
Abstract
An unresolved mystery in the field of spinal muscular atrophy (SMA) is why a reduction of the ubiquitously expressed Smn protein causes defects mostly in motoneurons. We addressed the possibility that this restricted vulnerability stems from elevated Smn expression in motoneurons. To explore this, we established an ex vivo zebrafish culture system of GFP-marked motoneurons to quantitatively measure Smn protein and smn mRNA levels as well as promoter activity in motoneurons versus other cell types. Importantly, we uncovered that Smn levels are elevated in motoneurons by means of transcriptional activation. In addition, we identified the ETS family transcription factor Etv5b to be responsible for increased smn transcription in motoneurons. Moreover, we established that the additional supply of Smn protein in motoneurons is necessary for proper axonogenesis in a cell-autonomous manner. These findings demonstrate the reliance of motoneurons on more Smn, thereby adding a novel piece of evidence for their increased vulnerability under SMA conditions.
Similar content being viewed by others


Introduction
SMA is the most common genetic cause of infant death and is characterized by the degeneration of lower α-motoneurons and subsequent muscle atrophy1. Reduced levels of Survival of Motor Neuron (Smn), a protein controlling snRNP assembly, are responsible for SMA2,3,4. Such reduction appears to be tolerable for most cells, while motoneurons are impaired. In addition, Schwann cells and neurons of the enteric nervous system are also affected in the SMA mouse model5,6, explaining why a motoneuron specific replenishment of Smn is only sufficient to restore functionality of the motor unit, but does not improve survival of SMA zebrafish and mice7,8,9,10,11,12. These findings suggest that cell-autonomous and non-cell-autonomous mechanisms together contribute to SMA. By complementing studies in mice, zebrafish offers a powerful system to understand the etiology of SMA. Accordingly, knockout or knockdown of smn in zebrafish embryos using genetic mutations, morpholino oligonucleotides (MO) or synthetic micro-RNAs results in developmental defects including morphological abnormalities of motoneuron axons13,14,15,16.
To explain the increased dependence of motoneurons on Smn, current hypotheses propose that motoneurons are more vulnerable to reduced splicing efficiency17,18,19, and/or that Smn is necessary for axonal localization of various mRNAs20,21,22,23. These models raise the possibility that motoneurons might express more Smn protein to satisfy an extra demand for this protein. Based on this, we thought to address the increased sensitivity of motoneurons in SMA by a novel hypothesis: are Smn levels indeed elevated in motoneurons compared with other cells, and if so, is such additional supply of Smn essential for their axonal development? Although previous studies had suggested that Smn levels are elevated in the motoneuron-rich ventral horn of rat and human spinal cords24,25, this question has never been addressed quantitatively at the single-cell level.
Results and Discussion
To assess the levels of Smn protein in motoneurons, we established an ex vivo culture system of dissociated zebrafish embryos coupled with Smn immunofluorescence (single-cell immunofluorescence, scIF; Fig. 1a–c). Motoneurons were identified by eGFP expression under control of the motoneuron-specific mnx1/hb9 promoter, allowing for direct comparison of Smn levels in motoneurons versus other cells. GFP positive cells developed Znp-1 positive axons after overnight incubation confirming their neuronal identity (Fig. 1c). The quantification of Smn levels was performed on cells fixed shortly after dissociation (see Methods for details). The specificity of the used Smn antibody was confirmed by scIF of smn MO injected embryos (Fig. S1a–e) and by Western blot analysis (Fig. S1f). Moreover, pre-adsorbing the antibody with the N-terminal part of human SMN resulted in almost no detectable signal in the scIF assay (Fig. S1g–k). Intriguingly, comparing Smn levels in GFP-positive and GFP-negative cells revealed that motoneurons contain on average 70% more Smn than other cells (Fig. 1d–f). This increase was not due to a difference in cell sizes, as motoneurons and surrounding cells have comparable sizes (Fig. S2a,b). We similarly observed slightly elevated levels of Smn in motoneuron cell bodies in cryo-sectioned intact spinal cords (Fig. S1l). To test whether such elevation is specific to motoneurons, we performed scIF in nkx2.2a:GFP embryos marking perineurial cells that originate from the ventral spinal cord and ensheath the motor nerve26. We found no difference in Smn levels of those cells compared to control cells (Fig. S2c–e), suggesting that the elevation of Smn levels is a hallmark of motoneurons.

