
Abstract
MoS2 nanosheet, a new two-dimensional transition metal dichalcogenides nanomaterial, has attracted significant attentions lately due to many potential promising biomedical applications. Meanwhile, there is also a growing concern on its biocompatibility, with little known on its interactions with various biomolecules such as proteins. In this study, we use all-atom molecular dynamics simulations to investigate the interaction of a MoS2 nanosheet with Villin Headpiece (HP35), a model protein widely used in protein folding studies. We find that MoS2 exhibits robust denaturing capability to HP35, with its secondary structures severely destroyed within hundreds of nanosecond simulations. Both aromatic and basic residues are critical for the protein anchoring onto MoS2 surface, which then triggers the successive protein unfolding process. The main driving force behind the adsorption process is the dispersion interaction between protein and MoS2 monolayer. Moreover, water molecules at the interface between some key hydrophobic residues (e.g. Trp-64) and MoS2 surface also help to accelerate the process driven by nanoscale drying, which provides a strong hydrophobic force. These findings might have shed new light on the potential nanotoxicity of MoS2 to proteins with atomic details, which should be helpful in guiding future biomedical applications of MoS2 with its nanotoxicity mitigated.
Similar content being viewed by others
Introduction
The most common two-dimensional (2D) nanomaterials are probably those carbon-based ones, such as graphene, graphyne and their derivatives, which have attracted tremendous interests in many fields including biomedicine since its discovery1,2,3,4,5,6,7. Novel 2D nanomaterials, such as MoS28, WS29 and WO310, are quickly catching up and emerge as a new research frontier. These materials have been featured with unparalleled structural amenability, exceptionally high specific surface area11, unusual size-dependent effects12 and excellent mechanical and electrical properties13,14. Therefore, there have been many attempts to take advantage of various 2D nanomaterials as delivery platforms, diagnostic agents, therapeutic nanodrugs and tissue engineering scaffolds15,16,17,18,19,20,21.
Accompanying with these promising biomedical applications, there is also a growing concern on the biosafety and cytotoxicity of these 2D nanomaterials22,23,24,25. Taking graphene as an example, its potential cytotoxicity has been raised widely in literature. Its adverse effects on tissues, cells and various biomolecules have been heavily investigated with various experimental techniques26. Meanwhile, recent theoretical studies have also revealed graphene’s (including graphene oxide) influence on the integrity of cell membranes27,28,29,30, as well as protein structures23,25. Based on these findings from both experiment and theory on the fundamental mechanisms, various strategies have been developed to enhance graphene’s biocompatibility, such as functionalization with organic molecules, lipids, polymers, peptides and proteins31,32,33,34,35,36.
More interestingly, MoS2 (molybdenum disulfide)8, a branch in 2D transition metal dichalcogenides nanomaterials, is receiving a significant amount of attention lately. It is believed that molybdenum disulfide might share similar physicochemical properties with graphene and can potentially replicate graphene’s success in biomedical applications. Recent studies indicate that it has strong antimicrobial and antifungal activity37,38. Meanwhile, a field effect biosensor has been proposed for tumor marker proteins using its unique direct band gap39. In addition, its high near-infrared (NIR) absorbance and extensive specific surface area makes it ideal as a novel photothermal-triggered drug delivery platform40. It is also proposed for cancer therapy through a combined approach with both photothermal and chemotherapeutic agents39,41. MoS2 can also be used as a contrast agent in X-ray tomography imaging with Mo’s excellent absorption ability40.
Despite these efforts, the detailed molecular interactions between MoS2 nanosheets and various biomolecules such as proteins remain largely unknown. In this work, we conduct all-atom molecular dynamics (MD) simulations to study the interaction of Villin Headpiece (HP35), a widely used model protein in folding studies, with MoS2. It is shown that MoS2 exhibits exceptionally robust denaturation capability to HP35, with the protein secondary structures all severely destroyed within a few hundred nanosecond simulations, indicating a potentially severe nanotoxicity. Both the aromatic and basic residues contribute to the initial protein anchoring on the surface of MoS2, which then trigger the successive protein unfolding process, with hydrophobic residues play a key role in the denaturing process.
Models and Methods
The HP35 is a protein that folds independently into a three-helix bundle, which has been widely used as a model scaffold in protein folding studies. The initial protein structure used in this study was downloaded from the Protein Data Bank with the PDB code 1YRF42. The simulation system consisted of a HP35 protein and a MoS2 nanosheet with a size of 6.735 nm × 6.600 nm. The MoS2 nanosheet parameters were adopted from a previous study (see Table S1 for details)43. The initial distance between the protein and the MoS2 nanosheet was set to be 0.8 nm. The complex system was then solvated in a rectangular water box (6.924 nm × 6.744 nm × 5.000 nm), containing 6,568 water molecules (21,728 atoms in total). Two chloride ions were also added to this water box to neutralize the system. This fully solvated complex was then simulated with molecular dynamics simulations, which are widely used in the studies of biomolecules44,45,46,47,48,49,50,51,52 and nanomaterials23,53,54,55,56,57.
