moreover, based on TCGA data, a significant correlation between DNA methylation and mRNA expression in ccRCC tumors exists (Supplementary Fig. S9B). In addition, immunohistochemical staining of OCT2 in primary ccRCC tumors and metastasis demonstrated that OCT2 was highly expressed in all metastases of ccRCC irrespective of the organ and its expression was found to be quite homogeneous in metastases of ccRCC. In contrast, OCT2 protein was not detectable in any of the five studied RCC cell lines. This finding results in important consequences because OCT2 is known to be involved in the uptake of anticancer agents (e.g. platinum drugs or the survivin inhibitor YM155) and is one of the key transporters that needs to be evaluated in drug development according to the International Transporter Consortium16,42. Based on our data, DNA methylation (Fig. 3C) and expression of several uptake transporters potentially relevant for novel cancer drugs are altered in RCC cell lines compared to primary tumors and metastases indicating that these cell lines do not sufficiently reflect conditions in vivo. Thus, in case of novel drugs developed for treatment of RCC in the future this discrepancy is of major importance for valid interpretation of preclinical data derived from RCC cell line experiments and to avoid misleading conclusions for further steps in the drug development process. Probably treatment of cells with demethylating agents might be promising in order to increase SLC transporter expression in cell lines, corroborated by the fact that exemplarily decitabine pretreatment results in higher sensitivity of cells to cisplatin (Supplementary Fig. S8). Generally, demethylating agents like 5-Azacytidine or decitabine have been approved for therapy of myelodyplastic syndrome, but have also been proposed as therapeutic option for solid tumors. Previously, Ricketts et al. provided preclinical evidence that treatment with demethylating agents might be useful for RCC19. Since metastases and primary tumors share similar methylation profiles, use of demethylating agents alone or in combination might be promising for treatment of metastatic patients. Combination therapy using decitabine and platinum drugs has previously been reported to be more effective in tumor models43, most likely because they exerted their effects through different mechanisms44. Recently, even an immune-based mechanism of action of DNA methyltransferase inhibitors has been proposed45. Thus, combination of for instance demethylating agents and new immune-checkpoint–inhibitors, such as those blocking programmed death 1 (PD-1), might be a promising option for treatment of ccRCC.
Although we provide the first data on DNA methylation in metastasis of ccRCC, our retrospective study has some limitations. Of course, we cannot exclude that even small differences between metastases and primary tumors in individual CpG sites might be of biological relevance, which were not statistically significant because of lack of statistical power due to the size of our cohort. The observed alterations in gene expression might not only be caused by epigenetic factors, but also by genetic factors known to play an important role in gene regulation. Genetic variants which might additionally alter variability of DNA methylation have not been systematically investigated, although consideration of SNPs present on the Illumina HumanMethylation 450 BeadChip revealed no significant impact of genetic variants (data not shown). As shown in Table 1, some of the patients received treatment before surgery for metastasis; however, statistical analyses including prior treatment indicated no influence (data not shown). Whether prior treatment decreases the heterogeneity of DNA methylation in RCC and metastasis as previously suggested for colorectal cancer, has not been studied. However, our immunohistochemical staining of OCT2 protein in metastases of different organs is rather homogeneous irrespective of prior treatment suggesting limited evidence of intra-tumor heterogeneity at least for OCT2. Moreover, we found a significant correlation of DNA methylation patterns derived from two regions of one tumor. Nevertheless, further studies investigating heterogeneity in more samples of each organ (e.g. lung) are needed.
In addition, further studies need to evaluate whether primary RCC cells derived from tumor or metastases tissue are more appropriate to study pharmacogene-related processes. However, data from primary renal proximal tubule cells demonstrated that the expression of drug transporters is down-regulated indicating also limited applicability of primary cells to study drug transport processes46,47. Spheroids, as well established three-dimensional (3D) models for drug screening, might represent an interesting alternative to improve preclinical drug response prediction48. In this context, patient derived tumor xenografts (PDX) might be used as well to study not only cancer biology, but also drug related processes49. There are first data regarding PDX models for RCC and especially for the ccRCC subtype50,51, indicating that PDX models preserve gene expression profiles of the primary tumors52. Only recently, Kim et al.53 provided evidence that PDX models (including models derived from a corresponding metastasis and single cells derived from PDX-tumor and metastasis) might be promising to design personalized therapeutic strategies for ccRCC. Whether DNA methylation profiles of pharmacogenes are maintained in these models needs to be evaluated, but such models offer great potential to study drug related processes.
