
Abstract
Tumors are formed by the abnormal proliferation of somatic cells with disordered growth regulation under the influence of tumorigenic factors. Recently, the theory of “cancer drivers” connects tumor initiation with several specific mutations in the so-called cancer driver genes. According to the differentiation of four basic levels between tumor and adjacent normal tissues, the cancer drivers can be divided into the following: (1) Methylation level, (2) microRNA level, (3) mutation level and (4) mRNA level. In this study, a computational method is proposed to identify novel lung adenocarcinoma drivers based on dysfunctional genes on the methylation, microRNA, mutation and mRNA levels. First, a large network was constructed using protein-protein interactions. Next, we searched all of the shortest paths connecting dysfunctional genes on different levels and extracted new candidate genes lying on these paths. Finally, the obtained candidate genes were filtered by a permutation test and an additional strict selection procedure involving a betweenness ratio and an interaction score. Several candidate genes remained, which are deemed to be related to two different levels of cancer. The analyses confirmed our assertions that some have the potential to contribute to the tumorigenesis process on multiple levels.
Similar content being viewed by others


Introduction
Tumors are defined as new creatures formed by the abnormal proliferation of somatic cells with disordered growth regulation under the influence of tumorigenic factors1. Around the world, tumors have been reported to be the second killer of human health, ranked only behind cardiovascular disease. However, it is still not clear how tumor tissues initiate and invade during the precancerous lesion stage2. Specific genetic alterations have been detected in tumor cells of different types. Some well-known genes, such as p53, K-Ras, etc., have been reported in various tumor types, which have been regarded as genomic markers for the given tumors and may be the original mutation related to tumor initiation and progression3,4.
In 2012, the theory of “cancer drivers” was first presented at the RAOF (Round Asia Oncology Forum), which connects tumor initiation with several specific mutations in the so-called cancer driver genes5. Such theory attributes tumor initiation to several original specific genomic alterations, which sequentially induce metabolic and functional disorders in somatic cells. As we know, based on the differentiation of four basic levels between the tumor and adjacent normal tissues, we can divide cancer driver genes into four clusters: (1) Methylated CpG site genes, (2) microRNA target genes, (3) somatic mutation genes and (4) mRNA genes.
First, the level of methylation of driver genes may change during tumor initiation. Generally, the methylation and demethylation of peculiar regions in chromosomes reflects the regulation of gene expression on the transcriptional level6. The demethylation of oncogenes and/or the methylation of tumor suppressors may induce the proliferation and genomic instability of tumor cells7. Such processes may be the driving procedures of tumor initiation and progression. In addition to methylation and demethylation, another level cancer driver genes contribute to is associated with microRNA expression8. microRNA is a small non-coding RNA molecule that contributes to post-transcriptional regulation by RNA silencing9. During tumor initiation and progression, the specific microRNA expression level changes and may further regulate its functional target genes10. Such microRNA target genes are regulated differently in the tumor versus normal tissues. Thus, the level of functional microRNAs may also reveal several cancer driver genes. Genomic instability is another basic characteristic of tumor cells. As a result of that, mutations exist extensively in malignant cells. Therefore, the third level for cancer driver genes to initiate tumors is mutations. Some cancer driver genes, such as p53 and K-Ras, include specific mutations which contribute to the structural and functional changes of their protein products and may further induce tumor initiation and progression11. Mutations not only occur in the exons of driver genes but in their regulatory sequences as well, such as promoters, enhancers, etc.12,13. These mutations may not alter the structures or functions of target proteins which are encoded by driver genes but may strongly improve or reduce the quantity of mRNAs and proteins14. Apart from that, some unique abnormal regulatory factors with mutations may also contribute to the regulation of cancer genes at the transcription and/or translation levels15.
Lung adenocarcinoma is a typical subtype of lung cancer that is highly related to specific genetic background16. As a crucial threat to human health, however, most studies on lung adenocarcinoma drivers are restricted to a single level (one of the four clusters as we have mentioned above) and the core driver factors that contribute to the initiation and progression of lung adenocarcinoma have not been fully revealed. In these studies, most progressions of lung adenocarcinoma associated drivers mainly concentrate to the mutations and copy number variations (CNVs) of particular oncogenes17,18. For example, it has been widely confirmed by in vitro and in vivo experiments that specific mutations of EGFR and KRAS may exactly drive the initiation and progression of lung adenocarcinoma, implying the key role of certain drivers for the tumorigenesis of lung adenocarcinoma19,20. Apart from that, on another level, a specific microRNA associated mutation (target site on KRT81) has also been reported to be associated with lung adenocarcinoma21. As stated above, the original specific genomic alterations may contribute to tumor initiation in multiple levels, implying the analysis of specific variant should be extended to multi-omics. However, few multi-level analysis (such as the combination of mutations and copy number variants) of lung adenocarcinoma drivers have been presented and reported. For the first time, based on TCGA database, our study concentrate on all the four levels of drivers as we have mentioned above and try to fill the gap of this research field.
