
Abstract
Ketamine is commonly used for anesthesia and as a recreational drug. In pregnant users, a potential neurotoxicity in offspring has been noted. Our previous work demonstrated that ketamine exposure of pregnant rats induces affective disorders and cognitive impairments in offspring. As the prefrontal cortex (PFC) is critically involved in emotional and cognitive processes, here we studied whether maternal ketamine exposure influences the development of the PFC in offspring. Pregnant rats on gestational day 14 were treated with ketamine at a sedative dose for 2 hrs, and pups were studied at postnatal day 0 (P0) or P30. We found that maternal ketamine exposure resulted in cell apoptosis and neuronal loss in fetal brain. Upon ketamine exposure in utero, PFC neurons at P30 showed more dendritic branching, while cultured neurons from P0 PFC extended shorter neurites than controls. In addition, maternal ketamine exposure postponed the switch of NR2B/2A expression, and perturbed pre- and postsynaptic protein expression in the PFC. These data suggest that prenatal ketamine exposure impairs neuronal development of the PFC, which may be associated with abnormal behavior in offsprings.
Similar content being viewed by others


Introduction
Ketamine is widely used clinically for anesthesia, especially in the developing world1. Up to 2% pregnant women require surgery, due to problems associated with pregnancy itself or to other medical conditions, and ketamine a common choice. This number is increasing, partly because of laparoscopic procedures and fetal surgery2. Furthermore, ketamine is a popular drug of abuse in diverse populations including pregnant women in the West3,4 and Southeast Asia5. As ketamine may have a prolonged effect on the central nervous system, it is important to investigate whether its use in pregnant subjects may affect neurodevelopment of their offspring.
General anesthetics may be toxic to the developing brain6,7,8,9, as evidenced by increased apoptotic cells, impaired neurogenesis, and neuroinflammation10. In rodents and primates, some anesthetics used in early-life might impair cognitive function, an effect that can last into adulthood11,12. These findings are supported by retrospective clinical data which indicate that the use of anesthetics during early life may be associated with late-onset learning disabilities8,13. Learning and memory function may not be the sole target of toxicity associated with anesthetics, and emotional disorders such as depression and anxiety have not been studied.
We previously reported that14, maternal exposure to ketamine for 2 hrs on gestational day 14 induced anxiety and depression-like behavior15, and cognitive impairment in offsprings. The mismatched expression of N-methyl-D-aspartate (NMDA) receptor subunits and decreased levels of brain-derived neurotrophic factor and postsynaptic density-95 protein (PSD-95) may be responsible for the abnormal behavior14. Emotion modulates memory and learning, interferes with decision making and provides the motivation for critical action16. Conversely, cognitive processing is an integral part of emotion: Anxiety and depression lead to information processing biases and cognitive dysfunction, which contribute to the onset and/or maintenance of symptoms17. Cognition and emotion are intricately intertwined, and both are implemented by overlapping networks in brain regions including the PFC, hippocampus and amygdala18. The hippocampus to PFC pathway plays a critical role in learning and memory19. The PFC receives input from all other cortical regions and hippocampus, and mediates planning and goal-directed motor, cognitive, and affective behavior20. Compared to other areas, the maturation of the PFC is protracted and therefore more susceptible to harmful factors, contributing to neuropsychiatric disorders21. As the abnormal development of the PFC is responsible for diverse emotional, cognitive, and mnemonic problems22, we tested whether the maternal exposure to ketamine impacts the development of PFC, and explored some potential mechanisms.
At low and subanesthetic doses, ketamine is a selective and potent antagonist of NMDA receptors23. In the brain, NMDA receptors are involved in neuronal development and circuit formation by mediating synaptic activity, organizing functional circuits and regulating dendritic growth24,25. Two NMDA receptor subunits, NR2A and NR2B, have specific effects during dendritic arbor development, and a switch in expression of NR2B to NR2A occurs during synaptogenesis25,26. The cytoplasmic C-terminus of NR2A and NR2B interacts with membrane-associated guanylate kinase members and PSD-95, which is essential for clustering and anchoring NMDA receptors at synapses27,28. In this study, we aimed to test the hypothesis that maternal ketamine exposure may disturb the temporal switch of NR2A/2B subunits and impact dendritic development via the PSD-95/NR2A/2B-NMDA receptors in the PFC.