The MD simulation was performed with the software package GROMACS (version 4.6.6)58. The VMD software59 was used to analyze and visualize the simulation results. We adopted CHARMM 27 force field60 and TIP3P water model61 for the protein and water molecules, respectively. Temperature was fixed at 300 K using v-rescale thermostat and the volume of the simulation box also remained constant during the simulation (NVT)62. Periodic boundary conditions were applied in all directions. To avoid the “artificial collapsing” of nanosheets with their mirror images due to the limited size of simulation box (which is due to the limited computational resource), the MoS2 nanosheet was frozen throughout the simulation process. The long-range electrostatic interactions were treated with PME method63 and the van der Waals (vdW) interactions were calculated with a cutoff distance of 1.0 nm. All solute bonds were maintained constant at their equilibrium values with the LINCS algorithm64 and water geometry was also constrained using the SETTLE algorithm65. During the production runs, a time step of 2.0 fs was used and data were collected every 20 ps. The total aggregated simulation time was larger than 2 μs.
Results and Discussions
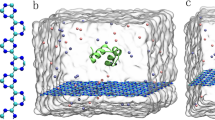
Our simulation shows that the MoS2 nanosheet can be highly detrimental to the native protein folds. In all simulations with protein HP35, we found that the characteristic helical structures of HP35 have been severely destroyed on MoS2 surface. As featured in the final snapshots from three independent runs at 500 ns (Fig. 1B), HP35 lost most of its native α-helical content (~80%). This is in high contrast with the control run with protein HP35 in bulk water without the MoS2 nanosheet (Fig. 1A), which displays only marginal difference from the initial crystal structure.

Simulations in bulk water and on MoS2 surface.
(A) A snapshot of the control simulation in the bulk water. The initial and final structures of HP35, respectively marked in yellow and cyan, are superimposed in order to show their structural consistency. Water box is depicted with white surface and two Cl− ions (red spheres) are introduced to neutralize the system. (B) Characteristic structures of HP35 on MoS2 surface. The initial fold of HP35 on MoS2 surface is shown in the bottom panel (see Figure S1 for more view angles) and the final denatured proteins obtained at t = 500 ns from three independent runs are displayed in the top. Mo and S atoms of MoS2 surface are depicted with pink and white van der Waals balls, respectively.
For a more quantitative measure on how the adsorption affects the protein tertiary structure, we then monitored the atomic contacts of HP35 on the MoS2 surface and the native contacts of the protein. Here, a contact with MoS2 surface was counted when any heavy atom of the sidechain of HP35 is within 0.6 nm of any atom of MoS2. With a same criterion on the cut-off distance, the fraction of native contacts of protein at time t, Q(t), was defined as the ratio of the total number of native contacts at time t to that at time zero (i.e., the total number of native contacts in the x-ray crystal structure), where only residue pairs apart at least 3 consecutive residues from each other were considered.
As shown in Fig. 2, HP35 seriously loses its native contact as the adsorption progresses. Especially, the extent of protein denaturation is directly proportional to that of surface adsorption (Fig. 2A), which strongly implies that MoS2 directly impacts on the protein tertiary structure. Accordingly, it is accompanied with deformation on the secondary structures as well as the local hydrogen bonding network (Fig. 2B,C). More specifically, for the first 15 ns after the onset, HP35 quickly lost its native contacts by about 30%, which was mainly led by the initial contacts from residues between the second and third helices (e.g., Asn60, Leu-61, Pro62 and Trp64; see Fig. 2D at 2.6 and 14.4 ns) with an end-on orientation (rather than side-on) (see Fig. 2D at t = 2.6 ns; this pattern was also found in the other two trajectories (Figure S2)). After about 135 ns of a metastable state from 15 to 150 ns, HP35 started further denaturation again until it reached a more stable state at ~410 ns. The loss of the protein native contacts and the increase of the contacts with MoS2 both display a stepwise manner (Fig. 2A), indicating a fragment- or residue-based rupture of the protein structure onto the MoS2 nanosheet (see Fig. 2C,D).

Structural dynamics of HP35 on MoS2 surface.