In this study, we investigated the specific driver factors of lung adenocarcinoma on four functional levels based on the gene expression, microRNA expression, DNA methylation and somatic mutation data of lung adenocarcinoma tissues and normal control samples from TCGA (The Cancer Genome Atlas)22. We first sought to search all the shortest paths (SP) connecting dysfunctional genes on different levels in a large network constructed by protein-protein interactions (PPI) and to identify new candidate cancer driver genes on these paths. Then, these genes were filtered by a permutation test and a strict selection procedure. The final obtained candidate genes were deemed to be related to two different levels, i.e., they can drive tumorigenesis on two levels. Furthermore, some candidate genes may occur more than once, meaning that they can drive tumorigenesis on multiple levels. The more levels a gene can drive tumor initiation, the more significant the gene may be.
Materials and Methods
Dataset
We downloaded the gene expression, microRNA expression, DNA methylation and somatic mutation data of lung adenocarcinoma tissues and normal control samples from TCGA (https://tcga-data.nci.nih.gov/docs/publications/luad_2014/)22.
The expression level of 20,531 genes in 230 tumor samples and 43 normal samples was measured with RNA-Seq and transformed into the log2 scale. The expression level of 1,046 microRNAs in 181 tumor samples and 32 normal samples was measured with microRNA-Seq and also transformed onto the log2 scale. The DNA methylation level of 485,577 CpG sites in 181 tumor samples and 21 normal samples was measured using the Infinium HumanMethylation450 BeadChip array. The somatic mutation data of 14,989 genes in 230 tumor samples were called with MuTect23.
Identification of the differentially expressed mRNA genes, microRNAs, methylated CpG sites and somatic mutation genes
We used the SAM (Significance Analysis of Microarrays) method24 to detect differentially expressed genes, microRNAs and the methylated CpG sites. There were 1,373 differentially expressed mRNA genes, 42 microRNAs and 295 methylated CpG sites with a SAM FDR (False Discovery Rate) less than 0.01 and a fold change greater than 5. The 42 microRNAs regulated 825 target genes based on at least three out of six microRNA-target databases, including miRBase25 (http://microrna.sanger.ac.uk/targets/v5/), TargetScan26 (http://www.targetscan.org/), miRanda27 (http://www.microrna.org/microrna/), TarBase28 (http://diana.cslab.ece.ntua.gr/tarbase/), mirTarget229 (http://mirdb.org/miRDB/download.html) and PicTar30 (http://pictar.mdc-berlin.de/). The 295 differentially expressed methylated CpG sites were annotated to 153 genes based on their chromosome locations from TCGA. The 197 somatic mutation genes with mutation frequencies greater than 10% were selected.
As a result, 153 methylated CpG site genes, 825 microRNA target genes, 197 somatic mutation genes and 1,373 differentially expressed mRNA genes were considered seed genes and comprised the gene sets G1, G2, G3 and G4, respectively. The symbols of these genes are provided in the Supplementary Material I.
Network construction
The constructed network was built according to the PPI information retrieved from STRING (Search Tool for the Retrieval of Interacting Genes/Proteins, http://www.string-db.org/, Version 9.1)31,32. The obtained file, ‘protein.links.v9.1.txt.gz’, contained large numbers of PPIs involving 1,133 organisms. In total, 2,425,314 human PPIs were extracted from this file by selecting lines starting with ‘9606.’. PPIs reported in STRING are obtained by the following sources: (1) genomic context, (2) high-throughput experiments, (3) (conserved) co-expression and (4) previous knowledge. Thus, they can widely measure the associations between proteins and have been used to deal with several protein related problems33,34,35,36,37,38,39,40. For each of the 2,425,314 human PPIs, there are two proteins represented by Ensembl IDs and a score, indicating the strength of the interaction ranging between 150 and 999. Proteins in an interaction with a high score have strong associations. The constructed network took all the proteins occurring in 2,425,314 human PPIs, totaling 20,770 proteins, as nodes and two nodes were connected by an edge if and only if the corresponding proteins could comprise an interaction. Clearly, each edge represented a human PPI. To indicate the score of each interaction, each edge should be assigned a weight. Because the range of the interaction score is between 150 and 999, i.e., the maximum value of interaction score is 999 and a model using the shortest path algorithm as a basic algorithm requires edges assigned low weights indicate strong associations between corresponding nodes, the weight of each edge in the constructed network was defined as 1,000 minus the interaction score of the corresponding interaction.
SP method for searching new candidate cancer drivers
It has been reported in some studies that two proteins in a PPI may share similar functions33,35,38,41,42,43. By further considering the interaction score, two proteins in a PPI with a high score are more likely to share similar functions. This can be further induced by the fact that if p1, p2,…,ps is a series of proteins such that pi and pi+1 can comprise a PPI with a high score (the corresponding edge in the network was assigned a low weight) and p1 and ps are two proteins encoded by dysfunctional genes, then p2,…,ps−1 may also be encoded by dysfunctional genes. By mapping p1, p2,…,ps to the network constructed in Section “Network construction”, the corresponding nodes can comprise the shortest path by the construction of the network. Because dysfunctional genes can be divided into four levels, comprising of four sets of seed genes denoted by G1, G2, G3 and G4, we searched candidate genes by pairing Gi and Gj (i ≠ j) to identify new genes that can drive tumorigenesis on two levels. Thus, for any Gi and Gj (i ≠ j), we searched all of the shortest paths connecting genes in Gi and Gj by Dijkstra’s algorithm44. Accordingly, we obtained six sets of the shortest paths. For each set, we extracted genes that occurred in at least one path as the candidate genes. Furthermore to distinguish them, a measurement, namely betweenness45, was conducted for each candidate gene, which is defined as the number of paths containing the gene. Betweenness is a measure of centrality of a vertex within a graph which counts the number of times a node acts as a bridge in the shortest path between two other nodes, which in this study can be used to judge whether the candidate genes can drive tumor initiation on two levels46. For convenience, the set consisting of candidate genes for Gi and Gj is denoted by  .
