
Abstract
PPARγ modulates energy metabolism and inflammation. However, its specific functions in the balance of immunity in vivo have been explored incompletely. In this study, by the age of 14 mo, PpargC/− mice with PPARγ expression at 25% of the normal level exhibited high autoantibody levels and developed mesangial proliferative glomerulonephritis, which resembled systemic lupus erythematosus (SLE)-like autoimmune disease. These symptoms were preceded by splenomegaly at an early age, which was associated with increases in splenocyte accumulation and B-cell activation but not with relocation of hematopoiesis to the spleen. The mechanism of splenic lymphocyte accumulation involved reduced sphingosine-1-phosphate receptor 1 (S1P1) expression and diminished migration toward S1P in the PpargC/− splenocytes, which impeded lymphocyte egression. Mechanistically, increased Th17 polarization and IL-17 signaling in the PpargC/− CD4+ T cells contributed to B-cell hyperactivation in the spleen. Finally, the activation of the remaining PPARγ in PpargC/− mice by pioglitazone increased S1P1 levels, reduced the Th17 population in the spleen and ameliorated splenomegaly. Taken together, our data demonstrated that reduction of Pparg expression in T-helper cells is critical for spontaneous SLE-like autoimmune disease development; we also revealed a novel function of PPARγ in lymphocyte trafficking and cross talk between Th17 and B cells.
Similar content being viewed by others


Introduction
Over several decades, the prevalence of metabolic and autoimmune diseases has increased in Western countries1,2. Notably, arthritis (referring to more than 100 rheumatic diseases) and obesity maps have shown considerable overlaps (http://www.cdc.gov/obesity/data/prevalence-maps.html and http://www.cdc.gov/arthritis/data_statistics/state-data-current.htm). Furthermore, familial partial lipodystrophy, a type of body fat loss, is associated with autoimmune diseases3,4. The association between dysregulated metabolic balance and autoimmune diseases suggests that common etiological factors underlie both conditions5. We hypothesize that peroxisome proliferator-activated receptor gamma (PPARγ) is one of these factors.
PPARγ is a transcription factor involved in adipocyte differentiation and glucose metabolism. It has also been implicated in modulating inflammation and immune responses. Among cell-specific knockout mouse models, PPARγ CD4+ T-cell–specific knockout mice have enhanced T-helper 17 (Th17) differentiation and are more susceptible to myelin oligodendrocyte glycoprotein (MOG)-induced experimental allergic encephalomyelitis (EAE)6. Macrophage-specific PPARγ knockout mice develop systemic lupus erythematosus (SLE) nephritis caused by deficient phagocytosis7. Among haploinsufficient mouse models, Pparg+/− B cells show increased proliferation and Pparg+/− mice are more susceptible to ovalbumin or methylated BSA-induced arthritis8. By contrast, Pparg+/− mice are susceptible to MOG-induced EAE, which is associated with an increase in T-cell proliferation and Th1 response9. Thus, PPARγ loss implicates the susceptibility of an individual to autoimmunity.
Because the influence of individual genes on autoimmune disease development involves multiple regulatory pathways, the conclusions obtained using cell-type–specific knockout models may be somewhat biased. Although most of the aforementioned studies were conducted in a cell-specific or haploinsufficient manner with the stimulation of specific antigens, the detailed regulation of the balance between tolerance and immunity by PPARγ might have been masked in those experimentally induced systems. Furthermore, subtle gene expression variations have been linked to autoimmune disease development in mouse models10,11. Moreover, clinical studies have shown that single-nucleotide polymorphisms, manifested as a modest change in gene expression, are often associated with autoimmunity12,13. Thus, a modest change in gene expression could shift the balance between tolerance and autoimmunity. A novel tool for revealing the actual functions of PPARγ in the development of autoimmunity without stimulating specific antigens is required.
In this study, we investigated the function of PPARγ in the humoral immune response by using mice with different levels of PPARγ expression (25%–100%) to titrate the PPARγ dose effects on the immune system. These PPARγ quantitative variant mouse strains differ only in the Pparg 3′-UTR sequence and produce normal PPARγ protein in all relevant tissues14,15,16. Thus, these PPARγ quantitative variants are useful for revealing the involvement of PPARγ in the complex immune system. Here, we reported that young mice with PPARγ expression at 25% of the normal level showed splenomegaly independent of extramedullary hematopoiesis compared with mice with ≥50% PPARγ expression. Because the disturbance and hyperactivation of the immune system are frequently associated with splenomegaly, we hypothesized that immunological homeostasis is disrupted at a certain low level of PPARγ expression, consequently enhancing humoral responses and resulting in autoimmunity.
Results
Spleen enlargement in PPARγ hypomorphic mice
Four mouse strains, PpargC/−, Pparg+/−, PpargC/+ and Pparg+/+ (wild-type [WT]), with 23%, 64%, 83% and 100% PPARγ expression in the spleen were generated by crossing Pparg+/− mice with PpargC/+ mice, which carry an allele of Pparg with a c-fos AU-rich element inserted in the 3′-UTR region (Fig. 1A)14,15. The reduction was confirmed by the immunofluorescent staining without the change of cellular localization (Fig. S1A). In addition, the ratio to PPARγ level of Ser273 phosphorylation, which is known to inhibit its transactivation17, was higher in PpargC/− splenocytes (Fig. S1B), suggesting that PPARγ activity may be even repressed in PpargC/− splenocytes. At 2–3 mo of age, among all littermates, only PpargC/− mice exhibited splenomegaly (Fig. 1A). The increase in spleen weight in PpargC/− mice compared with WT littermates occurred at all ages beginning at 1 mo in mice of both sexes and the differences became highly apparent after the age of 12 mo (Figs 1B and S1C,D). PpargC/− mice aged 4 mo showed a normal splenic architecture (Fig. 1C) but increased numbers of total splenocytes, B cells and T cells (Fig. 1D). However, the composition of immune cell populations in the spleen—including B and T cells and their subsets as well as dendritic cells (CD11C+FSC+), macrophages (Gr-1−CD11b+F4/80+), plasma cells (CD19lowCD138+), germinal center B cells (B220+PNAhigh) and activated CD4+ T cells (CD62LhighCD44high)—did not differ between WT and PpargC/− mice at both ages (Tables SI–II). Thus, splenomegaly in PpargC/− mice was associated with the increase in total cellularity, rather than with a specific increase in a certain subpopulation.