(A) Time profile of native contact Q of HP35 (black) and heavy atom contact number between HP35 sidechains and MoS2 (red). (B,C) Time profiles for hydrogen bond and α-helix ratios of HP35 on MoS2, respectively. (D) Key intermediate structures of HP35 along the representative simulation trajectory. Some key residues are highlighted with sticks marked in green and blue for carbons and nitrogens, respectively. Other representation schemes are same as those used in Fig. 1.
To further understand the initial binding dynamics and underlying mechanism, the time evolution of secondary structures and heavy atom contact numbers between each residue with MoS2 for all the three trajectories have been analyzed in detail (Figure S3). First, the detailed kinetics for each helix unfolding does differ somewhat from trajectory to trajectory (Figure S3, first column). That said, the protein was initially separated reasonably far away from the MoS2 nanosheet, but some of the major events and typical conformations still correlate quite well between different trajectories. For example, both run-1 and run-3 share very similar unfolding dynamics (Figure S3) and final configurations (Fig. 1B), with the second helix firstly unfold, followed by the third helix, while the first helix remained mostly intact till the end of the simulation. Also as shown in the second column of Figure S3, for all the three simulated trajectories, the second helix (from amino acid (AA) 54 to 61) absorbed onto the MoS2 surface first, followed by the first helix (AA 43 to 52) and then the third helix (AA 62 to 74).
Detailed analysis on the heavy atom contact number between each residue with MoS2 as the function of simulation time also reveal that the first contacting (driving) residues for the second α-helix are Arg-55, Phe-58, Asn-60, Leu-61, Leu-63 and Trp-64, with the hydrophobic (including aromatic) residue account for 67%, which is larger than its “intrinsic” proportion in the second α-helix (~57%, see Table S2), indicating hydrophobic residues are preferred in general during the adsorption process. Furthermore, the second α-helix contains the highest percentage of hydrophobic residues, followed by the first α-helix, then the third α-helix, which is also consistent with the above analysis that the hydrophobic residues have played a significant role in the early adsorption process.
To better understand the energetics responsible for the above process, we computed nonbonding interaction energies between HP35 and MoS2 during adsorption process. As shown in Fig. 3A, the time-profile clearly shows that the protein favors binding onto the MoS2 surface in terms of both electrostatic and dispersive van der Waals interaction energies. Although both interactions contribute to the binding energy as the atomic contacts with MoS2 increase, HP35 adsorption is mainly driven by the strong dispersion interaction between the protein and MoS2 surface (with the vdW interaction contributing 449.39 ± 7.79 kcal/mol and the electrostatic interaction 19.01 ± 5.16 kcal/mol).

Energy dynamics of HP35 adsorption on MoS2 surface.
(A) Time profiles for vdW (black) and electrostastic (red) interaction energies between HP35 and MoS2. (B) Time evolution of residue-specific vdW interaction energies of HP35 on MoS2 nanosheet.
We further quantified the time-profile of residue-specific vdW interaction energies of HP35 with MoS2 nanosheet (Fig. 3B). As shown in Fig. 2D, the very initial contacts occurred at t = 2 ns with residues Asn-60, Pro-62 and Trp-64, by which the second (Asn-60 and Pro-62) and third (i.e., Trp-64) α-helices then anchored onto the MoS2 surface. These events induced the contacts of neighboring Leu-63 and Arg-55 at t = 9 ns. Once Arg-55 was fully settled on the surface at t = 15 ns, the first α-helix also got anchored via Leu-42. This state lasted for the next ~150 ns (i.e., metastable state) until several N-terminal residues started to be in contact as well. After Trp-64 was further stabilized at t = 171 ns via a large conformation change in its indole sidechain, the adsorption began to propagate towards the C-terminus for the next 60 ns, which drew the whole third α-helix to lay down on the surface. Once the C-terminal Phe-76 maximized its surface interaction at t = 235 ns, no additional residue made contacts on the surface but the local adjustment continued until the system found its energy minimum at t = 410 ns where the C-terminal helix was fully denatured.
As indicated in analyses above that the vdW interactions dominate the adsorption energetics, we then further calculated the vdW energy contributions for each residue by averaging the last 100 ns over all three independent trajectories (Fig. 4). As shown in Fig. 4, most of the highly favorable residues, with its vdW energy <−20 kcal/mol, coincide with the aforementioned residues that are important in guiding the HP35 adsorption onto MoS2 surface, such as Arg-55, Phe-58, Trp-64, Lys-70, Lys-71 and Phe-76. Since MoS2 surface is largely hydrophobic with a water contact angle around 82° 66, it is not surprising that hydrophobic residues like Phe-58, Trp-64 and Phe-76 are found in this group. However, it is interesting to note that basic residues like Arg-55, Lys-70 and Lys-71 are also in the same group. Actually, they played a crucial role in tethering the protein onto the surface of MoS2. For example, Arg-55 was holding the second α-helix to the surface until it is fully denatured. This important role of basic residues such as Arg is consistent with the similar role they play in their interactions with graphene and carbon nanotubes67, where even without any electrostatic interactions the dispersion interactions with the solvent-exposed long aliphatic sidechains in Arg and Lys contribute favorably (of course, the π–stacking between the guanidinium group and aromatic rings also contributes in the Arg case there67).