.
Permutation test
For Gi and Gj (i ≠ j), we can obtain a set of candidate genes, making up the gene set  , by the method described in Section “SP method for searching new candidate cancer drivers”. However, not all of them have the potential to become novel driver genes. False positives are inevitable. Among them, some are produced by the construction of the network. Taking this into consideration, we randomly produced two groups of gene sets; each group contained 1,000 gene sets, denoted by
, by the method described in Section “SP method for searching new candidate cancer drivers”. However, not all of them have the potential to become novel driver genes. False positives are inevitable. Among them, some are produced by the construction of the network. Taking this into consideration, we randomly produced two groups of gene sets; each group contained 1,000 gene sets, denoted by  and
and  .
.  is the same size as Gi , while
is the same size as Gi , while  is the same size as Gj . For
is the same size as Gj . For  and
and  (k = 1,2, …, 1000), all the shortest paths connecting one gene in
(k = 1,2, …, 1000), all the shortest paths connecting one gene in  and one gene in
and one gene in  were searched in the constructed network. We counted the betweenness of the candidate genes in
were searched in the constructed network. We counted the betweenness of the candidate genes in  based on these paths. Thus, each candidate gene had one betweenness on Gi and Gj and 1,000 betweenness on
based on these paths. Thus, each candidate gene had one betweenness on Gi and Gj and 1,000 betweenness on  and
and  (k = 1,2, …,1000). Another measurement, namely permutation FDR, was computed for each candidate gene, which is defined as the ratio of the 1,000 betweenness on
(k = 1,2, …,1000). Another measurement, namely permutation FDR, was computed for each candidate gene, which is defined as the ratio of the 1,000 betweenness on  and
and  (k = 1,2, …,1000) that are larger than the betweenness on Gi and Gj. It can be seen that a candidate gene with a high permutation FDR is more likely to be a false positive produced by the construction of the network and should be excluded. Thus, candidate genes with a permutation FDR larger than or equal to 0.05 were discarded. The remaining candidate genes, making up the gene set
(k = 1,2, …,1000) that are larger than the betweenness on Gi and Gj. It can be seen that a candidate gene with a high permutation FDR is more likely to be a false positive produced by the construction of the network and should be excluded. Thus, candidate genes with a permutation FDR larger than or equal to 0.05 were discarded. The remaining candidate genes, making up the gene set  , were further evaluated by the method in the following section.
, were further evaluated by the method in the following section.
Further selection using betweenness and PPI
For Gi and Gj (i ≠ j), some candidate genes remained after executing the permutation test. However, some of them may have strong associations with cancer, while others have weak associations. To reflect this fact, we proposed some rules in this section and selected the important candidate genes based on these rules.
It has been elaborated that the betweenness of a candidate gene is the number of paths connecting genes in Gi and in Gj including the candidate gene. Clearly, a candidate gene with high betweenness suggests it has strong associations with genes in Gi and Gj, thereby having a high likelihood of being a novel cancer driver gene. To build a uniform rule, we must also consider the sizes of Gi and Gj because a candidate gene with a small betweenness for small Gi and Gj is not always less important than another candidate gene with a large betweenness for large Gi and Gj. Thus, we set a betweenness ratio R(g) to measure the importance of a candidate gene g based on its betweenness and sizes of Gi and Gj, which is defined as

It can be seen that a high betweenness ratio of a candidate gene means that the majority of the shortest paths connecting genes in Gi and in Gj contains the candidate gene. We set a threshold of 0.01 for the betweenness ratio to select important candidate genes.
Another rule was built based on the PPIs and their interaction scores. It has been reported that two proteins in a PPI with a high score are more likely to share similar functions33,47,48. Thus, for a candidate gene g, if g and genes in Gi and Gj can comprise PPIs with high interaction scores, g has strong associations with genes in Gi and Gj. Thus, we computed the following value, namely the min-max interaction score:

Similarly, a candidate gene with a high min-max interaction score implies that it has strong associations with at least one gene in Gi and at least one gene in Gj, indicating that it has a high linkage with cancer. Similar to the proportion mentioned in the paragraph above, we can also set a threshold of 400 for the min-max interaction score to select important candidate genes.
For the gene sets Gi and Gj, genes in  were filtered by the above two rules. The remaining genes constituted the set
were filtered by the above two rules. The remaining genes constituted the set  , which were deemed to be significant for two levels.
, which were deemed to be significant for two levels.
Results
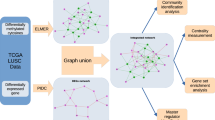
In this study, we proposed a computational method to identify candidate cancer driver genes that can drive tumor initiation on multiple levels. The flowchart of our method is shown in Fig. 1. The results of our method are described in the following sections.

Flowchart of our method.