Spleen enlargement in PPARγ hypomorphic mice.
(A) Percentage of splenic PPARγ expression, gross morphology of spleens and spleen-to-body weight ratio from WT, PpargC/+, Pparg+/− and PpargC/− mice at 2–3 mo of age by one-way ANOVA with Scheffe’s test. (B) Spleen weight at different ages. (C) Hematoxylin and eosin staining of spleen sections from 3-mo-old WT and PpargC/− mice. Scale bar, 500 μm. (D) Total splenic cellularity of 4- and 7-mo-old mice and total B cells and T cells in the spleens of 4-mo-old mice. Numbers inside bars indicate the number for each group. *p < 0.05; **p < 0.01; ***p < 0.001.
No signs of extramedullary hematopoiesis in young PpargC/− mice
To investigate whether extramedullary hematopoiesis causes splenomegaly in PpargC/− mice, we performed microcomputed tomography analysis on the distal metaphysis of femurs harvested from younger (2-mo-old) and older (9-mo-old) mice (Fig. S2A). Compared with their WT littermates, older PpargC/− mice exhibited significantly increased bone mineral density, trabecular volumes and trabecular numbers but decreased trabecular separation (Fig. S2B); however, these parameters did not differ among the younger mice. Furthermore, the percentage of Lin−cKit+Sca1+ hematopoietic stem cells (HSCs) in the spleen of the older PpargC/− mice was significantly higher than that in their WT littermates (Fig. S2C). The expression of transcription factors crucial for HSC differentiation and self-renewal—LMO2 and GATA1—was significantly higher in the spleen but lower in the bone marrow of the older PpargC/− mice (Fig. S2D); however, these parameters did not differ between the younger WT and PpargC/− mice. These data demonstrate that splenomegaly is independent of extramedullary hematopoiesis in younger PpargC/− mice.
Reduced sphingosine-1-phosphate receptor 1 expression and migration in the splenocytes of PPARγ hypomorphic mice
We next examined whether other immune organs of younger PpargC/− mice also demonstrated increased cellularity. The inguinal and mesenteric lymph nodes of 3-mo-old mice and the thymus of 6-wk-old PpargC/− mice showed normal cellularity (Fig. 2A). Moreover, the numbers of circulating leukocytes, lymphocytes, B cells (CD19+) and T cells (CD3+) were similar between WT and PpargC/− mice (Table SIII). The normal cellularity of leukocytes in blood and of the immune organs (except the spleen) suggested that the increase in the number of splenocytes in younger PpargC/− mice is caused by a trafficking defect.

Cell accumulation and proliferation in the spleen of PPARγ hypomorphic mice.
(A) Total cellularity of inguinal (ILN) and mesenteric (MLN) lymph nodes of 3-mo-old mice and thymus of 6-wks-old mice. (B) S1P1 mRNA level in the spleen of 3-mo-old mice. (C) Relationship between spleen weight and S1P1 mRNA level. (D) S1P1 mRNA and protein levels in B cells and CD4+ T cells from 3-mo-old mice. (E) Promoter activity of HEK293T cells transiently transfected with reporter constructs and treated with or without 80 μM pioglitazone (Piog) for 24 hours. (F) ChIP-PCR in WT splenocytes with Piog or GW9662 treatment for 24 hours. (G) ChIP-PCR in WT and PpargC/− splenocytes. Sequences containing the potential PPARγ binding site in S1P1 were amplified by real-time PCR. (H) Migration of splenocytes, B cells and CD4+ T cells in response to 100 nM S1P in Transwell migration assays. (I) Migration of splenocytes in response to 200 nM S1P after 24-hour treatment with 40 μM pioglitazone or 40 μM GW9662. (J) Percentages of cells incorporating BrdU for all lymphocytes, CD19+ B cells and CD4+ T cells of 3-mo-old mice. (K) Apoptotic cells (green) in the spleen of 3-mo-old mice detected by a TUNEL assay. Scale bar, 150 μm. Numbers inside bars or parentheses indicate the number for each group. *p < 0.05; **p < 0.01; ***p < 0.001.
We first examined the expression of CXCR4 and CXCR5, which are crucial in lymphocytes homing to central lymphoid tissues18, on B cells and CD4+ T cells and found none of them were different (Fig. S3A). The predominant lysophopholipid sphingosine-1-phosphate (S1P) receptor, S1P1, is essential for T- and B-cell egression from peripheral lymphoid organs19,20. Compared with WT mice, PpargC/− mice exhibited a decreased S1P1 mRNA level in the spleen (Fig. 2B). Furthermore, S1P1 expression levels were negatively correlated with spleen weight in PpargC/− mice (Fig. 2C). Splenic B cells and CD4+ T cells of PpargC/− mice also showed reduced S1P1 mRNA and protein levels (Fig. 2D). We performed experiments to confirm that PPARγ acts as a transcription factor in regulation of S1P1 expression. In both WT and PpargC/− splenocytes, pioglitazone (PPARγ agonist) and GW9662 (PPARγ antagonist) increased and reduced S1P1 levels, respectively (Fig. S3B). To further ascertain whether S1P1 expression is directly regulated by PPARγ, the S1p1 promoter and its adjacent sequences were analyzed in silico by using the MatInspector tool. A putative PPARγ responsive element (PPRE) was predicted downstream of the transcription start site at +16 to +38 (score: 0.898) of S1p1. We cloned this region for a reporter assay and observed that its luciferase activity was significantly enhanced by pioglitazone (Fig. 2E). Chromatin immunoprecipitation-PCR in WT splenocytes showed that pioglitazone increased the binding of PPARγ to this predicted PPRE in S1p1, whereas GW9662 abrogated it (Fig. 2F). Finally, PpargC/− splenocytes showed lesser binding of PPARγ to S1p1 than WT splenocytes did (Fig. 2G). These results suggest that PPARγ-mediated S1P1 expression involves direct transcriptional regulation.