Residue-specific vdW energy profile.
Dispersion energies for each residue of HP35 on MoS2 were averaged over the last 100 ns for each simulation trajectory.
To probe the role of interfacial water in mediating the adsorption process, some intermediate configurations in a representative trajectory are displayed in Figure S4. For the clarity MoS2 was not shown. At t = 5 ns, a small, partial, drying zone was formed as marked with the black circle, between the hydrophobic region (white region) and the MoS2 surface. The drying zone expanded to a larger area after ~50 ns. Meanwhile, there were also some water molecules bridging the partially positive-charged region (blue region) of the protein and the surface of MoS2 (marked with red circle; near the drying region). At t = 235 ns, the drying region spread to the entire “MoS2-contacted hydrophobic region”, accompanied by a much larger overall contacting surface. Interestingly, water molecules that reside at the interval between positively charged regions of protein and MoS2’s surface (highlighted by the red circle), can persist throughout the entire simulation length, which implies that water may act as lubricant for the binding of these hydrophilic regions to MoS2. Taken together, water indeed played a significant role in the binding process: (i) partial dewetting occurred at the interface between the hydrophobic region of protein and the surface of MoS2, which provided a strong driving force for the early adsorption; and (ii) water acted as lubricant to facilitate adsorption of hydrophilic zone of the protein with MoS2.
In order to further elaborate this point on key contributors, we narrowed down our focus by selecting the two most representative residues, Trp-64 and Arg-55 (one aromatic and one basic). Since water hydration is an important factor for the surface adsorption, water population in the first solvation shell (FSS) of each residue was taken into account in addition to the time-evolution of atomic contacts on MoS2 surface. At the beginning, Trp-64 was solvated by 18 waters in the bulk (Fig. 5A). This number was not changed much until t = 171 ns, even after the indole ring of Trp-64 started to contact with MoS2 surface at t = 3 ns. In this state, the indole ring was found to vertically contact on the surface through its edge with 8 atomic contacts on average. Due to this edge (indole ring)-to-face (MoS2 surface) contact mode, the water population in the FSS was not noticeably affected. However, once the indole sidechain lay down on the surface at t = 171 ns, about 12 water molecules left from FSS (nanoscale drying), while the atomic contact increased by 2. The face-to-face conformation was quite stable during our simulation till 500 ns. This nanoscale drying44,45,68,69,70 further enhanced the binding strength of the indole ring with the MoS2 surface, in addition to the strong dispersive interactions.

Surface adsorption dynamics for key residues, Trp-64 (A) and Arg-55 (B). Left panel: time-profiles for heavy atom contact (green curve) between the sidechain and MoS2 and water number in the FSS of the selected residues (black curve). Right panels: representative snapshots for key intermediate states along the adsorption process. The selected residues (64W and 55R) are depicted by sticks marked in green (carbons) and blue (nitrogens) and nearby residues (47F, 51F and 58F) by gray sticks. Water in FSS are depicted with red spheres. Other schemes are same as those used in Fig. 1.
Similarly to Trp-64, Arg-55 also displays a very stable adsorption on the MoS2 surface. However, the detailed process turned out to be more complicated. For the first 14 ns, Arg-55 was fully solvated with 18 waters in its FSS with no surface contact. After the onset of the adsorption, the full sidechain atoms were adsorbed within ~0.5 ns. A snapshot at t = 14.5 ns shows that guanidinium group prefers to have the face-to-face configuration toward MoS2 surface with a contact number of 7 (which remains the same till the end of simulation). In the meantime, about 5 water molecules were squeezed out from the interface between the residue and MoS2 surface until t = ~160 ns, after which the number of water in FSS slightly increased by ~2 for the next ~20 ns. The restoration was due to water intrusion to the place originally excluded by the salt-bridge formation with Asp-44 (see snapshot at t = 160 ns of Fig. 5B). Not long after the breakage, Phe-47 began to interact with Arg-55 (see snapshot at t = 235 ns of Fig. 5B), eventually forming a stable stacking via energetically favorable cation-π interaction71. By this coupling, Arg-55 became desolvated again by ~3 water molecules, meaning a total of 6 waters left from FSS of Arg-55 compared to that of the bulk.