(A) Four gene sets consisting of dysfunctional genes on four levels; (B) SP method to search candidates in a network. Yellow nodes represent dysfunctional genes on different levels and the dashed lines represent the shortest path connecting a and i, e and h are selected; (C) Six candidate gene sets obtained by the SP method; (D) Permutation test to filter some false positives. Two randomly produced sets {d, f} and {c, g} were shown in the network (highlighted in red and green), in detail, red nodes d and c replace yellow node a, while green nodes g and f replace yellow node i, dotted lines represent the shortest path connecting d and f, dashed-dotted lines represent the shortest path connecting c and g and e (highlighted in pink) is removed by the permutation test; (E) Six candidate gene sets filtered by the permutation test; (F) Six candidate gene sets filtered by further selection using betweenness and PPI.
Results of the SP method
As mentioned in Section “Identification of the differentially expressed mRNA genes, microRNAs, methylated CpG sites and somatic mutation genes”, we employed four gene sets G1, G2, G3 and G4 with different expression levels for cancer. For each pair, e.g., Gi and Gj (i ≠ j), we searched all the shortest paths connecting any gene in Gi with any gene in Gj in a network constructed in Section “Network construction” and extracted genes on these paths. The obtained six sets of candidate genes,  , are provided in the Supplementary Material II. The numbers of genes in these sets are listed in column 2 of Table 1. It can be seen that many candidate genes were included in each set, meaning that further filtering was necessary. Furthermore, the betweenness of each candidate gene in
, are provided in the Supplementary Material II. The numbers of genes in these sets are listed in column 2 of Table 1. It can be seen that many candidate genes were included in each set, meaning that further filtering was necessary. Furthermore, the betweenness of each candidate gene in  was calculated and also provided in the Supplementary Material II.
was calculated and also provided in the Supplementary Material II.
Results of the permutation test
To control for the false positives produced by the construction of the network in each candidate set  , a permutation test was adopted. A permutation FDR was calculated for each candidate gene in
, a permutation test was adopted. A permutation FDR was calculated for each candidate gene in  , which is also listed in the Supplementary Material II. By setting a threshold of 0.05 for the permutation FDR, we extracted a candidate gene subset
, which is also listed in the Supplementary Material II. By setting a threshold of 0.05 for the permutation FDR, we extracted a candidate gene subset  from
from  . The detailed genes in the six gene sets
. The detailed genes in the six gene sets  are provided in the Supplementary Material III and the numbers of genes in these sets are listed in column 3 of Table 1, from which we can see that the number of candidate genes decreased significantly and became close to the reality.
are provided in the Supplementary Material III and the numbers of genes in these sets are listed in column 3 of Table 1, from which we can see that the number of candidate genes decreased significantly and became close to the reality.
Results of further selection
To select the core genes in  , we calculated the betweenness ratio (cf. Equation 1) and the min-max interaction score (cf. Equation 2) for each candidate gene using a threshold of 0.01 for the betweenness ratio and a threshold of 400 for the min-max interaction score. These two measurements of each candidate gene are provided in the Supplementary Material III and the remaining genes are listed in the Supplementary Material IV. The number of genes in the gene sets
, we calculated the betweenness ratio (cf. Equation 1) and the min-max interaction score (cf. Equation 2) for each candidate gene using a threshold of 0.01 for the betweenness ratio and a threshold of 400 for the min-max interaction score. These two measurements of each candidate gene are provided in the Supplementary Material III and the remaining genes are listed in the Supplementary Material IV. The number of genes in the gene sets  is listed in column 4 of Table 1. Compared to the number of candidate genes listed in column 3 of Table 1, the candidate genes were again decreased significantly. It is believed that these candidate genes have few false positives and have strong associations with cancer. Some of them are discussed in the following section.
is listed in column 4 of Table 1. Compared to the number of candidate genes listed in column 3 of Table 1, the candidate genes were again decreased significantly. It is believed that these candidate genes have few false positives and have strong associations with cancer. Some of them are discussed in the following section.
Discussion
Analysis of candidate genes of two levels
Cancer driver genes as we have mentioned above have been widely reported to be the driving force for the tumorigenesis. According to our method, we obtained various genes that contribute to the initiation and progression of lung adenocarcinoma on at least two levels of the four basic driver levels (methylation, mutations, microRNA expression and expression/mRNA). The important candidates involving any two driver levels are listed in Tables 2, 3, 4, 5, 6, 7. Here, we only provide the brief analyses, the detailed analyses are provided in Supplementary Material V.
 (based on methylated CpG site genes and microRNA target genes) identified by our method.
(based on methylated CpG site genes and microRNA target genes) identified by our method. (based on methylated CpG site genes and somatic mutation genes) identified by our method.
(based on methylated CpG site genes and somatic mutation genes) identified by our method. (based on methylated CpG site genes and mRNA genes) identified by our method.
(based on methylated CpG site genes and mRNA genes) identified by our method. (based on microRNA target genes and somatic mutation genes) identified by our method.
(based on microRNA target genes and somatic mutation genes) identified by our method. (based on microRNA target genes and mRNA genes) identified by our method.
(based on microRNA target genes and mRNA genes) identified by our method. (based on somatic mutation genes and mRNA genes) identified by our method.