We then studied the function of PPARγ in splenocyte migration toward S1P by using a Transwell assay. PpargC/− splenocytes, B cells and CD4+ T cells showed significantly impaired migration toward S1P (Fig. 2H) and they lost their dose-dependent effect in response to the increasing dose of S1P (Fig. S3C–E). Pioglitazone increased migration in both WT and PpargC/− splenocytes, whereas GW9662 reduced it (Fig. 2I). Finally, the treatment of mouse embryonic fibroblasts with GW9662 downregulated the S1P1 levels (Fig. S3F). These data suggested that PPARγ regulates splenocyte migration, which is correlated with S1P1 levels. Therefore, the decreased S1P1 expression in the PpargC/− splenocytes likely reduces their egression from the spleen, resulting in their splenic accumulation.
Increased B-cell proliferation and apoptotic cells in the spleen of PPARγ hypomorphic mice in vivo
The increased cellularity of the spleen may also be associated with alterations in cell proliferation and apoptosis. An in vivo BrdU incorporation assay showed that the proliferation of total splenocytes and CD19+ B cells , but not CD4+ T cells, increased in PpargC/− mice compared with that in WT mice (Fig. 2J). However, the in vitro proliferation of B cells and CD4+ T cells, in response to various stimulations, was similar between WT and PpargC/− mice (Fig. S4A,B). A TUNEL assay demonstrated more TUNEL-positive cells in the spleen of PpargC/− mice than in that of WT mice (Fig. 2K). However, the apoptosis rates of B cells and CD4+ T cells did not differ in culture between the PpargC/− and WT mice (Fig. S4C,D). These results suggest that the increased B-cell proliferation and apoptosis in vivo are attributable to the microenvironment of the PpargC/− spleen.
Increased autoantibody production and SLE-like autoimmune disease development in older PPARγ hypomorphic mice
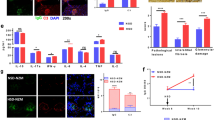
We next investigated the autoimmune status of PpargC/− mice. The serum levels of antibodies against dsDNA did not differ between PpargC/− and WT mice aged ≤8 mo. However, the sera from 14-mo-old PpargC/− mice showed significantly higher levels of antibodies against dsDNA (Fig. 3A), with significant increases in the levels of IgG1 and IgG2a (Fig. 3B). The sera of the older PpargC/− mice also showed detectable levels of antibodies against nuclei (Fig. 3C). Increased autoantibody production probably mediates the pathogenesis of autoimmune disease; for example, it likely induces SLE. Accordingly, 14-mo-old PpargC/− mice showed mesangial proliferative glomerulonephritis, characterized by glomerular cell proliferation, mesangial matrix expansion and enlarged glomeruli (Fig. 3D,E), despite normal urine albumin levels, daily urinary albumin excretion and serum creatinine levels (Fig. S5A). Immunofluorescent staining of kidney tissue revealed granular deposition of IgG, IgG2a, IgA and C1q in the glomerular mesangium of the older PpargC/− mice (Fig. 3F). Electron microscopic examination revealed electron-dense deposits in the expanded mesangium without involvement of the glomerular capillary loops (Fig. S5B). These results demonstrate that nuclear antigen-specific autoantibodies, immune complex deposition and glomerular injury in older PpargC/− mice, resembling SLE-like autoimmune diseases.

The development of autoimmune disorders in older PPARγ hypomorphic mice.
(A) Antibodies against dsDNA in the sera from mice at different ages. (B) IgG subclass of anti-dsDNA in the sera of 14-mo-old mice. (C) HEp-2 cells stained for anti-nuclear antibodies from the sera of 14-mo-old mice. Scale bar, 40 μm. (D) Representative periodic acid-Schiff (PAS)-stained glomerular morphology and (E) histomorphometric quantification of 14-mo-old mice. Scale bar, 20 μm. (F) Representative immunofluorescent images and quantifications of immune complex deposition for IgG, IgG2a, IgA and C1q in the glomeruli of 14-mo-old mice. Scale bar, 20 μm. Relative mean fluorescence intensity was analyzed using ImageJ software. Numbers inside bars indicate the number for each group. *p < 0.05; **p < 0.01; ***p < 0.001.
PPARγ deficiency in effector T-helper cells increases B-cell activation
Because B-cell proliferation and activation are associated with increased autoantibody production, we next tested whether the increased PpargC/− B-cell proliferation in vivo is associated with nearby PPARγ hypomorphic CD4+ T cells. Both mRNA and protein levels of PPARγ in the B cells and CD4+ T cells from the PpargC/− spleen were significantly lower (Fig. 4A), confirming the presence of the PPARγ hypomorph in both cell types. Because the PpargC/− spleen showed normal lymphocyte composition, equal numbers of WT and PpargC/− splenocytes were treated with anti-CD3/CD28 antibodies for comparing T-cell activation (Fig. 4B). After 3 d, the percentages of B cells and plasma cells in the PpargC/− splenocyte culture increased considerably (Fig. 4B). Moreover, after 7 d, the IgG levels in the PpargC/− splenocyte culture medium were significantly higher than those in the WT splenocyte culture medium.

Increased B-cell activation by PPARγ hypomorphic effector T-helper cells.