Results
Ketamine induces a widespread apoptosis in fetal brain
To assess apoptosis, TUNEL-positive cells were counted in different regions 8 hrs after ketamine administration and in control fetuses. The number of TUNEL positive cells was significantly larger in the ketamine than in the control group (Fig. 1), particularly in the ventral telencephalon (Fig. 1b,e,g, 116 ± 9 cells/mm2 vs. 70 ± 6 cells/mm2, t = 4.199, p = 0.001), hippocampal primordium (Fig. 1c,f,h, 82 ± 7 cells/mm2 vs. 60 ± 6 cells/mm2, t = 2.319, p = 0.039) and neocortex (Fig. 1a,d,i, 26 ± 2 cells/mm2 vs. 20 ± 2 cells/mm2, t = 2.381, p = 0.039). As necrotic cells also contain fragmented DNA and TUNEL cannot distinguish between apoptosis and necrosis, we also used anti-activated caspase-3 immunofluorescence to detect apoptotic profiles. We found that positive cells were significantly increased in the ketamine compared to the control group (Fig. 2), in the ventral telencephalon (Fig. 2b,e,g, 106 ± 8 cells/mm2 vs. 66 ± 5 cells/mm2, t = 4.257, p = 0.001), hippocampus primordium (Fig. 2c,f,h, 73 ± 6 cells/mm2 vs. 45 ± 4 cells/mm2, t = 4.224, p = 0.001) and neocortex (Fig. 2c,f,i, 21 ± 2 cells/mm2 vs. 15 ± 1 cells/mm2, t = 3.153, p = 0.011).

Embryonic day 14 coronal sections were stained with TUNEL. In control (a–c) and ketamine (d–f) groups, positive cells were widely distributed in the ventral telencephalon (vTel), hippocampal primordium (Hip) and neocortex (NCx). Positive cells in different brain areas were significantly increased in the ketamine compared to the control group (g): vTel; (h): Hip; (i): NCx). LV, lateral ventricle. *p < 0.05; **p < 0.01; n = 6 in each group.

Embryonic day 14 sections were stained with antibodies against cleaved caspase-3. In control (a–c) and ketamine (d–f) groups, positive cells were widely distributed in the ventral telencephalon (vTel), hippocampal primordium (Hip) and neocortex (NCx). Their densities were significantly increased in the ketamine compared to the control group in different areas ((g) vTel; (h) Hip; (i) NCx). LV, lateral ventricle. *p < 0.05; **p < 0.01; n = 6 in each group.
Ketamine causes neuronal loss in laminae II-III of the PFC in offspring
We previously found that ketamine-treated offsprings developed mental disorders in adulthood14. As the PFC is a key hub in neural networks that modulate emotional behavior29, we studied the architecture of the PFC at P0 and P30 using Nissl staining. Compared to the control group, cell density in laminae II-III of ketamine-treated offsprings was decreased by 14% at P0, and 32% at P30 (Fig. 3b–e,h; t = 2.354, p = 0.040 and t = 2.302, p = 0.044, respectively). However, there was no significant difference in cell density in lamina V of the PFC at P0 and P30 (Fig. 3b,c,f,g,i, t = 0.628, p = 0.544 and t = 0.645, p = 0.533, respectively). Thus, neurons in laminae II-III appear more susceptible to ketamine.

Coronal sections encompassing the PFC at P0 and P30, stained with cresyl violet. The boxed area indicated in panel a was chosen for analysis. At P0, the cytoarchitecture of the PFC was comparable in control (b) and ketamine (c) groups, but the cell density in laminae II-III was significantly decreased in the ketamine group (h). At P30, cell distribution was similar in laminae II-III (d,e) and V (f,g) of control (d,f) and ketamine-treated animals (e,g). In the ketamine group, the cell density was significantly decreased in laminae II-III (h) but not in lamina V (i), compared to the control group. PrL, prelimbic cortex; *p < 0.05; n = 6 in each group.
Ketamine exposure disturbs the maturation of pyramidal neurons in the PFC of offspring
To study pyramidal neurons, we performed Golgi–Cox impregnation and reconstructed basal and apical dendritic trees of 120 pyramidal neurons in each group (10 neurons per region and 6 pups per group) selected from laminae II-III and V of the PFC. Basal and apical dendrites, and the total dendritic branch length in neurons of lamina V were similar in the ketamine and control groups (data not shown). In contrast, in laminae II-III, neurons from ketamine-treated pups had more basal branches (Fig. 4a–c, n = 6, t = 2.495, p = 0.032) and longer total branch length than in the control group (Fig. 4a,b,d, t = 2.370, p = 0.039). This was confirmed by Sholl analysis (Fig. 4f, p = 0.009). Furthermore, the spine density was significantly increased in laminae II and III neurons from ketamine-treated offsprings compared with that in control samples (Fig. 4a’, t = 3.371, p = 0.007).

Six P30 brains in each group were processed for Golgi–Cox impregnation and pyramidal neurons in laminae II-III of the PFC were studied in the control (examples in a) and ketamine (examples in b) groups. The left panels of (a,b) show two examples of Golgi-impregnated neurons. The total branch number and dendritic length were significantly increased in the ketamine compared to the control group (c,d) n = 60 neurons in each group). In addition, the ketamine group had higher spine density than the control (a’, b’, (e) n = 60 dendrites in each group). Sholl analysis showed that the complexity of dendritic trees was higher in the ketamine than in the control group (f) Kolmogorov–Smirnov test). *p < 0.05; **p < 0.01.