While the hydrophobic interaction is important for the adsorption of Arg’s aliphatic chain, our analysis indicates that more complicated interactions are involved in this case with the guanidinium moiety. Due to the net positive charge, a similar dramatic nanoscale drying as seen in Trp-64 is not likely in Arg-55 (only 6 waters left from the FSS of Arg-55 as compared to 12 in Trp-64). Furthermore, the electrostatic interactions with Phe-47 via cation-π interaction also contributed to the stability. Overall, the stable binding of Arg-55 on MoS2 surface is contributed by both dispersion and electrostatic interactions.
Conclusion
In recent years, MoS2 has attracted a great deal of attention in the biomedical field due to its unique properties. Applications of MoS2 include the near-infrared (NIR) photothermal-triggered drug delivery platform, as well as photothermal and chemotherapy combined therapeutic agents of cancer. The MoS2 nanosheets can be used as contrast agents in X-ray computed tomography imaging and field-effect biosensors for label free sensitive detection of cancer marker proteins in solution. Although these applications have clearly demonstrated the importance of MoS2 nanosheets in biomedicine, their wider applications demand more studies on their biosafety, in particular the molecular origin of their potential nanotoxicity. Furthermore, the detailed molecular interactions between MoS2 nanosheets and biomolecules, such as proteins and membrane lipids, are still largely unclear. In this study, we examined the adsorption process of a model protein, Villin Headpiece (HP35), onto the surface of MoS2 using all-atom molecular dynamics (MD) simulations. We discovered that MoS2 exhibits exceptionally robust denaturation capability to Villin Headpiece, which is comparable to that of graphene nanosheet, where nearly 80% of α-helix content was destroyed after 500 ns simulation72.
As shown in all the three simulated trajectories, the overall structure of HP35 was severely destroyed. Both aromatic residues and basic residues were shown to play a very important role in anchoring the protein onto the surface of MoS2, which lead to successive profound denaturation. The strong direct dispersion interactions between key residues (such as Arg-55, Asn-60, Pro-62, Leu-63, Trp-64) and MoS2 were identified as the main driving force for the entire adsorption process (similar to the case of graphene nanosheet72). Meanwhile, water molecules at the interfacial region between some key residues (e.g. Trp-64) and MoS2 nanosheet were also found to play an important role due to a nanoscale drying, which accelerated the adsorption process. The similar important role of water was also identified previously in mediating the adsorption of amyloid fibrils onto graphene nanosheets73, as well as blood proteins onto carbon nanotubes74 and graphene nanosheets75,76.
Further studies on the nanotoxicity of MoS2 to other proteins, DNA, cell membranes, as well as cells, tissues and animal models are highly desired for a deeper understanding of its toxicity. We also envision more development efforts on applying MoS2 as a new type of 2D nanomaterials in biomedical applications such as antibacterial agents. These studies on MoS2 toxicity can also stimulate and facilitate the cytotoxicity studies of other related nanomaterials in this emerging field of nanotoxicology.
Additional Information
How to cite this article: Gu, Z. et al. Robust Denaturation of Villin Headpiece by MoS2 Nanosheet: Potential Molecular Origin of the Nanotoxicity. Sci. Rep. 6, 28252; doi: 10.1038/srep28252 (2016).
References
Lee, J. S., Joung, H. A., Kim, M. G. & Park, C. B. Graphene-based chemiluminescence resonance energy transfer for homogeneous immunoassay. ACS Nano 6, 2978–2983 (2012).
Ocsoy, I. et al. Nanotechnology in Plant Disease Management: DNA-Directed Silver Nanoparticles on Graphene Oxide as an Antibacterial against Xanthomonas perforans. ACS Nano 7, 8972–8980 (2013).
Singh, N. et al. Cicer alpha-galactosidase immobilization onto functionalized graphene nanosheets using response surface method and its applications. Food Chem. 142, 430–438 (2014).
Cha, C. Y., Shin, S. R., Annabi, N., Dokmeci, M. R. & Khademhosseini, A. Carbon-based nanomaterials: multifunctional materials for biomedical engineering. ACS Nano 7, 2891–2897 (2013).
Li, Z. H. et al. Towards graphyne molecular electronics. Nat. Commun. 6, 9 (2015).
Malko, D., Neiss, C., Vines, F. & Gorling, A. Competition for Graphene: Graphynes with Direction-Dependent Dirac Cones. Phys. Rev. Lett. 108, 4 (2012).
Kou, J. L., Zhou, X. Y., Lu, H. J., Wu, F. M. & Fan, J. T. Graphyne as the membrane for water desalination. Nanoscale 6, 1865–1870 (2014).
Coleman, J. N. et al. Two-Dimensional Nanosheets Produced by Liquid Exfoliation of Layered Materials. Science 331, 568–571 (2011).