(A) PPARγ mRNA and protein levels in B cells and CD4+ T cells from 3-mo-old mice. (B) Schematic of the splenocyte activation protocol. Percentages of B cells (CD19+) gating from lymphocytes and proliferating blast cells, percentages of plasma cells (CD19+ CD138+) gating from B cells (CD19+) measured by flow cytometry after anti-CD3/CD28 stimulation for 3 d and IgG production in the medium measured after anti-CD3/CD28 stimulation for 7 d. (C) Coculture of WT B cells with WT or PpargC/− CD4+ T cells, the same parameters as in B were measured. (D) Coculture of WT CD4+ T cells with WT or PpargC/− B cells, the same parameters as in B were measured. Splenocytes, B cells and CD4+ T cells were isolated from 4–6-mo-old mice. Numbers inside bars or parentheses indicate the number for each group. *p < 0.05; **p < 0.01; ***p < 0.001.
To further analyze the contribution of B cells and CD4+ T cells in B-cell hyperactivation in PpargC/− splenocytes, we isolated B cells and CD4+ T cells from PpargC/− and WT spleens to perform coculture experiments. A WT B-cell and PpargC/− CD4+ T-cell coculture generated higher percentages of B cells and plasma cells and higher levels of secreted IgG than did a WT B-cell and WT CD4+ T-cell coculture (Fig. 4C). By contrast, the activities of PpargC/− B cells and WT B cells cocultured with WT CD4+ T cells did not differ (Fig. 4D). Moreover, a PpargC/− B-cell and PpargC/− CD4+ T-cell coculture generated higher percentages of plasma cells and higher levels of secreted IgG than a PpargC/− B-cell and WT CD4+ T-cell coculture did (Fig. S6A). By contrast, the activities of PpargC/− B cells and WT B cells cocultured with PpargC/− CD4+ T cells did not differ (Fig. S6B).
We further elucidated the effect of the PPARγ hypomorph on B cells by examining the responses of PpargC/− B cells to T-cell independent (T-I) and dependent (T-D) antigens in vitro. Splenic B cells from PpargC/− and WT mice were stimulated with the T-I antigens LPS (T-I type I) and NP-Ficoll (T-I type II), or anti-CD40 plus IL-4 (mimic T-D antigen). Upon stimulation with both T-I antigens, PpargC/− B cells produced IgM and IgG levels similar to those of WT B cells (Fig. S6C). Furthermore, PpargC/− B cells showed a normal response mimicking T-D antigen stimulation for IgM and IgG production (Fig. S6D). PpargC/− B-cells also showed normal expression of Blimp-1 and Bcl-6, which are important for induction of plasma cells and germinal center reaction21,22, in response to NP-Ficoll and anti-CD40 plus IL-4 stimulation (Fig. S6E). These results suggested that PpargC/− B cells have normal intrinsic responses to mitogen and costimulatory signals. Thus, the increased B-cell activity in the PpargC/− spleen is likely caused by the presence of PPARγ hypomorphic effector T-helper cells, rather than reduced PPARγ expression in B cells.
Increased Th17 polarization in PPARγ hypomorphic mice
To determine the type of effector CD4+ T cell in the PpargC/− spleen that affects B cell activation, we stimulated splenic CD4+ T cells in vitro by using anti-CD3/CD28 antibodies and assessed T-helper cell differentiation according to signature cytokine production. Although the mRNA levels of IFN-γ and IL-4 did not differ between WT and PpargC/− CD4+ T cells, those of IL-17A and IL-17F were considerably higher in the stimulated PpargC/− CD4+ T cells (Fig. 5A). Similarly, IL-17A levels in the cultured medium of stimulated PpargC/− CD4+ T-cells was significantly higher. These results suggested that PpargC/− CD4+ T cells exhibit a greater capacity for Th17 polarization in response to anti-CD3/CD28 stimulation than WT cells do. Furthermore, the in vitro stimulation of PpargC/− CD4+ T cells under Th17 differentiation significantly increased the mRNA levels of IL-17A and IL-17F as well as the protein level of IL-17A (Fig. 5B). Furthermore, PpargC/− CD4+ T cells showed significantly higher levels of the transcription factor RORγt and lower levels of Foxp3 compared with WT CD4+ T cells, whereas the levels of T-bet and GATA-3 in CD4+ T cells were similar in both genotypes (Fig. 5C). Although Park et al. reported that PPARγ CD4+ T-cell–specific knockout mice have an increased number of follicular T-helper (Tfh) cells23, we observed that the number of Tfh cells, stained using PD-1 and CXCR5, was not altered in both young and older PpargC/− mice (Figs 5C and S7). We also observed increased IL-17A protein levels in the spleen of PpargC/− mice (Fig. 5D). These results indicate that Th17 polarization increases in the PpargC/− spleen and this increase is correlated with B-cell hyperactivation.

Increased Th17 population in the spleen of PPARγ hypomorphic mice.
(A) Relative expression of cytokines in the splenic CD4+ T cells and the protein levels of IL-17A in the medium stimulated with anti-CD3/CD28 for 3 d. (B) Relative expression of IL-17 in the splenic CD4+ T cells and the protein levels of IL-17A in the medium stimulated with TGF-β (5 ng/ml) and IL-6 (20 ng/ml) for 3 d. (C) Flow cytometric analysis of transcription factors (T-bet, GATA-3, RORγt and Foxp3) in the splenic CD4+ T cells and expression of PD-1 and CXCR5 on the splenic CD4+ T cells. (D) Immunoblotting of IL-17A in the spleen. Mice are 4–6-mo-old. Numbers inside bars or parentheses indicate the number for each group. *p < 0.05; **p < 0.01; ***p < 0.001.