Ketamine administration inhibits neurite growth in neuronal culture
To verify that anomalies of pyramidal neurons in animals exposed to ketamine in utero are due to a cell intrinsic defect, we cultured PFC neurons from control and ketamine-treated P0 pups. As shown in Fig. 5, after 4 days in vitro (DIV), neurons from both groups were comparable in their ability to extend neurites. However, quantitative analysis showed that the total neurite length was significantly shorter and the branching was significantly decreased in the ketamine-treated compared to the control group (Fig. 5m,n, t = 2.389, p = 0.020 and t = 2.246, p = 0.029, respectively). Similar results were obtained by Sholl analysis (Fig. 5o, p < 0.01). These data suggested that maternal administration of ketamine resulted in a cell autonomous decrease in neurite extension of PFC neurons in vitro.

P0 PFC neurons from pups whose mothers received ketamine at gestational day 14 or not (control) (n = 6 in each group) were cultured for 4 DIV, immunostained with anti-β-tubulin antibodies (a,b), prior to reconstruction using Imaris (examples in (c–g), control; (h–l), ketamine group). Quantitative analysis showed that the total neurite length and number were significantly lower in the ketamine than the control groups ((m,n); n = 60 neurons in each group). This difference was confirmed by Sholl analysis (o). *p < 0.05; **p < 0.01.
Expression of NR2B/NR2A, synaptophysin and PSD-95 is affected by maternal exposure of ketamine
We measured the expression of NR2B and NR2A, the presynaptic protein synaptophysin and the postsynaptic protein PSD-95 in the PFC at P0 and P30 (Fig. 6). The expression of NR2B was significantly higher in the control than the ketamine-treated group at P0 (Fig. 6a,c, 0.857 ± 0.038 vs. 0.680 ± 0.040, F = 10.24, p = 0.033). In contrast, the situation was different at P30, when NR2B levels were slightly lower in the ketamine than in the control group (Fig. 6a,c, the control vs. the ketamine: 0.367 ± 0.066 vs. 0.593 ± 0.044, F = 8.260, p = 0.045). NR2A protein was expressed a low level at P0, with no significant difference in the two groups (Fig. 6b,d, 0.287 ± 0.050 in control vs. 0.247 ± 0.046 in ketamine, F = 0.346, p = 0.588). Interestingly, the NR2A protein level was significantly decreased in the ketamine compared to the control at P30 (Fig. 6b,d, ketamine vs. control: 0.427 ± 0.035 vs. 0.620 ± 0.047, F = 10.745, p = 0.031). The level of synaptophysin was significantly higher in the control than the ketamine-treated group at both P0 and P30 (Fig. 6e,g, t = 3.590, p = 0.023 and t = 2.874, p = 0.014, respectively). In contrast, PSD-95 was weakly and similarly expressed in both groups at P0, but very significantly higher in ketamine-treated rats than control animals at P30 (Fig. 6f,h, t = 4.732, p = 0.009). In addition, to better define the distribution and localization of synaptic proteins, we performed double immunofluorescence using anti-PSD-95 and anti-synaptophysin (SY-38) antibodies on PFC sections from P30 brains. In accord with the results obtained with western blot, there was clearly higher fluorescence intensity for PSD-95 and less immunoreactivity of synaptophysin in the ketamine group compared to the control (Fig. 7c,d, t = 2.661, p = 0.024 and t = 2.859, p = 0.017, respectively).

PFC tissue from P0 and P30 rats were processed for western blots to detect proteins NR2B (a), NR2A (b), synaptophysin (SY-38, e) and PSD-95 (f). The NR2B protein levels in the ketamine group were significantly lower at P0 and higher at P30 compared to the corresponding control samples (a,c). The NR2A protein was expressed at a low level at P0 in both control and ketamine samples (b,d) but was significantly decreased in the ketamine compared to the control at P30 (b,d). The ketamine group expressed significantly less SY-38 at P0 and P30 relatively to controls (e,g). In contrast, there was no significant difference of PSD-95 levels at P0 and more PSD-95 protein in the ketamine group than in the control at P30 (f,h). ctrl-P0, control group at P0; ctrl-P30, control group at P30; ktm-P0, the ketamine group at P0; ktm-P30, ketamine group at P30. *p < 0.05; **p < 0.01, comparison between the control and ketamine group; #p < 0.05; ##p < 0.01, comparison between P0 and P30; n = 6 in each group.

The rat with location of the medial prefrontal cortex (PrL, a). Areas of prefrontal cortex as indicated in (b) and laminae II-III as indicated in (e–h) were selected for analysis. Paraffin coronal sections from P30 PFC were stained with anti-PSD-95 and anti-synatophysin (SY-38) antibodies. Fluorescent immunohistochemistry for PSD-95 (green) and SY-38 (green), and DAPI (blue). The signal was estimated by quantifying the density of immnofluorescence using Image J. The ketamine group showed a significant increase of PSD-95 immunoreactivity (c) and a significant decrease of synaptophysin (d) at P30 compared to the control. *p < 0.05; n = 6 in each group.