(based on somatic mutation genes and mRNA genes) identified by our method.For the levels of methylation diversity and microRNA expression abundance, 27 genes were predicted to driver the lung adenocarcinoma in such two levels. Among them, six of them have the evidences to support the claim and are listed in Table 2. Interacting with specific microRNAs such as microRNA-142-3p, functional genes like TCF3, MEN1, MLL, EFNA4, PBX1 and SHH have all been confirmed to contribute to tumorigenesis via methylation alteration and microRNA regulation (The detailed analysis of the important candidates can be seen in Supplementary Material V). Take TCF3 as an example. The methylation alteration of TCF3 has been confirmed to contribute to the proliferation of A549 cells, a typical lung adenocarcinoma cell line in vitro experiments, implying that such gene may also contribute to the initiation and progression of lung adenocarcinoma on such level49. As for the microRNA level, the interactions between TCF3 and a group of microRNAs (miR-590, miR-17 and miR-18) has been confirmed, validating that TCF3 may also contribute to the initiation and progression of lung adenocarcinoma on such level50. For the methylation diversity and mutation differentiation, there are still several genes (such as TCF3, MLL, MEN1, SHH, CTNNB1 and FZD1, listed in Table 3) that have been reported to participate in the tumor associated pathways (The detailed analysis of the important candidates can be seen in Supplementary Material V). The methylation and mutation status of such genes have been confirmed to be abnormal during the progression of lung adenocarcinoma and similar solid tumors. Take FZD1 as an example, FZD1 is a functional gene on our list, which has been widely reported to be related to various tumor subtypes51,52. The methylation status of this gene has been associated with prostate cancer and age-associated diseases53,54. There are only a few reports of FZD1-associated mutations. However, it has been proven that the mutations of FZD-1 may be crucial for specific diseases including tumors, validating our prediction55,56. Considering that the gene expression is regulated by specific methylation process in the genome, this level, corresponding to the mRNA level diversity between the tumor tissue and the adjacent normal tissue, is associated with the first level (methylation diversity). Genes like MEN1, TCF3 and SHH, listed in Table 4, are all crucial genes that contribute to the initiation and progression of tumors on multiple levels (The detailed analysis of the important candidates can be seen in Supplementary Material V). TCF3 as we have mentioned above turns out to be confirmed to contribute to lung adenocarcinoma on methylation alteration49. What’s more, considering the expression regulation function of microRNAs, such identified microRNA-associated cancer driver may also contribute to tumor genesis on mRNA level. Some of the candidate genes have also been predicted to be related to both microRNA expression differentiation and mutation diversity of malignant and somatic cells. Interacting with microRNA-365 and microRNA-27b, the most important candidates for these two levels are listed in Table 5. COL1A2, SHC1, FKBP1A, TTN and NGF are all crucial cancer driver genes that contribute to lung adenocarcinoma in their respective ways (The detailed analysis of the important candidates can be seen in Supplementary Material V). The fifth group of genes (including PTCH1, SHH, ITGA2, ITGA5, GRB2, EP300 and SMC3, listed in Table 6) contribute to the progression of lung adenocarcinoma on at least the microRNA regulation and mRNA expression level (The detailed analysis of the important candidates can be seen in Supplementary Material V). As for the last set of genes, such genes ITGA2, ITGA5, NOTCH1, PXN and DYNLL1, listed in Table 7, contributes to tumors on both the mutation and mRNA levels (The detailed analysis of the important candidates can be seen in Supplementary Material V). All of our predicted genes that contribute to at least two levels have been confirmed to be real driver genes by recent publications
Analysis of candidate genes of high frequencies
In Section “Results”, six sets of candidate genes were obtained that were deemed to induce tumor initiation and progression on two levels. We took the union operation of these six sets and obtained 110 candidate genes. Among them, some genes occurred many times, meaning that they may drive tumor initiation and progression on multiple levels. Thus, the frequencies of 110 candidate genes were counted and listed in the Supplementary Material VI. Because there were totally six sets of candidate genes, six is the maximum value of frequencies for each candidate gene. This section gives a detailed discussion of the genes with frequencies greater than three (half of the maximum frequency), which are listed in Table 8. These candidate genes occurred in more than half of the candidate gene sets and have been reported and confirmed to participate in and contribute to the process of tumorigenesis. Here, their brief discussion is provided. Readers can found the detailed analyses in Supplementary Material VII.
Genes occurred in more than three gene sets have been analyzed. Three genes, PTCH1, CTNNB1 and FYN, have been predicted to contribute to the initiation and progression of lung adenocarcinoma in all the six gene sets. Such three genes have all been regarded as core functional cancer driver genes. Associated with proliferation and adhesion associated pathways such a PI-3K cascade, such three genes not only participate in the initiation and proliferation of the tumor , but regulate the metastasis processes as well57 (The detailed analysis of such genes can be seen in the Supplementary Material VII). As for the five genes (BCAR1, SHH, NGF, VEGFA and GCG) that can be identified to be shared in five gene sets, they are also confirmed to be significant driver genes. Take BCAR1 as an example, such gene involves in crucial regulatory pathways like tyrosine kinase signaling pathways and further contribute to the survival, proliferation and invasion processes during tumorigenesis58 (The detailed analysis of such genes can be seen in the Supplementary Material VII). Quite more genes have been clustered in the group with the regulatory level frequency of four. Such genes regulate the abnormal pathways during the tumorigenesis processes such as the cell-cell adhesion regulation (CDH1), proliferation (STAT3, SRC) and chronic inflammatory reaction (LEP). All of such genes can be confirmed to be cancer driver genes by recent publications (The detailed analysis of such genes can be seen in the Supplementary Material VII).