Taken together, the present study not only provides new insight into the epigenetic landscape of ccRCC derived metastases with impact on therapy or diagnosis, but additionally elucidates limited benefit of RCC cell lines for investigating pharmacogene-related processes.
Materials and Methods
Study cohort
Metastases (n = 20) as well as primary tumor samples (n = 34) of ccRCC patients were obtained from the Departments of Urology and Pathology (University Hospital Tuebingen, Germany), as well as the Department of Pathology (Robert Bosch Krankenhaus, Stuttgart, Germany) (see Supplementary Methods). Patients’ characteristics, clinicopathological features, survival data as well as details about metastases are given in Table 1. Informed written consent was provided by each subject and use of the tissue was approved by the ethics committee of the University of Tuebingen, Germany. All methods were carried out in accordance with the approved guidelines.
Additionally, publicly available DNA methylation data of The Cancer Genome Atlas (TCGA) from three independent cohorts of RCC patients (n = 611) including primary ccRCC (KIRC), chromophobe (KICH) and papillary RCC (KIRP) were analyzed. Details about analyses of TCGA data is given in Supplementary Methods.
RCC cell lines
A-498, 786-O, Caki-1, and ACHN cell lines (purchased from CLS Cell Lines Service, Eppelheim, Germany), as well as the Caki-2 cell line (purchased from Sigma-Aldrich, Taufkirchen, Germany) were cultivated as previously described25. Cell lines were routinely tested for mycoplasma infection using a PCR-based test (Venor®GeM Classic, Minerva Biolabs GmbH, Berlin, Germany) and authentication of cell lines was performed using the PowerPlex® 21 System (Promega, Madison, USA) according to the manufacturer’s protocol.
Analyses of DNA methylation and gene expression
Genome-wide DNA methylation profiles of 20 metastases of ccRCC in different organs (see Table 1) and 34 primary ccRCC tumors of our cohort, as well as five RCC cell lines were generated using the Illumina Infinium HumanMethylation 450 BeadChip. Genomic DNA was purified from fresh-frozen ccRCC and metastasis tissue, as well as cell lines using standard procedures (QIAamp DNA Mini Kit). For three tumor samples, DNA was isolated from two regions of the tumor tissue and independently processed. Bisulfite conversion and further analyses using Illumina HumanMethylation 450 BeadChip were performed by Service XS (Service XS, Leiden, The Netherlands). Tumor and metastasis samples were randomly processed. Genome-wide mRNA expression analyses of tumor samples, metastases and RCC cell lines were performed using the Human Transcriptome Array 2.0 (Affymetrix). In brief, RNA was purified from fresh-frozen ccRCC and metastasis tissue, as well as cell lines using the mirVana™ miRNA Isolation Kit (Life Technologies) and microarrays were processed according to manufacturer’s procedure (Affymetrix). The accession number for data of metastases and primary ccRCC, as well as cell lines, at the European Genome-phenome Archive (EGA) (www.ebi.ac.uk/ega/home), which is hosted by the EBI and the CRG, is EGAS00001001176. Further details about data analyses are provided in Supplementary Methods.
DNA methylation, mRNA, and protein expression of OCT2/SLC22A2
For quantitative DNA methylation analyses of SLC22A2, matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) was applied as described26. mRNA levels were quantified using TaqMan technology and OCT2 protein was quantified through Western Blot technology using a validated polyclonal antibody against OCT2 as described previously26. Tissue microarray sections were processed and immunostained as previously described25,54. Details see Supplementary Methods.
Statistical analyses
Statistical analyses were performed with R-3.1.1 (http://www.r-project.org)55 or GraphPad Prism 5.0 (GraphPad Software, Inc., USA). P-values were adjusted for multiple testing by the Benjamini-Hochberg procedure56. All statistical tests were two-sided. Statistical significance level was defined as 5%. See Supplementary Data for details.