Discussion
The present study provides evidence that maternal administration of subanesthetic dose of ketamine during pregnancy in rats induces apoptosis and neuronal loss in fetal brain, as well as abnormalities of PFC layer II/III pyramidal neuron development. In the PFC, there are mismatched expression of NMDA receptor subunits and disturbed expression of pre- and postsynaptic proteins at P0 and P30. Together with our previous study, these results indicate that in utero ketamine exposure has detrimental effects on brain development, particularly in the hippocampus and PFC.
Anesthetics can be toxic for the developing brain, and two critical factors determine neurotoxicity: the stage of brain development at which the compound is administered, and the dose and/or duration of exposure30. Neurodevelopment proceeds at a different pace in different brain regions, so that vulnerability depends both on stage and region. Although the dose in our experiment was subanesthetic, sedative dose23 could induce cell apoptosis in fetal brain. This is consistent with a previous observation that ketamine exposure of fetal rhesus monkeys resulted in 2.2 times more neuronal loss than exposure at early postnatal stages6. Our data confirm that fetal brains are more vulnerable to ketamine than neonatal ones, with preferential neuronal loss in laminae II and III of PFC region. We previously showed that ketamine administration in utero impaired postnatal neurogenesis in the dentate gyrus and in the subventricular zone. It is therefore likely that ketamine induced neuronal loss by impacting the proliferation and/or apoptosis of neural progenitor cells, which remains to be demonstrated further.
The hippocampus and PFC play well-established roles in cognitive and mnemonic processes. Psychiatric disorders, such as schizophrenia, depression and post-traumatic stress syndrome, share common symptoms of cognitive impairment and emotional dysregulation and are associated with structural and pathophysiological changes in the hippocampus and PFC31. Our previous study showed that ketamine exposure impaired maturation of pyramidal cells in the CA3 hippocampal field of offspring (at P30)14. In contrast, the present study shows that pyramidal neurons in laminae II and III of the PFC of offspring (at P30) harbor the opposite profiles, exhibiting more branched and longer basilar dendrites, and increased spine density after ketamine exposure. This discrepancy is likely due to regional differences of anaesthesia-induced neurotoxic effect on synaptogenesis and neuroplasticity32. Moreover, our data are consistent with reports that psychoactive drugs such as amphetamine, cocaine or nicotine, increased spine density in PFC and nucleus accumbens33,34. For layer II/III cells, basal dendrites receive inputs from local sources and apical dendrites receive inputs from other cortical areas as well as nonspecific thalamic inputs35,36. Our results indicates that exposure of ketamine in utero may influence local synaptic circuits in PFC in adulthood.
In addition to inducing apoptosis, anesthetics interfere with synaptogenesis37,38. Most excitatory inputs are targeted to dendrites and the establishment of dendritic trees is a highly dynamic process characterized by extension and retraction of branches, followed by their stabilization and establishment of synaptic connections39. We investigated dendritic development at two developmental time points, early (cultured PFC neurons at P0) and mature stages (P30, Golgi staining). In cultured neurons derived from neonatal PFC, the total neurite length was significantly shorter and neurites less ramified in the ketamine than in the control group. This is consistent with a previous observations that ketamine induced neuronal damage in rat primary forebrain cultures40. Intriguingly, our in vivo data at P30 showed an opposite over-branched dendritic phenotype upon ketamine exposure. A possible explanation is that PFC neurons at P0 are at an early stage of dendritic development, characterized by dynamic branch extension, and this may be dampened by ketamine treatment. In contrast, neurons at P30 are at the end of those dynamic processes, when the overall effect of ketamine could result in dendritic over-branching. Given that Golgi impregnation does not work well at P0, other techniques, such as electron microscopy and electrophysiology-coupled imaging are required to test that hypothesis.
NMDA receptors are important for neuronal development and circuit formation24. The NR2B subunit is highly expressed in the embryonic and neonatal brain, whereas expression of the NR2A subunit increases during brain maturation41. This ‘developmental switch’ of the subunit content of synaptic NMDA receptors plays a key role in refining topographic projections, organizing functional circuits and regulating dendritic growth26. In control animals, we found that NR2B was abundantly expressed at P0 and significantly decreased at P30, while NR2A was expressed at a low level at P0 and significantly increased at P30, as reported42. However, the reduction of NR2B protein at P30 and the increase of NR2A expression were not as prominent in ketamine treated neurons. This blunted ‘developmental switch’ of NR2B/NR2A expression induced by ketamine may cause the impaired dendritic development of PFC neurons, and may be involved in the behavioral phenotype in offsprings. NMDA receptors are predominantly expressed at the postsynaptic membrane where they interact with components of the postsynaptic density that modulate their activity, particularly PSD-9543. PSD-95 regulates the size and strength of synapses44, the formation of synaptic assemblies45, and spine-maturation46. Synaptophysin is one of the most abundant synaptic vesicle integral proteins that regulates the formation and trafficking of synaptic vesicles47. Our Western blot and immunohistochemistry data showed that the expression of PSD-95 and synaptophysin at P0 and P30 was similar to what was reported elsewhere48,49. The higher levels of PSD-95 and NR2B expression in ketamine-treated than in the control samples presumably reflects increased synapse concentration, which is consistent with the overbranched dendritic phenotype in mature neurons39. The decreased expression of synaptophysin at P0 and P30 may reflect abnormal transmitter release in ketamine-treated offsprings.