As we have mentioned above, we identified a group of candidate cancer drivers that contribute to lung adenocarcinoma in multiple levels, which are all proved by recent literatures. Here, we may propose a new hypothesis for the initiation and progression of lung adenocarcinoma: the real core driver of lung adenocarcinoma (and maybe other cancers) may contribute to tumor genesis simultaneously on multiple levels. Considering the complicated regulatory system of human bodies, a single abnormal variation that contribute to the genomic alterations of a single level (e.g. mutations) may not be functional and significant enough to initiate the tumor genesis. The real core driver of cancer (lung adenocarcinoma) may contribute to tumor genesis on at least two levels to insure the initiation of malignant changes of normal cells. For example, the well-known typical core drivers for lung adenocarcinoma like EGFR all contribute to lung adenocarcinoma on multiple levels, though haven’t been identified and analyzed in the same publications19,59. All in all, based on our newly presented computational methods, we not only identified a group of novel cancer drivers for lung adenocarcinoma, but presented a new perspective for the underlying mechanisms of tumor genesis, providing a new sight into the initiation and progression of lung adenocarcinoma.
Conclusions
This contribution investigated the so-called cancer driver genes. A computational method was built to identify new potential candidate cancer driver genes. The analyses indicate that some of the obtained genes have the potential to drive tumorigenesis on multiple differentiation levels. It is hopeful that the findings presented in this study will promote the study of cancer driver genes and provide new insights into the investigation of tumor initiation. In this study, we used the protein information (protein-protein interaction) to investigate cancer driver genes. In future, we will consider adding some other information, such as microRNA related to cancer60, into our method, which may yield more useful information for the study of cancer driver gene.
Additional Information
How to cite this article: Chen, L. et al. Identification of novel candidate drivers connecting different dysfunctional levels for lung adenocarcinoma using protein-protein interactions and a shortest path approach. Sci. Rep. 6, 29849; doi: 10.1038/srep29849 (2016).
References
DeSantis, C. E. et al. Cancer treatment and survivorship statistics, 2014. CA Cancer J Clin 64, 252–271, doi: 10.3322/caac.21235 (2014).
Conteduca, V., Sansonno, D., Russi, S. & Dammacco, F. Precancerous colorectal lesions (Review). International journal of oncology 43, 973–984, doi: 10.3892/ijo.2013.2041 (2013).
Tschaharganeh, D. F. et al. p53-dependent Nestin regulation links tumor suppression to cellular plasticity in liver cancer. Cell 158, 579–592, doi: 10.1016/j.cell.2014.05.051 (2014).
Visani, M. et al. Multiple KRAS mutations in pancreatic adenocarcinoma: molecular features of neoplastic clones indicate the selection of divergent populations of tumor cells. Int J Surg Pathol 21, 546–552, doi: 10.1177/1066896912475073 (2013).
Liang, H. et al. Whole-exome sequencing combined with functional genomics reveals novel candidate driver cancer genes in endometrial cancer. Genome Res 22, 2120–2129, doi: 10.1101/gr.137596.112 (2012).
Paziewska, A. et al. DNA methylation status is more reliable than gene expression at detecting cancer in prostate biopsy. British journal of cancer 111, 781–789, doi: 10.1038/bjc.2014.337 (2014).
Cul’bová, M. et al. Methylation of selected tumor-supressor genes in benign and malignant ovarian tumors. Ceska Gynekol 76, 274–279 (2011).
Colangelo, T. et al. MicroRNA-130b Promotes Tumor Development and Is Associated with Poor Prognosis in Colorectal Cancer. Neoplasia 15, 1086–1099, doi: 10.1593/neo.13998 (2013).
Ota, H. et al. ADAR1 forms a complex with Dicer to promote microRNA processing and RNA-induced gene silencing. Cell 153, 575–589, doi: 10.1016/j.cell.2013.03.024 (2013).
Xiong, J., Du, Q. & Liang, Z. Tumor-suppressive microRNA-22 inhibits the transcription of E-box-containing c-Myc target genes by silencing c-Myc binding protein. Oncogene 29, 4980–4988, doi: 10.1038/onc.2010.241 (2010).
Xu, J. M. et al. KRAS mutations in tumor tissue and plasma by different assays predict survival of patients with metastatic colorectal cancer. Journal of experimental & clinical cancer research : CR 33, 104, doi: 10.1186/s13046-014-0104-7 (2014).
Reitman, Z. J., Pirozzi, C. J. & Yan, H. Promoting a new brain tumor mutation: TERT promoter mutations in CNS tumors. Acta neuropathologica 126, 789–792, doi: 10.1007/s00401-013-1207-5 (2013).
Gao, L., Yang, Q. H. & Xu, R. K. [Melatonin inhibits the proliferation of pituitary prolactin-secreting tumor by suppressing the enhancer elements mutation of PRL gene in the rat]. Sheng Li Xue Bao 57, 319–327 (2005).