(a–c) Single-cell immunofluorescent (scIF) experiments. 24 hpf pmnx1:eGFP embryos were dissociated and immunostained for Smn ((a) see also Methods). DIC image of cells following 3-hour (b) and 24-hour (c) incubation are shown with GFP in green. Note that the GFP signal in the cell body is oversaturated so that the weaker signal in the axon becomes visible. In (c) DNA is in blue and the motoneuron marker Znp1 (synaptotagmin) is in red; the axon growth cone is magnified in the corner of the images. Scale bars are 100 μm (a), 10 μm (low magnification in (b,c)) and 2 μm (high magnification in (c)). (d) scIF on pmnx1:eGFP embryos. Histone 2B (H2B), GFP and Smn signals are shown in Z-projected confocal sections. Cells marked by white rectangles are magnified on the right. Scale bars: 10 μm for low and 5 μm for high magnification. (e) To account for potential variability in the immunostaining, Smn levels were quantified relative to H2B. Diamonds denote GFP negative (GFP−) and GFP positive (GFP+) cells from one representative experiment. Blue bars indicate mean ± SD with significance values of *p < 0.05 and **p < 0.01. Exact values are (mean ± SD) 0.51 ± 0.14 (GFP−) and 0.87 ± 0.17 (GFP+), p = 0.001 with Wilcoxon Sum Rank Test. For more details, see Materials and Methods and Supplementary Table S1. (f) Average increase of Smn levels in motoneurons versus control cells. The exact value of enrichment is (mean ± SD): 1.67 ± 0.14. N = 3 experiments, n = number of analyzed cells.
What is the mechanism leading to higher Smn protein levels in motoneurons? To answer this question, we first quantified smn mRNA levels in motoneurons and other cell types using single molecule FISH (Fig. 2a) and found that smn mRNA is enriched in motoneurons to an extent similar to Smn protein (Fig. 2b,c, compare with Fig. 1f). To explore whether this increase at mRNA level is due to differential smn promoter activity, we created a transgenic zebrafish line, in which the putative zebrafish smn promoter drives expression of mCherry (psmn:mCherry, Fig. 2d). Quantification of mCherry levels established that the smn promoter is more active in pmnx1:eGFP positive motoneurons compared with neighboring cells (Fig. 2e–g). Suggestively, the activity of the murine Smn promoter is also elevated in cultivated cells derived from the spinal cord when compared with fibroblasts27, raising the possibility that the motoneuron-specific transcriptional activation of smn described here in zebrafish is evolutionary conserved.

(a) Single molecule FISH on pmnx1:eGFP embryos. DNA, GFP and smn mRNA signals are shown in Z-projected confocal sections. Scale bars: 10 μm for low and 5 μm for high magnification. (b) Quantification of smn mRNA signal from one representative experiment. Exact values are (mean ± SD): 127 ± 42 (GFP−) and 218 ± 61 (GFP+), p = 8.06 × 10−8 with Wilcoxon Sum Rank Test. (c) Average increase of smn mRNA levels in motoneurons versus control cells. The exact value of enrichment is 1.73 ± 0.09 (mean ± SD). N = 3 experiments, n = number of analyzed cells. (d) Putative smn promoter in zebrafish. Genomic situation (top) and mCherry construct (bottom). Arrows denote start of transcription. (e) scIF on pmnx1:eGFP × psmn:mCherry embryos. GFP and mCherry signals are shown. Scale bars: 10 μm for low and 5 μm for high magnification. (f) Quantification of mCherry signal from one representative experiment. Exact values are (mean ± SD): 8.9 ± 4 (GFP−) and 15 ± 4 (GFP+), p = 9.37 × 10−5 with Wilcoxon Sum Rank Test. (g) Average increase of mCherry levels in motoneurons versus control cells. The exact value of enrichment is 1.87 ± 0.27 (mean ± SD). N = 2 experiments, n = number of analyzed cells.
Next, we sought to identify the transcription factor driving additional smn transcription in motoneurons. The human, the murine and the zebrafish smn promoters contain several ETS transcription factor binding sites27,28 (Fig. S3a), and a member of this family, Pea3, is necessary for axon branching of motoneurons in mice29. Among the four Pea3 subfamily members in zebrafish (etv1, etv4, etv5a, etv5b)30,31, only etv5b shows a marked expression in the spinal cord (Fig. 3a–d and ZFIN database32). We therefore asked whether Etv5b is able to activate the smn promoter. Co-injection of etv5b mRNA and the psmn:mCherry plasmid lead to a robust increase of mCherry mRNA (Fig. 3e) and protein (Fig. S3b–g) in early embryos. This elevation was dependent on the two proximal ETS binding sites in the smn promoter (Fig. S3h,i). Apart from Etv5b, Etv4 was also able to activate the smn promoter (Fig. S4a) likely due to the high degree of conservation of the DNA-binding ETS domains present in this subfamily (Fig. S4b). Notably, etv5b may be controlling smn mRNA levels also in other cells and additional mechanisms may be at play in motoneurons. Altogether, our findings raised the possibility that Etv5b, acting in concert with unknown co-factor(s) present in motoneurons, is responsible for increased Smn levels in motoneurons. To test this, we knocked down etv5b as described previously33 (see also Fig. S5), and measured Smn protein levels in motoneurons. Importantly, unlike treatment with control morpholino (MO) (Fig. S2f), etv5b MO lead to a significant reduction of Smn in motoneurons (Fig. 3f–i), suggesting that Etv5b is needed for the additional Smn supply in motoneurons.