In summary, together with our previous study14, the present results show that exposure to ketamine in utero can impair the development of the hippocampus and the PFC, both of which might contribute to cognitive impairment and mood disorders in offspring. This information has important implications for the use of ketamine in anesthesia and as a recreational drug, especially during pregnancy.
Materials and Methods
Subjects
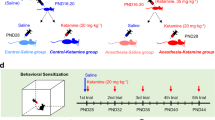
All experimental procedures were performed according to the guidelines that have been approved by the Ethics Committees of Jinan University, Guangzhou, China. The Sprague-Dawley rats, weighted 180–220 g, were housed with a 12 hr light/dark cycle and had access to water and food ad libitum. The vaginal plugs were checked in the morning and a positive one was defined as the gestational day 0. Ketamine injections were done on gestational day 14.
Anesthesia
On gestational day 14, 18 dams were randomly divided into control (n = 9) and ketamine (n = 9) groups. The establishment of animal models was described before14. Three dams per group were used for TUNEL assay 8 hrs after ketamine infusion whilst another six in each group were allowed to give birth naturally. Three P0 pups from each dam were used for neuronal culture and each experiment was repeated six times. P0 and P30 brains were used for histology and western blot. Golgi stain was carried out on P30 brains. A schematic representation of experimental protocols is shown in Fig. 8. The total dose of ketamine used in each dam was 144.2 ± 4.6 mg/kg (n = 9), and all anesthetized dams recovered fully without complications, such as respiratory depression, cardiac arrest, or miscarriage. The recovery time from anesthesia was 37 ± 4 min (n = 9), and the total anesthesia time (from initial intramuscular injection to recovery) was 176 ± 4 min. At P30, the body weight of rats in the ketamine group was less than that of the control group (71 ± 3 g vs. 88 ± 3 g; t = 3.770, p = 0.0003).

The flow chart of the experimental protocols.
Histology and immunohistochemistry
Ten paraffin coronal sections (5 μm) including the PFC (+2.7 mm from bregma and +3.7 mm from interaural landmark) were stained with 0.5% cresyl violet (n = 6 in each pup). Microphotographs were taken under a 40× objective (Leica, DM6000B, Germany). Nissl-stained cell numbers were counted in a blinded manner using Image J. (NIH, USA). For Immunohistochemistry, the sections were incubated with the following primary antibodies overnight at 4 °C: goat anti-cleaved caspase-3 antibody (L-18) (1:250, Cat.No.sc-1225, Santa Cruz, CA, Germany), mouse anti-Synaptophysin (1:1000, Cat.No.ab8049, Abcam, Cambridge, UK), mouse- anti-PSD-95 (1:1000, Cat.No. MAB1596, Millipore, USA). Signal was detected with Alexafluor 546- or 488-labeled fluorescent secondary antibodies (1:1000, Invitrogen, Carlsbad, CA, USA). Caspase-3 positive cells and PSD-95 and synaptophysin immunofluorescence intensity were evaluated using Image J (NIH, USA).
TUNEL analysis
Eight hrs after ketamine infusion, embryonic brains were fixed in 4% paraformaldehyde (PFA) overnight at 4 °C, then processed and embedded in wax. Ten to fifteen paraffin coronal sections (5 μm) of each brain (n = 6, 2 from each dam) were stained with a TUNEL kit (C1086, Beyotime institute of Biotechnology, China). Positive cells were examined under the microscope with a digital camera system (Leica).
Golgi stain
Golgi stain, tri-dimensional reconstruction and dendrite analysis were performed as described14. Frozen brain sections with 150 μm-thickness between +2.7 mm from bregma and +3.7 mm from interaural landmark were stained using a FD Rapid Golgi Stain kit (FD NeuroTechnologies, Inc. Columbia, USA) and 10 well individualized pyramidal neurons in superficial (II–III) and deep (V) laminaes were reconstructed using with the Imaris software (BitPlane AG, Zurich, Switzerland). To measure spine density, distal segments of dendrites of pyramidal neurons selected for reconstruction were photographed using a 100× objective for counting of spines in 40 μm segments, and the results were expressed as the number of spines /10 μm.