Additional Information
How to cite this article: Winter, S. et al. Methylomes of renal cell lines and tumors or metastases differ significantly with impact on pharmacogenes. Sci. Rep. 6, 29930; doi: 10.1038/srep29930 (2016).
References
Oosterwijk, E. et al. Basic research in kidney cancer. Eur Urol. 60, 622–633 (2011).
Rini, B. I., Campbell, S. C. & Escudier, B. Renal cell carcinoma. Lancet 373, 1119–1132 (2009).
Shuch, B. et al. Understanding Pathologic Variants of Renal Cell Carcinoma: Distilling Therapeutic Opportunities from Biologic Complexity. Eur Urol. 67, 85–97 (2015).
Buttner, F. et al. Survival Prediction of Clear Cell Renal Cell Carcinoma Based on Gene Expression Similarity to the Proximal Tubule of the Nephron. Eur Urol. (2015).
Rini, B. et al. A 16-gene assay to predict recurrence after surgery in localised renal cell carcinoma: development and validation studies. Lancet Oncol. 16, 676–685 (2015).
Wei, J. H. et al. A CpG-methylation-based assay to predict survival in clear cell renal cell carcinoma. Nat Commun. 6, 8699 (2015).
Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 499, 43–49 (2013).
Li, B. et al. Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature. 513, 251–255 (2014).
Beuselinck, B. et al. Efflux pump ABCB1 single nucleotide polymorphisms and dose reductions in patients with metastatic renal cell carcinoma treated with sunitinib. Acta Oncol. 53, 1413–1422 (2014).
Diekstra, M. H. et al. CYP3A5 and ABCB1 polymorphisms as predictors for sunitinib outcome in metastatic renal cell carcinoma. Eur Urol. 68, 621–629 (2015).
van Erp, N. P. et al. Pharmacogenetic pathway analysis for determination of sunitinib-induced toxicity. J Clin Oncol. 27, 4406–4412 (2009).
Gleeson, M. P., Hersey, A., Montanari, D. & Overington, J. Probing the links between in vitro potency, ADMET and physicochemical parameters. Nat Rev Drug Discov. 10, 197–208 (2011).
Waring, M. J. et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat Rev Drug Discov. 14, 475–486 (2015).
Wilding, J. L. & Bodmer, W. F. Cancer cell lines for drug discovery and development. Cancer Res. 74, 2377–2384 (2014).
Barretina, J. et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 483, 603–607 (2012).
Giacomini, K. M. et al. Membrane transporters in drug development. Nat Rev Drug Discov. 9, 215–236 (2010).
Giacomini, K. M. et al. International Transporter Consortium commentary on clinically important transporter polymorphisms. Clin Pharmacol Ther. 94, 23–26 (2013).
Morrissey, K. M., Stocker, S. L., Wittwer, M. B., Xu, L. & Giacomini, K. M. Renal transporters in drug development. Annu Rev Pharmacol Toxicol. 53, 503–529 (2013).
Ricketts, C. J. et al. Methylation profiling and evaluation of demethylating therapy in renal cell carcinoma. Clin Epigenetics. 5, 16 (2013).
Varley, K. E. et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 23, 555–567 (2013).
Gillet, J. P. et al. Redefining the relevance of established cancer cell lines to the study of mechanisms of clinical anti-cancer drug resistance. Proc Natl Acad Sci USA 108, 18708–18713 (2011).
Nestor, C. E. et al. Rapid reprogramming of epigenetic and transcriptional profiles in mammalian culture systems. Genome Biol. 16, 11 (2015).
Habano, W. et al. Analysis of DNA methylation landscape reveals the roles of DNA methylation in the regulation of drug metabolizing enzymes. Clin Epigenetics. 7, 105 (2015).
Fisel, P., Schaeffeler, E. & Schwab, M. DNA Methylation of ADME Genes. Clin Pharmacol Ther. 99, 512–527 (2016).
Fisel, P. et al. DNA methylation of the SLC16A3 promoter regulates expression of the human lactate transporter MCT4 in renal cancer with consequences for clinical outcome. Clin Cancer Res. 19, 5170–5181 (2013).