(a–d) Whole-mount in situ hybridization of etv5b expression in 24 hpf embryos. Expression pattern in the entire embryo (a), in magnified lateral (b) and dorsal (c) views as well as in a transverse section (d) of the trunk. In the latter case, yellow arrowhead denote signal in the motoneuron region. Scale bars: 100 μm (a) and 50 μm (b–d). nc, notochord. (e) RT-PCR in control (C - only psmn:mCherry) versus etv5b overexpressing (OE - etv5b mRNA and psmn:mCherry) embryos at early gastrula stage. (f,g) scIF of control (f) and etv5b MO (g) cells from pmnx1:eGFP embryos. H2B, GFP and Smn signals are shown in Z-projected confocal sections. GFP+ cells marked by white rectangles are magnified on the right. Scale bars: 10 μm for low and 5 μm for high magnification. (h) Relative Smn signal in control and in etv5b MO cells from one representative experiment. Exact values are (mean ± SD): 0.99 ± 0.25 (control) and 0.7 ± 0.14 (etvb5 MO), p = 0.003 with Wilcoxon Sum Rank Test. (i) Average relative Smn levels in control and in etv5b MO motoneurons. The exact value of decrease is 0.75 ± 0.04 (mean ± SD). N = 3 experiments, n = number of analyzed cells.
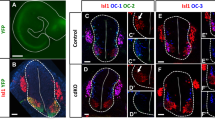
Is the extra pool of Smn essential for motoneurons? We addressed this major point by analyzing motor axon morphology following etv5b MO treatment and found prominent branching and truncation defects (Fig. 4a–c). Of note, these defects are less pronounced than those in smn morphants13,14, which is in agreement with Smn not being completely absent from motoneurons following etv5b MO knockdown. To explore whether the defects observed upon etv5b knockdown arise in a cell-autonomous fashion (that is due to lower Smn levels in motoneurons), we performed the same experiment in pmnx1:mCherry-Smn embryos, which express Smn exclusively in motoneurons (Fig. 4e). In such etv5b MO injected embryos, the axon defects were ameliorated (Fig. 4d–f). Thus, the motoneuron-specific restoration of Smn levels is sufficient to suppress defects caused by etv5b knockdown, thereby establishing that elevated Smn levels in motoneurons are of functional relevance.

(a,b) Z-projections of Znp1 stained motor axons on one side of an embryo after injection of control MO (a) and etv5b MO (b). Scale bar: 50 μm. Arrowheads mark different defects. (c) Quantification of axon defects. Normal, mild, moderate and severe axon morphology in control MO and etv5b MO injected embryos. N = 2 experiments (control MO, in total of 17 embryos) and 5 experiments (etv5b MO, in total of 42 embryos), n = number of axons analyzed. Significance is determined by comparing the occurrences of moderate and severe defects. Exact values in percentages of affected axons are (mean ± SD): 71 ± 7.8 (control MO, normal, 23 ± 10.6 (control MO, mild), 5 ± 2.1 (control MO, moderate), 1 ± 0.7 (control MO, severe), 38 ± 12.2 (etv5b MO, normal), 24 ± 4.4 (etv5b MO, mild), 21 ± 8.5 (etv5b MO, moderate) and 17 ± 5.8 (etv5b MO severe), p = 0.001 with unpaired Student’s t-test. (d,e) Z-projection of Znp1 stained motor axons on one side of control (d) and pmnx1:mCherry-Smn (e) embryos injected with etv5b MO. mCherry signal is shown below the Znp1 panel. (f) Quantification of axon morphology phenotypes in control and pmnx1:mCherry-Smn (mCh-S) embryos injected with etv5b MO. N = 3 experiments, n = number of axons analyzed in total of 22 (control MO) and 20 embryos (etv5b MO). Significance is determined by comparing the occurrences of moderate and severe defects. Exact values in percentages of affected axons are (mean ± SD): 32 ± 11.3 (control etv5b MO normal), 24 ± 5.9 (control etv5b MO mild), 28 ± 7 (control etv5b MO moderate), 16 ± 1.4 (control etv5b MO severe), 67 ± 9 (mCh-S etv5b MO normal), 21 ± 8.3 (mCh-S etv5b MO mild), 11 ± 0.8 (mCh-S etv5b MO moderate), 2 ± 0.9 (mCh-S etv5b MO severe), p = 0.009 with unpaired Student’s t-test.