Fredriksson, N. J., Ny, L., Nilsson, J. A. & Larsson, E. Systematic analysis of noncoding somatic mutations and gene expression alterations across 14 tumor types. Nat Genet 46, 1258–1263, doi: 10.1038/ng.3141 (2014).
Conti, A. et al. Expression of the tumor necrosis factor receptor-associated factors 1 and 2 and regulation of the nuclear factor-kappaB antiapoptotic activity in human gliomas. J Neurosurg 103, 873–881, doi: 10.3171/jns.2005.103.5.0873 (2005).
Normanno, N. & Cree, I. A. Genomics driven-oncology: challenges and perspectives. BMC Cancer 15, 141, doi: 10.1186/s12885-015-1147-7 (2015).
Liang, Y., Wakelee, H. A. & Neal, J. W. Relationship of Driver Oncogenes to Long-Term Pemetrexed Response in Non–Small-Cell Lung Cancer. Clin Lung Cancer 16, 366–373, doi: 10.1016/j.cllc.2014.12.009 (2015).
Jin, Y. et al. ROS1 gene rearrangement and copy number gain in non-small cell lung cancer. Virchows Arch 466, 45–52, doi: 10.1007/s00428-014-1679-2 (2015).
Zhang, Y. et al. The prognostic and predictive value of solid subtype in invasive lung adenocarcinoma. Sci Rep 4, 7163, doi: 10.1038/srep07163 (2014).
Qiu, T., Guo, H., Zhao, H., Wang, L. & Zhang, Z. Next-generation sequencing for molecular diagnosis of lung adenocarcinoma specimens obtained by fine needle aspiration cytology. Sci Rep 5, 11317, doi: 10.1038/srep11317 (2015).
Lee, S. Y. et al. A genetic variation in microRNA target site of KRT81 gene is associated with survival in early-stage non-small-cell lung cancer. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO 26, 1142–1148, doi: 10.1093/annonc/mdv100 (2015).
Cancer Genome Atlas Research, N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550, doi: 10.1038/nature13385 (2014).
Banerji, S. et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature 486, 405–409, doi: 10.1038/nature11154 (2012).
Tusher, V. G., Tibshirani, R. & Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA 98, 5116–5121, doi: 10.1073/pnas.091062498 (2001).
Griffiths-Jones, S., Grocock, R. J., van Dongen, S., Bateman, A. & Enright, A. J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res 34, D140–144, doi: 10.1093/nar/gkj112 (2006).
Friedman, R. C., Farh, K. K., Burge, C. B. & Bartel, D. P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19, 92–105, doi: 10.1101/gr.082701.108 (2009).
Betel, D., Wilson, M., Gabow, A., Marks, D. S. & Sander, C. The microRNA.org resource: targets and expression. Nucleic Acids Res 36, D149–153, doi: 10.1093/nar/gkm995 (2008).
Sethupathy, P., Corda, B. & Hatzigeorgiou, A. G. TarBase: A comprehensive database of experimentally supported animal microRNA targets. RNA 12, 192–197, doi: 10.1261/rna.2239606 (2006).
Wang, X. & El Naqa, I. M. Prediction of both conserved and nonconserved microRNA targets in animals. Bioinformatics 24, 325–332, doi: 10.1093/bioinformatics/btm595 (2008).
Krek, A. et al. Combinatorial microRNA target predictions. Nat Genet 37, 495–500, doi: 10.1038/ng1536 (2005).
von Mering, C. et al. STRING: a database of predicted functional associations between proteins. Nucleic Acids Res 31, 258–261 (2003).
Franceschini, A. et al. STRING v9. 1: protein-protein interaction networks, with increased coverage and integration. Nucleic acids research 41, D808–D815 (2013).
Hu, L. L. et al. Predicting functions of proteins in mouse based on weighted protein-protein interaction network and protein hybrid properties. PLoS ONE 6, e14556 (2011).
Chen, L. et al. Application of the shortest path algorithm for the discovery of breast cancer related genes. Current Bioinformatics 11, 51–58 (2014).
Gao, P., Wang, Q. P., Chen, L. & Huang, T. Prediction of Human Genes Regulatory Functions Based on Proteinprotein Interaction Network. Protein and Peptide Letters 19, 910–916 (2012).
Chen, L., Chu, C., Kong, X., Huang, G. & Huang, T. A Hybrid Computational Method for the Discovery of Novel Reproduction-Related Genes. PLoS ONE 10, e0117090 (2015).
Jiang, Y. et al. Identifying Gastric Cancer Related Genes Using the Shortest Path Algorithm and Protein-Protein Interaction Network. BioMed Research International 2014, 371397 (2014).
Chen, L., Chu, C., Kong, X., Huang, T. & Cai, Y. Discovery of New Candidate Genes Related to Brain Development Using Protein Interaction Information. PLoS ONE 10, e0118003 (2015).
Chen, L. et al. Mining for novel tumor suppressor genes using a shortest path approach. Journal of Biomolecular Structure and Dynamics 34, 664–675, doi: 10.1080/07391102.2015.1042915 (2016).
Zhang, J., Yang, J., Huang, T., Shu, Y. & Chen, L. Identification of novel proliferative diabetic retinopathy related genes on protein-protein interaction network. Neurocomputing (2016).