Sie, E. J. et al. Valley-selective optical Stark effect in monolayer WS2 . Nat. Mater. 14, 290–294 (2015).
Sun, W. et al. High Surface Area Tunnels in Hexagonal WO3 . Nano Lett. 15, 4834–4838 (2015).
Wang, C. et al. A novel hydrazine electrochemical sensor based on the high specific surface area graphene. Microchim. Acta 169, 1–6 (2010).
Katsnelson, M. I. Graphene: carbon in two dimensions. Mater. Today 10, 20–27 (2007).
Ajayan, P. M. Nanotubes from carbon. Chem. Rev. 99, 1787–1799 (1999).
Dresselhaus, M., Dresselhaus, G., Eklund, P. & Saito, R. Carbon nanotubes. Phys. World 11, 33–38 (1998).
Zhang, M. X. et al. Graphene oxide based theranostic platform for T-1-weighted magnetic resonance imaging and drug delivery. ACS Appl. Mater. Interfaces 5, 13325–13332 (2013).
Wang, C. Y. et al. Multifunctional chitosan magnetic-graphene (CMG) nanoparticles: a theranostic platform for tumor-targeted co-delivery of drugs, genes and MRI contrast agents. J. Mat. Chem. B 1, 4396–4405 (2013).
Sun, X. M. et al. Nano-graphene oxide for cellular imaging and drug delivery. Nano Res. 1, 203–212 (2008).
Kou, Z. Y. et al. A promising gene delivery system developed from PEGylated MoS2 nanosheets for gene therapy. Nanoscale Res. Lett. 9, 9 (2014).
Ma, Y. et al. 3D graphene network@WO3 nanowire composites: a multifunctional colorimetric and electrochemical biosensing platform. Chem. Commun. 50, 11135–11138 (2014).
Yang, G. B. et al. Two-dimensional magnetic WS2@Fe3O4 nanocomposite with mesoporous silica coating for drug delivery and imaging-guided therapy of cancer. Biomaterials 60, 62–71 (2015).
Ge, J. et al. A WS2 nanosheet based sensing platform for highly sensitive detection of T4 polynucleotide kinase and its inhibitors. Nanoscale 6, 6866–6872 (2014).
Zhou, R. H. & Gao, H. J. Cytotoxicity of graphene: recent advances and future perspective. Wiley Interdiscip. Rev.-Nanomed. Nanobiotechnol. 6, 452–474 (2014).
Zuo, G. H., Kang, S. G., Xiu, P., Zhao, Y. L. & Zhou, R. H. Interactions Between Proteins and Carbon-Based Nanoparticles: Exploring the Origin of Nanotoxicity at the Molecular Level. Small 9, 1546–1556 (2013).
Zuo, G. H., Huang, Q., Wei, G. H., Zhou, R. H. & Fang, H. P. Plugging into proteins: poisoning protein function by a hydrophobic nanoparticle. ACS Nano 4, 7508–7514 (2010).
Chong, Y. et al. Reduced Cytotoxicity of Graphene Nanosheets Mediated by Blood-Protein Coating. ACS Nano (2015).
Nel, A. E. et al. Understanding biophysicochemical interactions at the nano-bio interface. Nature Materials 8, 543–557 (2009).
Tu, Y. S. et al. Destructive extraction of phospholipids from Escherichia coli membranes by graphene nanosheets. Nat. Nanotechnol. 8, 594–601 (2013).
Li, Y. F. et al. Graphene microsheets enter cells through spontaneous membrane penetration at edge asperities and corner sites. Proc. Natl. Acad. Sci. USA 110, 12295–12300 (2013).
Guo, R., Mao, J. & Yan, L.-T. Computer simulation of cell entry of graphene nanosheet. Biomaterials 34, 4296–4301 (2013).
Mao, J., Guo, R. & Yan, L.-T. Simulation and analysis of cellular internalization pathways and membrane perturbation for graphene nanosheets. Biomaterials 35, 6069–6077 (2014).
Luo, Z. M. et al. Preparation of Highly Dispersed Reduced Graphene Oxide Decorated with Chitosan Oligosaccharide as Electrode Material for Enhancing the Direct Electron Transfer of Escherichia coli. ACS Appl. Mater. Interfaces 7, 8539–8544 (2015).
Muthoosamy, K. et al. Exceedingly biocompatible and thin-layered reduced graphene oxide nanosheets using an eco-friendly mushroom extract strategy. Int. J. Nanomed. 10, 1505–1519 (2015).
Hu, W. B. et al. Protein Corona-Mediated Mitigation of Cytotoxicity of Graphene Oxide. ACS Nano 5, 3693–3700 (2011).