In conclusion, our data support a hypothesis where a transcriptional mechanism ensures elevated Smn levels in motoneurons, potentially due to a higher demand for Smn in those cells. We uncovered that this extra pool of Smn is essential for the proper axonal morphology of motoneurons in a cell-autonomous manner. Conceptually, these findings can explain why the reduction of Smn in SMA leads to more profound defects in motoneurons compared with other cells (see model in Fig. 5). Interestingly, normal human motoneurons were reported to express lower levels of full-length SMN from the SMN2 locus when compared to other cells34. This was explained by inefficient exon 7 inclusion in SMN2 particularly in this cell type and suggests that distinct mechanisms might act together to cause increased motoneuron vulnerability.

Under healthy conditions in motoneurons, basal smn transcription (orange circle) is enhanced by Etv5b (green oval) acting together with other unknown factor(s) (yellow diamond) leading to more Smn protein (purple) in those cells. More Smn could be needed in motoneurons to meet an increased demand for the splicing machinery and/or for axonal mRNA localization. In the event of reduced Smn levels (that is in SMA), SMA-insusceptible cells are able to tolerate such a reduction, while motoneurons suffer from the cumulative reduction of multiple processes, thereby rendering them more vulnerable.
Methods
Generation and maintenance of transgenic zebrafish lines
All experiments were performed in accordance with relevant guidelines and regulations, and approved by the National University of Singapore (NUS) Institute of Animal Care and Use Committee (IACUC) (protocol numbers 082/10; R13-5714; BR15-0119). The 2.6 kb mnx1 promoter was kindly provided by Dirk Meyer (University of Innsbruck)35. Transgenic lines were generated using I-Sce I meganuclease as previously described36. In the pmnx1:eGFP construct, eGFP was cloned with a Kozak sequence (GCCACC) right after the mnx1 promoter. To generate the pmnx1:mCherry-smn construct, mCherry was cloned to the 5′ end of the smn cDNA via a flexible (SGGG)3 linker and subcloned into the pBSKII SK(+) plasmid. The zebrafish smn promoter was cloned by amplifying a 1547 bp region upstream of the start codon of smn (the DNA region between smn and the upstream gene, hspb11) with the following primer pairs:
Fwd: GCAGCATCCAGCATTTCGAG
Rev: GGGGATGTATGAAAATTTTAAATATTTCCC.
Subsequently, the psmn:mCherry construct was subcloned into the pBSKII SK(+) plasmid. The ΔETSpsmn:GFP plasmid was generated by modifying the psmn:mCherry plasmid with swapping the wild type smn promoter with the mutated version of the promoter and mCherry with GFP, respectively. The mutated ETS sites are as follows: GGAA –> TTTT and ATCC –> TTTC, see also Fig. S3G. The pnkx2.2a:GFP line was generated in Bruce Appel’s lab (University of Colorado, Denver)26.
Morpholino knockdown, RT-PCR and capped mRNA transcription
MOs were obtained from GeneTools. Control standard MOs were utilized as negative control, the anti-smn MO was used as described in17. The splice blocking anti-etv5b MO is described in33, and was used at a concentration of 125 μM.
To address the efficiency of the etv5b knockdown, RNA was isolated from 10–12 embryos at 24 hpf using the standard Trizol-Chloroform method followed by reverse transcription (RevertAid First Strand cDNA Synthesis Kit, Fermentas) with random hexamer primers. The exon spanning primer pairs amplifying the region between exon 10 and 11 were as follows:
etv5b exon 9–10 Fwd: GCAATGGTGAGGGTTTTGGG
etv5b exon 11–12 Rev: GCTCTCCAGCAACCTTTACC
In order to generate etv5b and etv4 mRNA, both cDNAs were cloned into the pCS2+ plasmid after amplification with Phusion Taq polymerase using the following primer pairs:
etv5b:
Fwd: GAGAGTGCGATACCCACTCG
Rev: TCCATTTATGGATGTTTATTTATCCTGTACC.
etv4:
Fwd: TGAAGCAAAGAAGAGGTCTCGAGC
Rev: AGCAATCTCTTGAACCACAGTATTGTG
SP6-driven mRNA transcription was performed with the mMESSAGE mMACHINE kit (Ambion) for 2 hours at 37 °C. Thereafter, A-tailing was achieved by the Poly(A) Tailing Kit (Applied Biosystems) and the resulting mRNA was injected at a concentration of 300 ng/μl into one-cell stage embryos together with the psmn:mCherry and ΔETSpsmn:GFP plasmids at concentration of 50 ng/μl.