Neuronal cultures and neurite analysis
Cerebral cortices from PFC were chopped into pieces and digested with 0.25% trypsin (w/v) in HBSS at 37 °C for 30 min. The reaction was stopped by fetal bovine serum (Cat.No.10099133, Gibco). Samples were triturated by gentle pipetting and cells were harvested by centrifugation at 1000 rpm for 5 min, and seeded on polylysine-coated coverslips at a density of 1.0 × 105 cells/ml. The culture medium was neurobasal-A medium (Cat.No.10888022, Invitrogen) supplemented with 2% B27 (Cat.No.17504044, Invitrogen). After 4 DIV, cells were fixed with 4% PFA and immunostained with rabbit anti-β-tubulin (1:1000, Cat.No.2128, Cell Signaling Technology). The images of 10 well developed neurons were captured and neurites were analyzed using the Imaris software (BitPlane AG). The complexity of total dendritic trees was estimated using Sholl analysis50.
Western blotting
Samples from PFC (n = 6/group) were dissociated in lysis buffer (containing protease inhibitors, 50 mM Tris–HCl, pH 7.6) on ice for 30 min and homogenized via ultrasonification (Ningbo scientz biotechnology CO. LTD, Ningbo, China). After centrifugation at 12,000 g for 10 min at 4 °C, supernatants were pooled and protein concentrations were measured with a BCA assay kit (Beyotime Institute of Biotechnology, China). Thirty μg protein samples were mixed with gel loading buffer (50 mM Tris-HCl, 10% SDS, 10% glycerol, 10% 2-mercaptoethanol, 2 mg/ml bromophenol blue) in a 1:1 ratio, boiled for 5 min and subjected to SDS-PAGE gels. The separated proteins were transferred to polyvinylidene fluoride membranes and incubated with mouse anti-Synaptophysin (1:1000, Cat.No.ab8049, Abcam, Cambridge, UK), rabbit anti-NR2B (1:1000, Cat.No.06-600, Millipore, USA), rabbit anti-NR2A (1:1000, Cat.No.4205, Cell Signaling Technology, USA), mouse anti-PSD-95 (1:1000, Cat.No.MAB1596, Millipore, USA), rabbit anti-GAPDH (1:1000, Cat.No.5174, Cell Signaling Technology, USA) and rabbit anti-β-tubulin (1:1000, Cat.No.2128, Cell Signaling Technology, USA) overnight at 4 °C. Signal was detected using HRP-conjugated rabbit or mouse antibodies followed by chemiluminescence using a Super Signal West Pico kit (Pierce) and Hyper film ECL (Amersham Biosciences). All experiments were carried out at least in triplicate. Autoradiography films were scanned and signals were quantified using Image J (NIH, USA). Density of each band was normalized to reference controls (β-tubulin or GAPDH).
Statistical analysis
Data were presented as mean ± SEM. The homogeneity of variance was verified with Levene’s test and single comparison between both groups was made using unpaired two-tailed Student t test. Data on expression of NR2B and NR2A were analyzed by two-way ANOVA, and significant main effects of two different timepoints were followed by one-way ANOVA for each group. The parametric Bonferroni and nonparametric Kruskal–Wallis H tests were used to analyze dendrite branching with the Sholl method. Results were considered significant (* or #) at p < 0.05 and highly significant (** or # #) at p < 0.01. All analyses were performed using SPSS 16.0 software (SPSS Inc., Chicago, IL, USA).
Additional Information
How to cite this article: Zhao, T. et al. Prenatal ketamine exposure causes abnormal development of prefrontal cortex in rat. Sci. Rep. 6, 26865; doi: 10.1038/srep26865 (2016).
References
Craven, R. Ketamine. Anaesthesia 62 Suppl 1, 48–53 (2007).
Cheek, T. G. & Baird, E. Anesthesia for nonobstetric surgery: maternal and fetal considerations. Clin Obstet Gynecol 52, 535–545 (2009).
Bokor, G. & Anderson, P. D. Ketamine: an update on its abuse. J Pharm Pract 27, 582–586 (2014).
Degenhardt, L. & Dunn, M. The epidemiology of GHB and ketamine use in an Australian household survey. Int J Drug Policy 19, 311–316 (2008).
Dargan, P. I. & Wood, D. M. Recreational drug use in the Asia Pacific region: improvement in our understanding of the problem through the UNODC programmes. J Med Toxicol 8, 295–299 (2012).
Brambrink, A. M. et al. Ketamine-induced neuroapoptosis in the fetal and neonatal rhesus macaque brain. Anesthesiology 116, 372–384 (2012).
Jevtovic-Todorovic, V. et al. Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J Neurosci 23, 876–882 (2003).
Kalkman, C. J. et al. Behavior and development in children and age at the time of first anesthetic exposure. Anesthesiology 110, 805–812 (2009).