Kundu, A., Nandi, S., Das, P. & Nandi, A. K. Fluorescent Graphene Oxide via Polymer Grafting: An Efficient Nanocarrier for Both Hydrophilic and Hydrophobic Drugs. ACS Appl. Mater. Interfaces 7, 3512–3523 (2015).
Lundqvist, M. et al. Nanoparticle size and surface properties determine the protein corona with possible implications for biological impacts. Proc. Natl. Acad. Sci. USA 105, 14265–14270 (2008).
Wan, S. et al. The “Sweet” Side of the Protein Corona: Effects of Glycosylation on Nanoparticle-Cell Interactions. ACS Nano 9, 2157–2166 (2015).
Karwowska, E., Kostecki, M., Sokołowska, A., Chodun, R. & Zdunek, K. Peculiar Role of the Metallic States on the Nano-MoS2 Ceramic Particle Surface in Antimicrobial and Antifungal Activity. International Journal of Applied Ceramic Technology, 1–6 (2014).
Yang, X. et al. Antibacterial activity of two-dimensional MoS2 sheets. Nanoscale 6, 10126–10133 (2014).
Wang, L. et al. Functionalized MoS2 Nanosheet-Based Field-Effect Biosensor for Label-Free Sensitive Detection of Cancer Marker Proteins in Solution. Small 10, 1101–1105 (2014).
Yin, W. et al. High-Throughput Synthesis of Single-Layer MoS2 Nanosheets as a Near-Infrared Photothermal-Triggered Drug Delivery for Effective Cancer Therapy. ACS Nano 8, 6922–6933 (2014).
Wang, S. et al. Biocompatible PEGylated MoS2 nanosheets: Controllable bottom-up synthesis and highly efficient photothermal regression of tumor. Biomaterials 39, 206–217 (2015).
Chiu, T. K. et al. High-resolution x-ray crystal structures of the villin headpiece subdomain, an ultrafast folding protein. Proc. Natl. Acad. Sci. USA 102, 7517–7522 (2005).
Varshney, V. et al. MD simulations of molybdenum disulphide (MoS2): Force-field parameterization and thermal transport behavior. Computational Materials Science 48, 101–108 (2010).
Zhou, R. H., Huang, X. H., Margulis, C. J. & Berne, B. J. Hydrophobic collapse in multidomain protein folding. Science 305, 1605–1609 (2004).
Liu, P., Huang, X. H., Zhou, R. H. & Berne, B. J. Observation of a dewetting transition in the collapse of the melittin tetramer. Nature 437, 159–162 (2005).
Eleftheriou, M., Germain, R. S., Royyuru, A. K. & Zhou, R. Thermal denaturing of mutant lysozyme with both the OPLSAA and the CHARMM force fields. J. Am. Chem. Soc. 128, 13388–13395 (2006).
Fitch, B. G. et al. Blue Matter: Strong scaling of molecular dynamics on Blue Gene/L. 846–854 (Springer : Berlin Heidelberg,, 2006).
Zhou, R., Das, P. & Royyuru, A. K. Single mutation induced H3N2 hemagglutinin antibody neutralization: a free energy perturbation study. J. Phys. Chem. B 112, 15813–15820 (2008).
Das, P., King, J. A. & Zhou, R. Aggregation of gamma-crystallins associated with human cataracts via domain swapping at the C-terminal beta-strands. Proc. Natl. Acad. Sci. USA 108, 10514–10519 (2011).
Das, P., Li, J., Royyuru, A. K. & Zhou, R. Free energy simulations reveal a double mutant avian H5N1 virus hemagglutinin with altered receptor binding specificity. J. Comput. Chem. 30, 1654–1663 (2009).
Li, J. et al. Hydration and dewetting near graphite-CH(3) and graphite-COOH plates. J. Phys. Chem. B 109, 13639–13648 (2005).
Xia, Z. et al. Molecular dynamics simulations of Ago silencing complexes reveal a large repertoire of admissible ‘seed-less’ targets. Sci. Rep. 2, 569 (2012).
Tu, Y. S. et al. Water-mediated signal multiplication with Y-shaped carbon nanotubes. Proc. Natl. Acad. Sci. USA 106, 18120–18124 (2009).
Li, J. Y. et al. Electrostatic gating of a nanometer water channel. Proc. Natl. Acad. Sci. USA 104, 3687–3692 (2007).
Das, P. & Zhou, R. Urea-induced drying of carbon nanotubes suggests existence of a dry globule-like transient state during chemical denaturation of proteins. J. Phys. Chem. B 114, 5427–5430 (2010).