For semi-quantitative RT-PCR, 20 early gastrula stage embryos were collected for RNA isolation and half of the isolated total RNA was reverse-transcribed using random hexamer primers with the above-mentioned kit. 1 μl of the cDNA was amplified for 23 (mCherry), 25 (etv5b and etv4) and 30 (gapdh) cycles using the following primer pairs:
gapdh Fwd: GATTGCCGTTCATCCATCTT
gapdh Rev: TCCATTTCTCACAAACAGAGGA
etv5b Fwd: GCAATGGTGAGGGTTTTGGG
etv5b Rev: GCTCTCCAGCAACCTTTACC
etv4 Fwd: CCCCTCAGGAATCTGAAGACCTC
etv4 Rev: TCTTCTGCTCATAGGCACTGGC
mCherry Fwd: CCACC ATGGTGAGCAAGGG
mCherry Rev: GCGAATTAAAAAACCTCCC
Embryo dissociation, single cell immunofluorescence (scIF) and immunostaining on spinal cord sections
24 hpf embryos were dechorionated followed by deyolking in Ringer’s solution and then dissociated to single cells using a Papain Dissociation System Kit (Worthington) according to the manufacturer’s instructions. The dissociation was performed in a thermoshaker at 1100 rpm set to 30 °C for 30 minutes, with pipetting the mixture 3–4 times every 10 minutes to promote dissociation. Following the reconstitution of cells in Leibovitz medium containing 2% FBS, the cells were counted in a BioRad TC20 machine. 150,000 cells in 50 μl were plated on poly-L-lysine slides (Fisherbrand, for a 3-hour culture) or on Laminin-coated coverslips (Neuvitro, for a 24-hour culture) and incubated at 30 °C. Of note, when working on slides, control and treated cells were plated in droplets on opposing ends of the same slide well-separated from each other to minimize artifacts in subsequent steps arising from slide-to-slide variances. When the cells appeared attached to the surface (approximately after 1 hour of incubation), 200 μl (for slides) or 1 ml (for coverslips in 12-well plates) of culture medium (described in37) was added.
Following incubation, cells were washed twice in PBS in a 50 ml glass container (for slides) or in a 12-well plate (for coverslips) and then fixed in 4% PFA (Sigma) in PBS for 5 minutes at room temperature. Following a PBS wash, the cells were submerged into PBS containing 0.1% Triton-X (Sigma) for 5 minutes at room temperature. Blocking was performed by PBS/0.1% Triton-X containing 5% BSA for 2 hours at room temperature. The primary antibodies α-Smn (Novus Biological, NB-1001936 or NB100-1936AF647 1/100) and α-Histone2B (LifeSpan BioSciences, LS-B2035, 1/100) were diluted in PBS and applied in 50 μl/coverslip or 100 μl/slide and incubated at 4 °C overnight. The cells were washed 3 times with PBS/0.1% Tween-20 (PBS-T, Tween 20 from Sigma) for 5 minutes each at room temperature followed by a final wash in PBS for 5 minutes at room temperature. The secondary antibodies Alexa-633-conjugated α-mouse (Life Technologies, A21052, 1/500) and Alexa-405-conjugated α-rabbit (Life Technologies, A31556, 1/500) were diluted in PBS and applied in 50 μl/coverslip and 100 μl/slide, then incubated at room temperature for 2–4 hours. The cells were washed 3 times with PBS-T for 5 minutes each at room temperature followed by a final wash in PBS for 5 minutes at room temperature and mounted in Mowiol (Sigma). In experiments with DNA staining, DAPI was added to the final PBS-T wash at a concentration of 1 μg/ml.
The slides were imaged using a Leica LSM510 confocal microscope with 1.2 μm thick optical slices. We utilized laser intensities and digital gain that did not cause oversaturation of the signal measured by a range indicator built in the Zen 2009 software, where oversaturated pixels appear red. Thereafter, the same setup was used for all images of an experiment to avoid oversaturation. Maximum intensity projections were performed and the signal intensities were measured by manually drawing the boundaries of a given cell using ImageJ. Typically, the signal intensities of a motoneuron with strong gfp expression (that are considered primary motoneurons38) and 8–10 neighboring cells were quantified per field. In experiments where Smn levels were quantified relative to H2B, we divided the Smn grey values with those of H2B. When pooled values are displayed, the ratios of control vs. treated values from each set of experiment were taken and averaged (with control values being 1) thereby correcting for experiment-to-experiment variation.
Cell size measurements were performed with Image J by quantifying the total cell areas on the DIC channel of Z-projected confocal sections. Typically, the cell area of one gfp positive cell and 8–10 neighboring cells were quantified per field.
In experiments with the psmn:mCherry line, mCherry expression in fixed cells was recorded using a Nikon Eclipse 90i fluorescent microscope equipped with a Nikon DS Ri1 camera in the middle plane avoiding apparent oversaturation of the signal and the fluorescent intensities were quantified with ImageJ. The intensities of the background (i.e. areas on the slide void of cells) were subtracted from the intensities measured in cells.
Immunostaining of in vivo motoneurons in 24 hpf pmnx1:eGFP embryos (fixed in 4%PFA for 4 hours at room temperature) was performed on 16 μm thick sections of the posterior trunk using a standard cryo-sectioning protocol. The sections were dried for 1 hour at 37 °C, washed with PBS-T for 5 minutes at room temperature and permeabilized in PBS containing 0.5% Triton-X for 20 minutes at room temperature. Thereafter, the slices were fixed and processed the same way as described above for scIF and 3 μm thick optical slices were imaged with the LSM510.