Schaeffeler, E. et al. DNA methylation is associated with downregulation of the organic cation transporter OCT1 (SLC22A1) in human hepatocellular carcinoma. Genome Med. 3, 82 (2011).
Aryee, M. J. et al. DNA methylation alterations exhibit intraindividual stability and interindividual heterogeneity in prostate cancer metastases. Sci Transl Med. 5, 169ra10 (2013).
Marzese, D. M. et al. Epigenome-wide DNA methylation landscape of melanoma progression to brain metastasis reveals aberrations on homeobox D cluster associated with prognosis. Hum Mol Genet. 23, 226–238 (2014).
Moarii, M. et al. Epigenomic alterations in breast carcinoma from primary tumor to locoregional recurrences. PLoS One. 9, e103986 (2014).
Carmona, F. J. et al. A comprehensive DNA methylation profile of epithelial-to-mesenchymal transition. Cancer Res. 74, 5608–5619 (2014).
Vanharanta, S. et al. Epigenetic expansion of VHL-HIF signal output drives multiorgan metastasis in renal cancer. Nat Med. 19, 50–56 (2013).
FDA Draft Guidance. Drug Interaction studies - Study design, data analysis, implications for dosing, and labeling recommendations. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/Ucm292362.pdf. (2012).
Aoki, M. et al. Kidney-specific expression of human organic cation transporter 2 (OCT2/SLC22A2) is regulated by DNA methylation. Am J Physiol Renal Physiol. 295, F165–F170 (2008).
Hocher, B. et al. Maternal eNOS Deficiency Determines a Fatty Liver Phenotype of the Offspring in a Sex Dependent Manner. Epigenetics. 0 (2016). [Epub ahead of print]
Yonezawa, A., Masuda, S., Yokoo, S., Katsura, T. & Inui, K. Cisplatin and oxaliplatin, but not carboplatin and nedaplatin, are substrates for human organic cation transporters (SLC22A1-3 and multidrug and toxin extrusion family). J Pharmacol Exp Ther. 319, 879–886 (2006).
Dengler, M. A. et al. Discrepant NOXA (PMAIP1) transcript and NOXA protein levels: a potential Achilles’ heel in mantle cell lymphoma. Cell Death Dis. 5, e1013 (2014).
Malouf, G. G. et al. DNA Methylation Signature Reveals Cell Ontogeny of Renal Cell Carcinomas. Clin Cancer Res. (2016). [Epub ahead of print]
Aceto, N. et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 158, 1110–1122 (2014).
Bottos, A. & Hynes, N. E. Cancer: Staying together on the road to metastasis. Nature. 514, 309–310 (2014).
Smiraglia, D. J. et al. Excessive CpG island hypermethylation in cancer cell lines versus primary human malignancies. Hum Mol Genet. 10, 1413–1419 (2001).
Antequera, F., Boyes, J. & Bird, A. High levels of de novo methylation and altered chromatin structure at CpG islands in cell lines. Cell. 62, 503–514 (1990).
Giacomini, K. M. & Huang, S. M. Transporters in drug development and clinical pharmacology. Clin Pharmacol Ther. 94, 3–9 (2013).
Appleton, K. et al. Phase I and pharmacodynamic trial of the DNA methyltransferase inhibitor decitabine and carboplatin in solid tumors. J Clin Oncol. 25, 4603–4609 (2007).
Qin, T. et al. Epigenetic synergy between decitabine and platinum derivatives. Clin Epigenetics. 7, 97 (2015).
Dear, A. E. Epigenetic Modulators and the New Immunotherapies. N Engl J Med. 374, 684–686 (2016).
Fisel, P., Renner, O., Nies, A. T., Schwab, M. & Schaeffeler, E. Solute carrier transporter and drug-related nephrotoxicity: the impact of proximal tubule cell models for preclinical research. Expert Opin Drug Metab Toxicol. 10, 395–408 (2014).
Tasnim, F. & Zink, D. Cross talk between primary human renal tubular cells and endothelial cells in cocultures. Am J Physiol Renal Physiol. 302, F1055–F1062 (2012).
Friedrich, J., Seidel, C., Ebner, R. & Kunz-Schughart, L. A. Spheroid-based drug screen: considerations and practical approach. Nat Protoc. 4, 309–324 (2009).