Enhanced Th17 function promotes B-cell activation through IL-17 signaling in PPARγ hypomorphic mice
To determine the relationship between enhanced Th17 polarization and B-cell hyperactivation in PpargC/− mice, the conditioned medium from activated WT or PpargC/− CD4+ T cells was added to the WT B-cell and WT CD4+ T-cell coculture (Fig. 6A). The conditioned medium from the activated PpargC/− CD4+ T cells showed increased B-cell proliferation, plasma cell percentages and IgG production compared with the medium from the activated WT CD4+ T cells (Fig. 6B–D). Depletion of IL-17A by antibodies in the conditioned medium of activated PpargC/− CD4+ T cells reversed these changes, demonstrating that IL-17A is an effective mediator of increased B-cell activation in PpargC/− mice.

Increased B-cell activation by the IL-17 secreted from PPARγ hypomorphic CD4+ T cells.
(A) Schematic of the WT B-cell activation protocol. Incubation of B cells and CD4+ T cells (both from WT mice) with the conditioned medium (CM) from anti-CD3/CD28-activated WT and PpargC/− CD4+ T cells with or without IL-17A depletion. (B) B-cell proliferation was analyzed by flow cytometry and the proliferation index was quantified by FlowJo software. (C) Percentage of plasma cells and (D) IgG production were determined. B cells and CD4+ T cells were isolated from 4–6-mo-old mice. *p < 0.05; **p < 0.01; ***p < 0.001 by one-way ANOVA with Scheffe’s test for (B–D). (E) Schematic of the adoptive transfer protocol. WT or PpargC/− CD4+ T cells were mixed with WT B cells and transferred into NOD/SCID mice. The recipient mice were stimulated with anti-CD3 antibodies (40 μg/d) for 5 d and analyzed 11 d after the end of stimulation. (F) Relative expression of IL-17A in the splenic CD4+ T cells. (G) Splenic CD4+ T cells were assessed for IL-17A production after PMA/inomycin restimulation through intracellular staining. (H) Percentage of plasma cells in splenocytes measured by flow cytometry. (I) IgG, IgG1, IgG2a and IgG3 levels in the sera. Numbers inside bars indicate the number for each group. *p < 0.05 and **p < 0.01 by Student’s t test for (F–I).
To further test these findings in vivo, we mixed WT or PpargC/− CD4+ T cells with WT B cells, transferred them into NOD/SCID mice, which lack T and B cells and stimulated the mice with anti-CD3 antibodies (Fig. 6E). PPARγ hypomorph in T cells increased IL-17A mRNA and protein levels in CD4+ T cells, the percentage of plasma cells in the splenocytes and levels of circulating IgG, IgG1 and IgG2a (Fig. 6F–I). These results suggested that observed Th17 polarization and lupus like phenotypes in the aged PpargC/- mice are modulated by PPARγ in a T cell-intrinsic fashion.
Pioglitazone ameliorates splenomegaly in PPARγ hypomorphic mice
We further evaluated whether activation of the remaining PPARγ sufficiently reverses the changes in PpargC/− mice. Treatment with low-dose pioglitazone for 2 mo starting at 2 mo of age increased S1P1 levels and reduced the Th17 population in the spleen of PpargC/− mice (Fig. 7A,B). Furthermore, CD4+ T cells isolated from pioglitazone-treated PpargC/− mice exhibited a lower potential for induction of B-cell activity than those isolated from nontreated control PpargC/− mice did (Fig. 7C). Consequently, pioglitazone treatment reversed splenomegaly in PpargC/− mice (Fig. 7D). Furthermore, treatment with high-dose pioglitazone for 6 wk significantly reduced serum IgG2a levels of older PpargC/− mice (Fig. 7E) and attenuated the levels of both IgG and IgG2a against nuclei (Fig. 7F). These results suggest that the activation of 25% PPARγ expression in PpargC/− mice promotes S1P1 expression in splenocytes and prevents B-cell hyperactivation by attenuating Th17 polarization; these activities collectively contribute to the decrease in spleen weight and, finally, amelioration of the SLE-like autoimmune disease.

Effects of PPARγ activation in PPARγ hypomorphic mice.
(A) Immunoblotting of S1P1 and (B) percentage of IL-17A+ CD4+ T cells in the spleen of PpargC/− mice treated with 20 mg/kg/d pioglitazone (Piog) for 2 mo starting at 2 mo of age. (C) Percentage of CD19+ B cells and secreted IgG levels from the coculture of WT B-cells with CD4+ T cells from control and Piog-treated PpargC/− mice. (D) Spleen weight and spleen-to-body weight ratio in Piog-treated PpargC/− mice. (E) The IgG2a levels in and (F) HEp-2 cells stained for anti-nuclear antibodies (IgG and IgG2a) from the sera of PpargC/− mice treated with 60 mg/kg/d Piog for 6 wks starting at 14 mo of age. Numbers inside bars or parentheses indicate the number for each group. *p < 0.05 and **p < 0.01.
Discussion
Studies have reported that PPARγ deficiency in either adipocytes or hematopoietic cells reduces bone marrow cellularity and promotes extramedullary hematopoiesis in the spleen, leading to splenomegaly24,25. By contrast, in the current study, younger PPARγ hypomorphic PpargC/− mice developed splenomegaly without changes in the bone architecture or relocation of hematopoiesis to the spleen. In these mice, S1P1 expression and migration toward S1P of PpargC/− splenocytes were decreased, suggesting that hindered lymphocyte egression is a mechanism for their splenic accumulation. We also observed increased in vivo B-cell proliferation in the spleen, which may have also contributed to splenomegaly in the younger PpargC/− mice. Consequently, the enhanced B-cell activation occurred partly because of the elevated IL-17 levels resulting from the increased Th17 polarization of PpargC/− CD4+ T cells and not because of the reduction of PPARγ levels in B cells. These factors, along with an increased number of apoptotic cells in the spleen—suggestive of impaired apoptotic cell clearance—likely contributed to the spontaneous development of SLE-like autoimmune disease in the older PpargC/− mice.