Whole-mount in situ hybridization and single molecule FISH
The whole-mount in situ hybridization was performed as previously reported39. The following primer pairs were used to clone a 500 bp long fragment of etv5b cDNA into the pCS2+ vector for riboprobe generation:
Fwd: AAAACTCCACCCTCCTGCTTCAGC
Rev: AGTCAAAGCCCACCGTTTGC
Fluorescent Quasar 570-conjugated probes against smn mRNA were designed and purchased from Biosearch Technologies and were used at a concentration of 1 μM. Dissociated cells grown on coverslips were fixed and probed with smFISH probes following the protocol for adherent cells provided by Biosearch Technologies (https://www.biosearchtech.com/assets/bti_stellaris_protocol_adherent_cell.pdf). The smn probes together with the mouse anti-GFP antibody (1:200, Invitrogen) were incubated at 37 °C overnight. The anti-mouse Alexa 488 conjugated secondary antibody (A11029, Life Technologies) was diluted in Wash buffer A in 1:1000 and incubated for 45 minutes at 37 °C.
The cells were imaged with an LSM510 confocal microscope using the 543 nm laser with open pinholes and optical sections of 2 microns avoiding oversaturation of the signal similarly as mentioned above. Maximum Z projections followed by manual drawing the boundaries of a cell and quantification of fluorescent intensities were performed with ImageJ. The intensities of the background (i.e. areas with no cells) were subtracted from the intensities measured in cells.
Whole-mount immunofluorescence
The immunofluorescence was analyzed after standard PFA-methanol fixation. Embryos were incubated with the following primary antibodies: mouse α-ZNP-1 (1/100, ZIRC) and rabbit α-RFP (1/100, MBL, PM005); and with the following secondary antibodies: Alexa-568-conjugated α-rabbit (1/500, A11036, Life Technologies) and Alexa-488-conjugated α-mouse (1/500, A11029, Life Technologies) both at 4 °C overnight.
The embryos were imaged with a Nikon Eclipse 90i fluorescent microscope equipped with a Nikon DS Ri1 camera using 10 μm thick optical sections. The axon defects were assessed qualitatively as mild (a branching defect occurring at the last third of the axon), moderate (branching and/or truncation at the middle third of the axon) and severe (branching and/or truncation at the first third of the axon). Typically, 6–7 axon pairs above the yolk extension were examined in each embryo.
Western blot and pre-adsorption experiments
10–30 dechorionated 24 hpf embryos were deyolked in Ringers solution and following centrifugation, the embryos were lysed in standard RIPA buffer on ice in a volume of 2 μl/embryo. Sample buffer was added, and the lysate was heated up at 95 °C for 5 minutes and loaded right away or stored at −20 °C. Following electrophoresis in a 12% poly-acrylamide gel, the proteins were transferred to PVDF membrane using wet transfer. Blocking was performed with 5% milk powder (Fluka) in TBS containing 0.1% Tween-20 (TBS-T) for 1 hour at room temperature. Primary antibodies were the same as the ones used in the scIF protocol with dilutions of 1:250 (α -Smn) and 1:500 (α -H2B). Secondary antibodies (Sigma) were applied in 1:5000 for 2 hours at room temperature. The chemiluminescent detection was performed with the SuperSignal West Pico Kit (Thermo scientific).
The pre-adsorption experiments were performed using a protein containing the first 160 amino acids of human SMN in complex with His-tagged GEMIN2 (referred to as antigen; a kind gift from Utz Fischer, University of Wuerzburg). We made use of Ni-NTA columns (Qiagen) following the manufacturer’s instructions. After equilibration with PBS, 300 μg of the antigen diluted in PBS was bound to the column for 1 hour at 4 °C. A control column was incubated with PBS void of antigen. After washing with PBS, 5 μg of the antibody diluted in PBS was applied to the column and incubated overnight at 4 °C. To ensure efficient binding of the antibody to the antigen, the antibody solution was spun through the column three times during this incubation. Thereafter, we used the flow-through for Western blot and scIF.
Statistical analysis
Experiments were performed in triplicates, unless otherwise stated. Statistical analysis of all cell culture experiments was performed using Wilcoxon Sum Rank Test (also known as Mann-Whitney U Test), while unpaired Student’s t-test was used in the case of the axon morphology experiments, both executed in Excel. Pooled data are displayed as mean ± SD with significance values of *p < 0.05 and **p < 0.001. All values and significance measurements used in this study are displayed in Supplementary Table S1.
Additional Information
How to cite this article: Spiró, Z. et al. Transcriptional enhancement of Smn levels in motoneurons is crucial for proper axon morphology in zebrafish. Sci. Rep. 6, 27470; doi: 10.1038/srep27470 (2016).