Zhou, Z. & Ma, D. Anaesthetics-induced neurotoxicity in developing brain: an update on preclinical evidence. Brain Sci 4, 136–149 (2014).
Jevtovic-Todorovic, V. et al. Anaesthetic neurotoxicity and neuroplasticity: an expert group report and statement based on the BJA Salzburg Seminar. Br J Anaesth 111, 143–151 (2013).
Paule, M. G. et al. Ketamine anesthesia during the first week of life can cause long-lasting cognitive deficits in rhesus monkeys. Neurotoxicol Teratol 33, 220–230 (2011).
Zheng, H. et al. Sevoflurane anesthesia in pregnant mice induces neurotoxicity in fetal and offspring mice. Anesthesiology 118, 516–526 (2013).
Ing, C. et al. Long-term differences in language and cognitive function after childhood exposure to anesthesia. Pediatrics 130, e476–e485 (2012).
Zhao, T. et al. Ketamine administered to pregnant rats in the second trimester causes long-lasting behavioral disorders in offspring. Neurobiol Dis 68, 145–155 (2014).
Clancy, B. et al. Web-based method for translating neurodevelopment from laboratory species to humans. Neuroinformatics 5, 79–94 (2007).
Davidson, R. J. & Irwin, W. The functional neuroanatomy of emotion and affective style. Trends Cogn Sci 3, 11–21 (1999).
Crocker, L. D. et al. Relationships among cognition, emotion, and motivation: implications for intervention and neuroplasticity in psychopathology. Front Hum Neurosci 7, 261 (2013).
Miller, G. A. Mistreating Psychology in the Decades of the Brain. Perspect Psychol Sci 5, 716–743 (2010).
Floresco, S. B., Seamans, J. K. & Phillips, A. G. Selective roles for hippocampal, prefrontal cortical, and ventral striatal circuits in radial-arm maze tasks with or without a delay. J Neurosci 17, 1880–1890 (1997).
Godsil, B. P., Kiss, J. P., Spedding, M. & Jay, T. M. The hippocampal-prefrontal pathway: the weak link in psychiatric disorders? Eur Neuropsychopharmacol 23, 1165–1181 (2013).
Kolb, B. et al. Experience and the developing prefrontal cortex. Proc Natl Acad Sci USA 109 Suppl 2, 17186–17193 (2012).
Heidbreder, C. A. & Groenewegen, H. J. The medial prefrontal cortex in the rat: evidence for a dorso-ventral distinction based upon functional and anatomical characteristics. Neurosci Biobehav Rev 27, 555–579 (2003).
Chizh, B. A. Low dose ketamine: a therapeutic and research tool to explore N-methyl-D-aspartate (NMDA) receptor-mediated plasticity in pain pathways. J Psychopharmacol 21, 259–271 (2007).
Sepulveda, F. J. et al. Differential roles of NMDA Receptor Subtypes NR2A and NR2B in dendritic branch development and requirement of RasGRF1. J Neurophysiol 103, 1758–1770 (2010).
Gambrill, A. C. & Barria, A. NMDA receptor subunit composition controls synaptogenesis and synapse stabilization. Proc Natl Acad Sci USA 108, 5855–5860 (2011).
Ewald, R. C., Van Keuren-Jensen, K. R., Aizenman, C. D. & Cline, H. T. Roles of NR2A and NR2B in the development of dendritic arbor morphology in vivo . J Neurosci 28, 850–861 (2008).
Elias, G. M., Elias, L. A., Apostolides, P. F., Kriegstein, A. R. & Nicoll, R. A. Differential trafficking of AMPA and NMDA receptors by SAP102 and PSD-95 underlies synapse development. Proc Natl Acad Sci USA 105, 20953–20958 (2008).
Elias, G. M. & Nicoll, R. A. Synaptic trafficking of glutamate receptors by MAGUK scaffolding proteins. Trends Cell Biol 17, 343–352 (2007).
Drevets, W. C., Price, J. L. & Furey, M. L. Brain structural and functional abnormalities in mood disorders: implications for neurocircuitry models of depression. Brain Struct Funct 213, 93–118 (2008).
Wang, C. Advanced pre-clinical research approaches and models to studying pediatric anesthetic neurotoxicity. Front Neurol 3, 142 (2012).
Godsil, B. P., Kiss, J. P., Spedding, M. & Jay, T. M. The hippocampal-prefrontal pathway: the weak link in psychiatric disorders? Eur Neuropsychopharmacol 23, 1165–1181 (2013).
Jevtovic-Todorovic, V. et al. Anaesthetic neurotoxicity and neuroplasticity: an expert group report and statement based on the BJA Salzburg Seminar. Br J Anaesth 111, 143–151 (2013).
Robinson, T. E. & Kolb, B. Structural plasticity associated with exposure to drugs of abuse. Neuropharmacology 47 Suppl 1, 33–46 (2004).