S1P has a major function in the immune system through binding with the receptors S1P1–S1P526,27. Among these, S1P1 is expressed at the highest levels in lymphocytes and is required for lymphocytes to egress from lymphoid organs19. The relatively high levels of S1P in circulation enable lymphocytes egression into blood, which is mediated through S1P119,20. Studies on kidney mesangial cells have shown that PPARγ agonists increase sphingosine kinase 1 levels, leading to increased intracellular S1P levels28. Similarly, in mesangial cells, PPARγ activation also upregulates S1P1 expression29. However, the involvement of similar S1P1 regulation by PPARγ in lymphocyte trafficking has never been explored thus far. In this study, we observed reduced S1P1 expression in splenic B cells and CD4+ T cells of PpargC/− mice, which is caused by reduced binding of PPARγ to the genomic S1p1 sequence. PpargC/− B cells and CD4+ T cells showed a significantly attenuated migratory response toward S1P. Moreover, pharmacological activation of PPARγ increased S1P1 expression and ameliorated splenomegaly. These findings support the involvement of a lymphocytic PPARγ–S1P1 axis in lymphocyte egression from the spleen.
In PpargC/− mice, the normal aging process was sufficient to cause SLE-like phenotypes. The function of PPARγ in lymphocyte proliferation and autoimmune diseases has been documented in various mouse models6,7,8,9; however, most of these studies have induced autoimmunity experimentally and evaluated only the function of PPARγ within individual immune cell types. Thus, they could not replicate the complex interaction of different immune cell types. Here, we devised an in vitro coculture system and demonstrated that B cells cocultured with PpargC/−, but not WT, CD4+ T cells became hyperactive regardless of the Pparg genotype of the B cells. In the spleen, B-cell follicles are surrounded by T-cell areas and activated CD4+ T cells move toward the T-B border for helping B-cell activation30. Particularly, Th17 cells act as B-cell helpers through the secretion of IL-1731. Although PPARγ deficiency promotes Th17 differentiation and CD4+ T-cell–mediated autoimmunity development6, whether this T-cell imbalance also affects B-cell activation and contributes to B-cell–mediated autoimmunity remains unclear. Consistently, we observed that PpargC/− CD4+ T cells exhibited increased polarization toward Th17 cells. The removal of IL-17A from the coculture medium of B cells and CD4+ T cells attenuated its ability to increase B-cell activity, demonstrating that the higher IL-17A levels resulting from increased Th17 polarization are essential for increasing B-cell activity in PpargC/− mice. Therefore, in our working model, PpargC/− spleen creates a microenvironment where sequestered lymphocytes increase cross talk between PpargC/− B cells and Th17-polarized CD4+ T cells through the secretion of IL-17, resulting in enhanced B-cell activation.
The mechanism by which PPARγ affects the Th17 polarization program and IL-17 production has been explored previously. PPARγ activation negatively regulates RORγt transcription by preserving the silencing mediator for retinoid and thyroid hormone receptors (SMRT) corepressor in the RORγt promoter6. In addition, PPARγ activation induces SOCS3 expression and interferes with the STAT3 signaling pathway, which is essential for the transcription of IL-17A, IL-17F and RORγt32. Therefore, PPARγ antagonism may increase Th17 polarization and IL-17 production through direct induction of RORγt or indirect induction of STAT3. These studies showed that PPARγ activation by an agonist suppressed IL-17 expression. By contrast, after blockade by using an antagonist, PPARγ loses the ability to conduct SOCS3 on inhibition of RORγt expression and IL-17 secretion32. Furthermore, treatment with a PPARγ antagonist restores expression of RORγt and IL-1733. The findings together indicate that Th17 polarization and IL-17 production are negatively regulated by PPARγ in an indirect fashion.
SLE pathogenesis is complex, involving dysregulation of multiple arms of the immune system, including defective lymphocyte trafficking, B-cell activation and impaired apoptotic cell clearance34,35,36. Although our results suggested that increased Th17 and B-cell cross talk and its consequent overproduction of autoantibodies causes SLE-like autoimmune disease development in older PpargC/− mice, increases in the number of apoptotic cells in the spleen cannot be neglected. Rőszer et al. demonstrated that mice with macrophage-specific deletion of PPARγ develop SLE nephritis because of impaired apoptotic cell clearance7; although their mice developed severe nephritis at an early age (4–6 mo), they showed normal immune cell expansion and B-cell activity without splenomegaly. Moreover, the role of macrophage activation in SLE pathogenesis has been proposed37 and our results showed that the basal and LPS-stimulated expression of IL-1β, iNOS and MCP-1 was higher in PpargC/− peritoneal macrophages (Fig. S8). Therefore, the SLE-like autoimmune diseases developed in the older PpargC/− mice were likely caused by a combination of symptoms associated with immune dysregulations.
We noticed that the symptoms of SLE-like nephritis in older PpargC/− mice are relatively mild. Based on the classification by the International Society of Nephrology and the Renal Pathology Society (ISN/RPS)38, we speculated that the glomerulonephritis of older PpargC/− mice is morphologically compatible with Class II lupus nephritis. Under electron microscope, we did not find effacement of podocyte foot processes in older PpargC/− mice (Fig. S5C), which is consistent with the absence of proteinuria and normal serum creatinine.
Our current results showed some discrepancies with those reported previously. For example, Setoguchi et al. reported that PPARγ haploinsufficiency enhances the proliferative response of isolated Pparg+/− B cells, but not T cells, in culture. By contrast, we observed that the in vitro proliferation of both PpargC/− B cells and CD4+ T cells as well as Pparg+/− spleen weight were normal. Although the reasons for this discrepancy remain unclear, it may be attributable to subtle differences in the genetic background: our mice were from the F1 generation of crossed 129S6 and C57BL6/J mice, whereas those used by Setoguchi et al. were from an ICR outbred background. Notably, although both 129 and C57BL/6 are nonautoimmune strains, a certain combination of their genomes can contribute to the expression of autoimmune phenotypes39,40. Thus, complex contributions of genetic background could not be disregarded in comparison of different sets of experiments.