References
Werdnig, G. Two early infantile hereditary cases of progressive muscular atrophy simulating dystrophy, but on a neural basis. 1891. Arch Neurol 25, 276–278 (1971).
Kashima, T. & Manley, J. L. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat Genet 34, 460–463, doi: 10.1038/ng1207 (2003).
Lefebvre, S. et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80, 155–165 (1995).
Monani, U. R. et al. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet 8, 1177–1183 (1999).
Gombash, S. E. et al. SMN deficiency disrupts gastrointestinal and enteric nervous system function in mice. Hum Mol Genet 24, 5665, doi: 10.1093/hmg/ddv292 (2015).
Hunter, G., Aghamaleky Sarvestany, A., Roche, S. L., Symes, R. C. & Gillingwater, T. H. SMN-dependent intrinsic defects in Schwann cells in mouse models of spinal muscular atrophy. Hum Mol Genet 23, 2235–2250, doi: 10.1093/hmg/ddt612 (2014).
McGovern, V. L. et al. SMN expression is required in motor neurons to rescue electrophysiological deficits in the SMNDelta7 mouse model of SMA. Hum Mol Genet 24, 5524–5541, doi: 10.1093/hmg/ddv283 (2015).
Martinez, T. L. et al. Survival motor neuron protein in motor neurons determines synaptic integrity in spinal muscular atrophy. J Neurosci 32, 8703–8715, doi: 10.1523/JNEUROSCI.0204-12.2012 (2012).
Paez-Colasante, X. et al. Improvement of neuromuscular synaptic phenotypes without enhanced survival and motor function in severe spinal muscular atrophy mice selectively rescued in motor neurons. PLoS One 8, e75866, doi: 10.1371/journal.pone.0075866 (2013).
Park, G. H., Maeno-Hikichi, Y., Awano, T., Landmesser, L. T. & Monani, U. R. Reduced survival of motor neuron (SMN) protein in motor neuronal progenitors functions cell autonomously to cause spinal muscular atrophy in model mice expressing the human centromeric (SMN2) gene. J Neurosci 30, 12005–12019, doi: 10.1523/JNEUROSCI.2208-10.2010 (2010).
Hua, Y. et al. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 478, 123–126, doi: 10.1038/nature10485 (2011).
Hao le, T., Duy, P. Q., Jontes, J. D. & Beattie, C. E. Motoneuron development influences dorsal root ganglia survival and Schwann cell development in a vertebrate model of spinal muscular atrophy. Human molecular genetics 24, 346–360, doi: 10.1093/hmg/ddu447 (2015).
McWhorter, M. L., Monani, U. R., Burghes, A. H. & Beattie, C. E. Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. The Journal of cell biology 162, 919–931, doi: 10.1083/jcb.200303168 (2003).
Winkler, C. et al. Reduced U snRNP assembly causes motor axon degeneration in an animal model for spinal muscular atrophy. Genes & development 19, 2320–2330, doi: 10.1101/gad.342005 (2005).
Giacomotto, J., Rinkwitz, S. & Becker, T. S. Effective heritable gene knockdown in zebrafish using synthetic microRNAs. Nat Commun 6, 7378, doi: 10.1038/ncomms8378 (2015).
Hao le, T. et al. Temporal requirement for SMN in motoneuron development. Human molecular genetics 22, 2612–2625, doi: 10.1093/hmg/ddt110 (2013).
See, K. et al. SMN deficiency alters Nrxn2 expression and splicing in zebrafish and mouse models of spinal muscular atrophy. Hum Mol Genet 23, 1754–1770, doi: 10.1093/hmg/ddt567 (2014).
Zhang, Z. et al. Dysregulation of synaptogenesis genes antecedes motor neuron pathology in spinal muscular atrophy. Proc Natl Acad Sci USA 110, 19348–19353, doi: 10.1073/pnas.1319280110 (2013).
Huo, Q. et al. Splicing changes in SMA mouse motoneurons and SMN-depleted neuroblastoma cells: evidence for involvement of splicing regulatory proteins. RNA Biol 11, 1430–1446, doi: 10.1080/15476286.2014.996494 (2014).
Rossoll, W. et al. Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. The Journal of cell biology 163, 801–812, doi: 10.1083/jcb.200304128 (2003).
Rossoll, W. et al. Specific interaction of Smn, the spinal muscular atrophy determining gene product, with hnRNP-R and gry-rbp/hnRNP-Q: a role for Smn in RNA processing in motor axons? Hum Mol Genet 11, 93–105 (2002).
Dombert, B., Sivadasan, R., Simon, C. M., Jablonka, S. & Sendtner, M. Presynaptic localization of Smn and hnRNP R in axon terminals of embryonic and postnatal mouse motoneurons. PLoS One 9, e110846, doi: 10.1371/journal.pone.0110846 (2014).