Muhammad, A. et al. Prenatal nicotine exposure alters neuroanatomical organization of the developing brain. Synapse 66, 950–954 (2012).
Spruston, N. Pyramidal neurons: dendritic structure and synaptic integration. Nat Rev Neurosci 9, 206–221 (2008).
Wallis, J. D. Cross-species studies of orbitofrontal cortex and value-based decision-making. Nat Neurosci 15, 13–19 (2012).
Aligny, C. et al. Ketamine alters cortical integration of GABAergic interneurons and induces long-term sex-dependent impairments in transgenic Gad67-GFP mice. Cell Death Dis 5, e1311 (2014).
Walker, S. M., Westin, B. D., Deumens, R., Grafe, M. & Yaksh, T. L. Effects of intrathecal ketamine in the neonatal rat: evaluation of apoptosis and long-term functional outcome. Anesthesiology 113, 147–159 (2010).
Emoto, K. Signaling mechanisms that coordinate the development and maintenance of dendritic fields. Curr Opin Neurobiol 22, 805–811 (2012).
Liu, F. et al. Ketamine-induced neuronal damage and altered N-methyl-D-aspartate receptor function in rat primary forebrain culture. Toxicol Sci 131, 548–557 (2013).
Liu, X. B., Murray, K. D. & Jones, E. G. Switching of NMDA receptor 2A and 2B subunits at thalamic and cortical synapses during early postnatal development. J Neurosci 24, 8885–8895 (2004).
Takai, H., Katayama, K., Uetsuka, K., Nakayama, H. & Doi, K. Distribution of N-methyl-D-aspartate receptors (NMDARs) in the developing rat brain. Exp Mol Pathol 75, 89–94 (2003).
Sans, N. et al. NMDA receptor trafficking through an interaction between PDZ proteins and the exocyst complex. Nat Cell Biol 5, 520–530 (2003).
Ehrlich, I. & Malinow, R. Postsynaptic density 95 controls AMPA receptor incorporation during long-term potentiation and experience-driven synaptic plasticity. J Neurosci 24, 916–927 (2004).
Garner, C. C., Nash, J. & Huganir, R. L. PDZ domains in synapse assembly and signalling. Trends Cell Biol 10, 274–280 (2000).
Nikonenko, I. et al. PSD-95 promotes synaptogenesis and multiinnervated spine formation through nitric oxide signaling. J Cell Biol 183, 1115–1127 (2008).
Mutch, S. A. et al. Protein quantification at the single vesicle level reveals that a subset of synaptic vesicle proteins are trafficked with high precision. J Neurosci 31, 1461–1470 (2011).
Glantz, L. A., Gilmore, J. H., Hamer, R. M., Lieberman, J. A. & Jarskog, L. F. Synaptophysin and postsynaptic density protein 95 in the human prefrontal cortex from mid-gestation into early adulthood. Neuroscience 149, 582–591 (2007).
Chang, L. R. et al. Different expression of NR2B and PSD-95 in rat hippocampal subregions during postnatal development. Microsc Res Tech 72, 517–524 (2009).
SHOLL, D. A. Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat 87, 387–406 (1953).
Acknowledgements
This work was supported by grants from Project of Internation as well as Hong Kong, Macao & Taiwan Science and Technology Cooperation Innovation Platform in Universities in Guangdong Province (2013gjhz0002, L. Zhou), National Basic Research Program of China (973 Program, 2014CB542205, L. Zhou), National Natural Science Foundation of China (81503172, T. Zhao) and Programme of Introducing Talents of Discipline to Universities (B14036).
Author information
Authors and Affiliations
Contributions
L.Z., T.Z., C.L., X.S. and D.M. designed research; T.Z. and C.L. performed experiments; T.Z., C.L., W.W. and H.Z. analyzed data; T.Z. wrote the paper; L.Z., X.S. and D.M. critically revised the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhao, T., Li, C., Wei, W. et al. Prenatal ketamine exposure causes abnormal development of prefrontal cortex in rat. Sci Rep 6, 26865 (2016). https://doi.org/10.1038/srep26865
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep26865
This article is cited by
-
Prenatal Exposure to General Anesthesia Drug Esketamine Impaired Neurobehavior in Offspring
Cellular and Molecular Neurobiology (2023)
-
Ketamine impairs growth cone and synaptogenesis in human GABAergic projection neurons via GSK-3β and HDAC6 signaling
Molecular Psychiatry (2022)
-
Development of prefrontal cortex
Neuropsychopharmacology (2022)
-
Neonatal Isoflurane Exposure in Rats Impairs Short-Term Memory, Cell Viability, and Glutamate Uptake in Slices of the Frontal Cerebral Cortex, But Not the Hippocampus, in Adulthood
Neurotoxicity Research (2022)
-
A synthetic peptide rescues rat cortical neurons from anesthetic-induced cell death, perturbation of growth and synaptic assembly
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