PPARγ CD4+ T-cell specific knockout mice with nearly no detectable PPARγ protein in CD4+ T cells have increased Tfh cells, as well as other helper T-cell subsets, such as Th1, Th2 and Th1723. However, in our study, PpargC/− mice with PPARγ hypomorph in CD4+ T cells showed increased Th17 polarization without altering the development of Th1, Th2 and Tfh cells. The discrepancy between two studies may stem from the PPARγ level in CD4+ T cells. In addition, because PpargC/− mice exhibit the reduction of PPARγ levels in all cell types, the contribution of PPARγ hypomorph in the cell types other than CD4+ T cells cannot be neglected. Nevertheless, these results suggest that the development of Th17 cells is more sensitive to the PPARγ level.
The changes in the PPARγ expression levels in our series of Pparg mutants were within the human physiological range as a consequence of polymorphic variations. Studies on the association between PPARG polymorphisms and the severity of autoimmune diseases have revealed mixed results. For example, the most common PPARG polymorphism, Pro12Ala, which exhibits decreased transcriptional activity41, is associated with several autoimmune diseases such as psoriatic arthritis42 and rheumatoid arthritis43. However, the polymorphism is also associated with delayed onset of multiple sclerosis44 and with protection against Graves orbitopathy45. Another common PPARG polymorphism, C161T, was associated with the longer survival of Japanese patients with IgA nephropathy46. In addition, treatment with PPARγ agonists reduces symptoms of autoimmune diseases6,47,48. Glucocorticoid is widely applied in SLE therapy; compared with the inhibitory effects of glucocorticoids on T cells, B cells are less affected and antibody production is largely preserved after short-term or low-dose glucocorticoid administration49,50. Notably, Th17 cells may be relatively resistant to the effects of glucocorticoids51,52. Therefore, in contrast to those of pioglitazone treatment, the effects of glucocorticoid treatment on SLE-like autoimmune diseases may vary. Our study thus highlights pioglitazone as a potential candidate for SLE therapy.
A recent clinical study showed that familial partial lipodystrophic patients with PPARγ E157D mutation, which severely reduces target gene transcription, have an increased risk of autoimmune diseases4. Thus, our work exemplifies how clinical observations can be mechanistically dissected through a basic investigation in a murine model. In conclusion, the titrated reduction of Pparg gene expression in mice demonstrated that the level of PPARγ required for maintaining normal immunity is 25%–50% of that in the WT. Our study also demonstrated that PPARγ hypomorphism causes excessive B-cell response with the aid of Th17 in the T-cell dependent humoral immune response. Thus, our study defines a novel function of PPARγ in lymphocyte trafficking and in cross talk between Th17 and B cells and indicates that decreased expression of the metabolism-related factor PPARγ is a risk factor for autoimmune disease, particularly in association with SLE and related autoimmune diseases.
Methods
Mice
Generation of mice carrying the modified Pparg locus has been described15. Pparg+/+ (WT) and PpargC/− mice were F1 littermates from the mating of PpargC/+ mice on a C57BL/6J background with Pparg+/− mice on a 129S6 background53. Mice were bred and housed in the animal facility of National Cheng Kung University (NCKU). Mice were treated with pioglitazone (20 mg/kg/d) via oral gavage for 2 mo. NOD/SCID mice were purchased from NCKU Laboratory Animal Center. All animal studies were performed according to protocols approved by the Institutional Animal Care and Use Committee of NCKU.
Microcomputed tomography
Right femurs of 2-mo-old and 9-mo-old mice were dissected free of soft tissue, fixed in formalin and scanned by a microcomputed tomography scanner (Skyscan 1076) and analyzed using a software (Skyscan NV, Aartselaar, Belgium). Image acquisition was performed at 48 kV and 200 μA, with a 0.6° rotation between frames. Micromorphological information was obtained from image stacks, allowing three-dimensional parameters of bone microstructure to be calculated, including trabecular bone volume, bone mineral density, trabecular number and trabecular thickness. Morphometric parameters measured by a CT-analyzer have been validated on both virtual objects and aluminum foil and wire phantoms.
Identification of PPRE and promoter activity assays
Mouse S1p1 promoter and its adjacent sequences were analyzed using a MatInpector tool for prediction of PPARγ-binding sites54. The fragment of the mouse S1p1 5′-flanking region (−834 to +173 bp) was cloned into pGL3-basic vector containing a luciferase reporter system. HEK293 T cells were transfected with reporter plasmids using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). Subsequently, cells were incubated with pioglitazone (80 μM) for an additional 24 hours and luciferase activity was measured using a Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA).
ChIP assay
The procedure for ChIP was described previously55. In brief, PPARγ protein was fixed with DNA by 1% formaldehyde for 10 minutes. Cells were harvested and sonicated to fragment DNA (average size of 200~500 bp). PPARγ antibody (Cell Signaling, Beverly, MA, USA) was used to pull down the PPARγ protein and DNA complexes. The potential PPARγ binding site was amplified using the primers 5′-CGTTTGCCTGGAGAAATACCA-3′ and 5′-GACTGAGCTGCGGAGAGCTT-3′.
Migration assays
Splenocytes were cultured and allowed to migrate through 5 μm pore size Transwell inserts (Merck Millipore, Darmstadt, Germany). The lower wells contained 50 to 200 nM S1P (Sigma-Aldrich, St. Louis, MO, USA). After 3 hours of incubation, the migrated cell from each well were counted with a microscope or analyzed for B-cells and CD4+ T-cells by flow cytometry.