Saal, L., Briese, M., Kneitz, S., Glinka, M. & Sendtner, M. Subcellular transcriptome alterations in a cell culture model of spinal muscular atrophy point to widespread defects in axonal growth and presynaptic differentiation. RNA 20, 1789–1802, doi: 10.1261/rna.047373.114 (2014).
Bechade, C. et al. Subcellular distribution of survival motor neuron (SMN) protein: possible involvement in nucleocytoplasmic and dendritic transport. Eur J Neurosci 11, 293–304 (1999).
Lefebvre, S. et al. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet 16, 265–269, doi: 10.1038/ng0797-265 (1997).
Kucenas, S. et al. CNS-derived glia ensheath peripheral nerves and mediate motor root development. Nat Neurosci 11, 143–151, doi: 10.1038/nn2025 (2008).
Boda, B. et al. Survival motor neuron SMN1 and SMN2 gene promoters: identical sequences and differential expression in neurons and non-neuronal cells. Eur J Hum Genet 12, 729–737, doi: 10.1038/sj.ejhg.5201217 (2004).
Monani, U. R., McPherson, J. D. & Burghes, A. H. Promoter analysis of the human centromeric and telomeric survival motor neuron genes (SMNC and SMNT). Biochim Biophys Acta 1445, 330–336 (1999).
Livet, J. et al. ETS gene Pea3 controls the central position and terminal arborization of specific motor neuron pools. Neuron 35, 877–892 (2002).
Kudoh, T. et al. A gene expression screen in zebrafish embryogenesis. Genome Res 11, 1979–1987, doi: 10.1101/gr.209601 (2001).
Roussigne, M. & Blader, P. Divergence in regulation of the PEA3 family of ETS transcription factors. Gene Expr Patterns 6, 777–782, doi: 10.1016/j.modgep.2006.01.008 (2006).
Sprague, J., Doerry, E., Douglas, S. & Westerfield, M. The Zebrafish Information Network (ZFIN): a resource for genetic, genomic and developmental research. Nucleic Acids Res 29, 87–90 (2001).
Mao, J., McGlinn, E., Huang, P., Tabin, C. J. & McMahon, A. P. Fgf-dependent Etv4/5 activity is required for posterior restriction of Sonic Hedgehog and promoting outgrowth of the vertebrate limb. Dev Cell 16, 600–606, doi: 10.1016/j.devcel.2009.02.005 (2009).
Ruggiu, M. et al. A role for SMN exon 7 splicing in the selective vulnerability of motor neurons in spinal muscular atrophy. Mol Cell Biol 32, 126–138, doi: 10.1128/MCB.06077-11 (2012).
Arkhipova, V. et al. Characterization and regulation of the hb9/mnx1 beta-cell progenitor specific enhancer in zebrafish. Developmental biology 365, 290–302, doi: 10.1016/j.ydbio.2012.03.001 (2012).
Rembold, M., Lahiri, K., Foulkes, N. S. & Wittbrodt, J. Transgenesis in fish: efficient selection of transgenic fish by co-injection with a fluorescent reporter construct. Nat Protoc 1, 1133–1139, doi: 10.1038/nprot.2006.165 (2006).
Chen, Z. et al. Primary neuron culture for nerve growth and axon guidance studies in zebrafish (Danio rerio). PLoS One 8, e57539, doi: 10.1371/journal.pone.0057539 (2013).
Eisen, J. S., Myers, P. Z. & Westerfield, M. Pathway selection by growth cones of identified motoneurones in live zebra fish embryos. Nature 320, 269–271, doi: 10.1038/320269a0 (1986).
Welten, M. C. et al. ZebraFISH: fluorescent in situ hybridization protocol and three-dimensional imaging of gene expression patterns. Zebrafish 3, 465–476, doi: 10.1089/zeb.2006.3.465 (2006).
Acknowledgements
We thank the DBS Center for Bioimaging Sciences (CBIS) for support with confocal microscopy, Utz Fischer for providing the purified human SMN protein and for advice on the pre-adsorption experiments, Roman Carrasco for suggestions on statistical analyses and Husvinee Sundaramurthi for help with cryo-sectioning. We thank Utz Fischer, Kalina Duszka and Viktor Menachem Sarusie for critical comments on the manuscript. This project is funded by a grant from Ministry of Health/National Medical Research Council of Singapore (CBRG12nov097). AK receives a scholarship from the NUS Graduate School for Integrative Sciences and Engineering (NGS), ST receives a scholarship from the Department of Biological Sciences (DBS) at NUS.
Author information
Authors and Affiliations
Contributions
Z.S., A.K., S.T. and K.S. generated transgenic zebrafish lines, Z.S. performed the experiments, Z.S. and C.W. designed the experiments, interpreted the data and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Spiró, Z., Koh, A., Tay, S. et al. Transcriptional enhancement of Smn levels in motoneurons is crucial for proper axon morphology in zebrafish. Sci Rep 6, 27470 (2016). https://doi.org/10.1038/srep27470
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep27470
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