Cassidy, J. W., Caldas, C. & Bruna, A. Maintaining Tumor Heterogeneity in Patient-Derived Tumor Xenografts. Cancer Res. 75, 2963–2968 (2015).
Hasmim, M. et al. Isolation and characterization of renal cancer stem cells from patient-derived xenografts. Oncotarget. (2015).
Schuller, A. G. et al. The MET Inhibitor AZD6094 (Savolitinib, HMPL-504) Induces Regression in Papillary Renal Cell Carcinoma Patient-Derived Xenograft Models. Clin Cancer Res. 21, 2811–2819 (2015).
Sivanand, S. et al. A validated tumorgraft model reveals activity of dovitinib against renal cell carcinoma. Sci Transl Med. 4, 137ra75 (2012).
Kim, K. T. et al. Application of single-cell RNA sequencing in optimizing a combinatorial therapeutic strategy in metastatic renal cell carcinoma. Genome Biol. 17, 80 (2016).
Fisel, P. et al. MCT4 surpasses the prognostic relevance of the ancillary protein CD147 in clear cell renal cell carcinoma. Oncotarget. 6, 30615–30627 (2015).
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. (2014).
Benjamini Y & Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. 289–300 (1995).
Acknowledgements
We gratefully acknowledge Monika Elbl, Andrea Jarmuth, Heidi Köhler and Ursula Waldherr for excellent technical assistance. The results shown here are in part based upon data generated by the TCGA Research Network. We would like to thank The Cancer Genome Atlas initiative, all tissue donors, and investigators who contributed to the acquisition and analyses of the samples used in this study. Information about TCGA and the investigators and institutions who constitute the TCGA research network can be found at http://cancergenome.nih.gov/. This work was supported by the Robert Bosch Foundation (Stuttgart, Germany), the DFG SFB grant [DFG SFB685 C05], Ferdinand-Eisenberger-grant of the German Society of Urology [RaS1/FE-14], the German Cancer Consortium (DKTK) and German Cancer Research Center (DKFZ), Heidelberg, Germany, and the ICEPHA Graduate School Tuebingen-Stuttgart, Germany.
Author information
Authors and Affiliations
Contributions
Conception and design: S.W., P.F., M. Schwab and E.S.; Development of methodology: S.W., P.F., D.D’A., F.B., S.R., J.H., M. Scharpf, U.H., H.v.d.K., F.F. and E.S.; Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): P.F., D.D’A., S.R., J.H., G.O., S.K., J.B., A.S., U.H., H.v.d.K., F.F., M. Schwab and E.S.; Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis, histopathological review): S.W., P.F., F.B., D.D’A., S.R., S.K., A.N., J.B., G.O., M. Scharpf, U.H., H.v.d.K., K.J., A.A., A.H., M. Schwab and E.S.; Writing, review, and/or revision of the manuscript: S.W., P.F., F.B., D.D’A., S.R., J.H., S.K., A.N., A.S., J.B., K.J., A.A., A.H., M. Schwab and E.S.; Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): S.W., F.B., S.R., J.H., S.K., J.B., M. Schwab and E.S.; Study supervision: J.B. and M. Schwab.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Winter, S., Fisel, P., Büttner, F. et al. Methylomes of renal cell lines and tumors or metastases differ significantly with impact on pharmacogenes. Sci Rep 6, 29930 (2016). https://doi.org/10.1038/srep29930
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep29930
This article is cited by
-
A novel molecular signature identifies mixed subtypes in renal cell carcinoma with poor prognosis and independent response to immunotherapy
Genome Medicine (2022)
-
Integrative -omics and HLA-ligandomics analysis to identify novel drug targets for ccRCC immunotherapy
Genome Medicine (2020)
-
The impact of estimated tumour purity on gene expression-based drug repositioning of Clear Cell Renal Cell Carcinoma samples
Scientific Reports (2019)
-
Clinical utility of the S3-score for molecular prediction of outcome in non-metastatic and metastatic clear cell renal cell carcinoma
BMC Medicine (2018)
-
Renal cell carcinoma
Nature Reviews Disease Primers (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