Guo, C., Luo, Y., Zhou, R. & Wei, G. Probing the self-assembly mechanism of diphenylalanine-based Peptide nanovesicles and nanotubes. ACS Nano 6, 3907–3918 (2012).
Xiu, P. et al. Urea-induced drying of hydrophobic nanotubes: comparison of different urea models. J. Phys. Chem. B 115, 2988–2994 (2011).
Hess, B., Kutzner, C., van der Spoel, D. & Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 (2008).
Humphrey, W., Dalke, A. & Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996).
Mackerell, A. D., Feig, M. & Brooks, C. L. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. Journal Of Computational Chemistry 25, 1400–1415 (2004).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 7 (2007).
Darden, T., York, D. & Pedersen, L. Particle mesh ewald - an n.log(n) method for ewald sums in large systems. Journal Of Chemical Physics 98, 10089–10092 (1993).
Hess, B., Bekker, H., Berendsen, H. J. C. & Fraaije, J. LINCS: A linear constraint solver for molecular simulations. Journal of computational chemistry 18, 1463–1472 (1997).
Miyamoto, S. & Kollman, P. A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 13, 952–962 (1992).
Chow, P. K. et al. Wetting of Mono and Few-Layered WS2 and MoS2 Films Supported on Si/SiO2 Substrates. ACS Nano 9, 3023–3031 (2015).
Gu, Z. L. et al. The role of basic residues in the adsorption of blood proteins onto the graphene surface. Sci Rep 5, 11 (2015).
Hua, L., Huang, X., Liu, P., Zhou, R. & Berne, B. J. Nanoscale dewetting transition in protein complex folding. J. Phys. Chem. B 111, 9069–9077 (2007).
Krone, M. G. et al. Role of water in mediating the assembly of Alzheimer amyloid-beta Abeta16-22 protofilaments. J. Am. Chem. Soc. 130, 11066–11072 (2008).
Das, P., Kapoor, D., Halloran, K. T., Zhou, R. & Matthews, C. R. Interplay between drying and stability of a TIM barrel protein: a combined simulation-experimental study. J. Am. Chem. Soc. 135, 1882–1890 (2013).
Ma, J. C. & Dougherty, D. A. The cation-π interaction. Chemical reviews 97, 1303–1324 (1997).
Zuo, G., Zhou, X., Huang, Q., Fang, H. & Zhou, R. Adsorption of Villin Headpiece onto Graphene, Carbon Nanotube and C60: Effect of Contacting Surface Curvatures on Binding Affinity. J. Phys. Chem. C 115, 23323–23328 (2011).
Yang, Z. et al. Destruction of amyloid fibrils by graphene through penetration and extraction of peptides. Nanoscale 7, 18725–18737 (2015).
Gu, Z. et al. Surface Curvature Relation to Protein Adsorption for Carbon-based Nanomaterials. Sci Rep 5 (2015).
Chong, Y. et al. Reduced Cytotoxicity of Graphene Nanosheets Mediated by Blood-Protein Coating. ACS Nano 9, 5713–5724 (2015).
Gu, Z. et al. The role of basic residues in the adsorption of blood proteins onto the graphene surface. Sci. Rep. 5, 10873 (2015).
Acknowledgements
We thank Bruce Berne, Bo Zhou, Hongsuk Kang and Xuanyu Meng for helpful discussions. This work was partially supported by the National Natural Science Foundation of China under Grant Nos 11374221, 11574224, 11404233 and 81402518 and Changzhou High-Level Medical Talents Training Project (2016C2BJ016). RZ acknowledges the support from IBM Blue Gene Science Program (W1258591, W1464125, W1464164). A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) and Jiangsu Provincial Key Laboratory of Radiation Medicine and Protection.
Author information
Authors and Affiliations
Contributions
R.Z. and J.L. conceived and designed the research. Z.Y., J.R.Y., J.L., S.-g.K. and R.Z. co-wrote the manuscript. Z.G. and Z.Y. carried out the molecular dynamics simulations. Z.G., Z.Y., J.R.Y., J.L. and R.Z. analyzed the data. All authors discussed the results and commented on the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Gu, Z., Yang, Z., Kang, Sg. et al. Robust Denaturation of Villin Headpiece by MoS2 Nanosheet: Potential Molecular Origin of the Nanotoxicity. Sci Rep 6, 28252 (2016). https://doi.org/10.1038/srep28252
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep28252
This article is cited by
-
Moderate binding of villin headpiece protein to C3N3 nanosheet reveals the suitable biocompatibility of this nanomaterial
Scientific Reports (2023)
-
Biofunctionalization of graphene and its two-dimensional analogues and synthesis of biomimetic materials: a review
Journal of Materials Science (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