Ng, K. L., Ciou, J. S. & Huang, C. H. Prediction of protein functions based on function-function correlation relations. Comput Biol Med 40, 300–305, doi: DOI 10.1016/j.compbiomed.2010.01.001 (2010).
Li, B. Q., Huang, T., Liu, L., Cai, Y. D. & Chou, K. C. Identification of Colorectal Cancer Related Genes with mRMR and Shortest Path in Protein-Protein Interaction Network. PLoS One 7, e33393, doi: 10.1371/journal.pone.0033393 (2012).
Jiang, M. et al. Identification of hepatocellular carcinoma related genes with k-th shortest paths in a protein-protein interaction network. Mol Biosyst 9, 2720–2728, doi: 10.1039/c3mb70089e (2013).
Gormen, T. H., Leiserson, C. E., Rivest, R. L. & Stein, C. (MIT press Cambridge, MA, 1990).
Kitsak, M. et al. Betweenness centrality of fractal and nonfractal scale-free model networks and tests on real networks. Physical review. E, Statistical, nonlinear and soft matter physics 75, 056115 (2007).
Cukierski, W. J. & Foran, D. J. Using Betweenness Centrality to Identify Manifold Shortcuts. Proc IEEE Int Conf Data Min 2008, 949–958 (2008).
Gao, Y. F. et al. Predicting Metabolic Pathways of Small Molecules and Enzymes Based on Interaction Information of Chemicals and Proteins. PLoS ONE 7, e45944 (2012).
Sharan, R., Ulitsky, I. & Shamir, R. Network-based prediction of protein function. Mol Syst Biol 3, 88 (2007).
Barnie, P. A. et al. CpG-oligodeoxynucleotides suppress the proliferation of A549 lung adenocarcinoma cells via toll-like receptor 9 signaling and upregulation of Runt-related transcription factor 3 expression. Biomed Rep 2, 374–377, doi: 10.3892/br.2014.257 (2014).
Hu, J. et al. MiR-145 regulates cancer stem-like properties and epithelial-to-mesenchymal transition in lung adenocarcinoma-initiating cells. Tumour Biol 35, 8953–8961, doi: 10.1007/s13277-014-2158-8 (2014).
Zhang, H. et al. Interference of Frizzled 1 (FZD1) reverses multidrug resistance in breast cancer cells through the Wnt/beta-catenin pathway. Cancer letters 323, 106–113, doi: 10.1016/j.canlet.2012.03.039 (2012).
Hung, T. H. et al. FZD1 activates protein kinase C delta-mediated drug-resistance in multidrug-resistant MES-SA/Dx5 cancer cells. Int J Biochem Cell Biol 53, 55–65, doi: 10.1016/j.biocel.2014.04.011 (2014).
Devaney, J. M. et al. Identification of novel DNA-methylated genes that correlate with human prostate cancer and high-grade prostatic intraepithelial neoplasia. Prostate Cancer P D 16, 292–300, doi: 10.1038/pcan.2013.58 (2014).
Salpea, P. et al. Postnatal development- and age-related changes in DNA-methylation patterns in the human genome. Nucleic Acids Research 40, 6477–6494, doi: 10.1093/nar/gks312 (2012).
Katoh, M. Molecular cloning and characterization of WRCH2 on human chromosome 15q15. International journal of oncology 20, 977–982 (2002).
Kaykas, A. et al. Mutant Frizzled 4 associated with vitreoretinopathy traps wild-type Frizzled in the endoplasmic reticulum by oligomerization. Nature Cell Biology 6, 52–U13, doi: 10.1038/ncb1081 (2004).
Elias, D. & Ditzel, H. J. Fyn is an important molecule in cancer pathogenesis and drug resistance. Pharmacological research : the official journal of the Italian Pharmacological Society 100, 250–254, doi: 10.1016/j.phrs.2015.08.010 (2015).
Grebenchtchikov, N. et al. Development of an ELISA for measurement of BCAR1 protein in human breast cancer tissue. Clin Chem 50, 1356–1363, doi: 10.1373/clinchem.2003.029868 (2004).
Schneeberger, V. E. et al. Inhibition of Shp2 suppresses mutant EGFR-induced lung tumors in transgenic mouse model of lung adenocarcinoma. Oncotarget 6, 6191–6202, doi: 10.18632/oncotarget.3356 (2015).
Zeng, X., Zhang, X. & Zou, Q. Integrative approaches for predicting microRNA function and prioritizing disease-related microRNA using biological interaction networks. Briefings in bioinformatics 17, 193–203, doi: 10.1093/bib/bbv033 (2016).
Acknowledgements
This study was supported by the National Natural Science Foundation of China (31371335), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA12050201) and the Hi-Tech Research and Development Program of China (2014AA01A302).
Author information
Authors and Affiliations
Contributions
T.H., M.Z., Y.J. and Y.-D.C. conceived the ideas and the study. L.C., Y.J. and Y.-D.C. performed the experiments. L.C., Y.-H.Z., T.H. and M.Z. analyzed the results. L.C. and Y.-H.Z. wrote the manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Chen, L., Huang, T., Zhang, YH. et al. Identification of novel candidate drivers connecting different dysfunctional levels for lung adenocarcinoma using protein-protein interactions and a shortest path approach. Sci Rep 6, 29849 (2016). https://doi.org/10.1038/srep29849
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep29849
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