Histopathology
Kidneys were fixed in 4% paraformaldehyde and stained with a periodic acid-Schiff reagent. Immune complex deposits were analyzed on cryosections, which were fixed in acetone and incubated with Alexa 488-conjugated antibodies against IgG (Invitrogen, Carlsbad, CA, USA) and IgG2a (Abcam, Cambridge, MA, USA), or CruzFluor 488-conjugated antibodies against IgA and C1q (Santa Cruz, Dallas, TX, USA). Fluorescence intensity was analyzed using ImageJ software. For the analysis of cell ultrastructure, transmission electron microscopy was performed from mouse kidney cortex with a Hitachi 7000 TEM.
BrdU and CFSE labeling
Mice were intraperitoneally injected with BrdU (4 mg/injection) twice daily for 5 d. Splenocytes were stained with antibodies against BrdU (BD Pharmingen, San Diego, CA, USA). For the carboxyfluorescein succinimidyl ester (CFSE) labeling, splenocytes (5 × 106 cells) were labeled with 5 μM CFSE (Sigma-Aldrich, St. Louis, MO, USA) and then washed.
Lymphocyte proliferation and IgG production
B cells and CD4+ T cells were isolated with anti-mouse CD19+ microbeads and anti-mouse CD4+ microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany). For mitogen stimulation, B cells were cultured with LPS (10 μg/ml, Sigma-Aldrich, St. Louis, MO, USA), anti-IgM F(ab′)2 (30 μg/ml, Jackson Immunoresearch, West Grove, Pennsylvania, USA) or anti-CD40 antibodies (10 μg/ml, BD Bioscience, San Jose, CA, USA) and CD4+ T cells were cultured with anti-CD3 (4 μg/ml) and anti-CD28 antibodies (4 μg/ml). In the coculture assay, CFSE-labeled B cells and CD4+ T cells were cocultured on microtiter wells pre-coated with anti-CD3/CD28 antibodies. After 3 d of co-culture, the cells were stained with CD19-PE (eBioscience, San Diego, CA, USA) and CD138-APC (BD Bioscience, San Jose, CA, USA) to determine the percentage of plasma cells in CFSE+ B cells. After 7 d of coculture, the supernatant was collected to determine IgG production by ELISA (Bethyl Laboratories, Montgomery, TX, USA).
In vitro T-cell differentiation
CD4+ T cells were stimulated with anti-CD3/CD28 antibodies (4 μg/ml) for 3 d and analyzed for cytokine expression. For the Th17 differentiation, CD4+ T cells were stimulated with TGF-β (5 ng/ml) and IL-6 (20 ng/ml) for 3 d. The levels of secreted IL-17A (eBioscience, San Diego, CA, USA) in the supernatant were determined by ELISA.
Flow cytometry
For the cell surface staining, cells were labeled with various antibodies (eBioscience, San Diego, CA, USA) and analyzed by a FACS-Calibur instrument (BD Bioscience, San Jose, CA, USA). For the intracellular staining of cytokines, after 5 hours of incubation with PMA (50 ng/ml) and ionomycin (1 μg/ml) in the presence of brefeldin A (10 μg/ml), cells were stained with anti-CD4 antibodies, followed by fixation and permeabilization (eBioscience, San Diego, CA, USA) and intracellular staining with T-bet, GATA-3 and RORγt (eBioscience, San Diego, CA, USA).
RNA isolation and real-time PCR
Total RNA was extracted using REzol (PROtech, Taipei, Taiwan). mRNA levels were analyzed with real-time quantitative RT-PCR (Applied Biosystems, Foster City, CA) with β-actin as the reference gene in each reaction.
Detection of autoantibodies
The serum levels of autoantibodies against dsDNA were determined by ELISA (Alpha Diagnostic International, San Antonio, TX, USA). For detection of anti-nuclear antibodies, HEp-2 cells were fixed and incubated with the mouse sera at a dilution of 1:200 and followed by Alexa 488-conjugated anti-mouse IgG antibodies (Invitrogen, Carlsbad, CA, USA).
Western blot analysis
Total proteins (20 μg) was separated by SDS-PAGE, transferred to PVDF membranes and probed with antibodies against PPARγ (Cell Signaling, Danvers, MA, USA), PPARγ Ser273, IL-17A (Santa Cruz, Dallas, TX, USA), S1P1 (Abcam, Cambridge, MA, USA) and α-tubulin (Sigma-Aldrich, St. Louis, MO, USA).
Adoptive transfer
CD4+ T cells from WT or PpargC/− mice and B cells from WT mice were mixed at 1:1 ratio. These cells were transferred in NOD/SCID mice (2 × 107 cells/mouse). The recipient mice were stimulated with anti-CD3 antibodies (40 μg/d) for 5 d. Eleven days after the end of stimulation, splenocytes from the recipient mice were isolated for mRNA and flow cytometric analyses. Sera from recipient mice were collected and IgG, IgG1, IgG2a and IgG3 antibodies were measured by using ELISA.
Statistical analysis
Values are reported as mean ± SEM. Statistical analyses were conducted by Student’s t test or one-way ANOVA followed by Scheffe’s multiple comparison test. Differences were considered to be statistically significant at P < 0.05.
Additional Information
How to cite this article: Liu, Y.-H. et al. Quantitative PPARγ expression affects the balance between tolerance and immunity. Sci. Rep. 6, 26646; doi: 10.1038/srep26646 (2016).
References
Sellner, J. et al. The increasing incidence and prevalence of female multiple sclerosis–a critical analysis of potential environmental factors. Autoimmun. Rev. 10, 495–502 (2011).
Ahima, R. S. & Lazar, M. A. Physiology. The health risk of obesity–better metrics imperative. Science 341, 856–858 (2013).
Owen, K. R. et al. Mesangiocapillary glomerulonephritis type 2 associated with familial partial lipodystrophy (Dunnigan-Kobberling syndrome). Nephron. Clin. Pract. 96, c35–38 (2004).
Campeau, P. M. et al. Clinical and molecular characterization of a severe form of partial lipodystrophy expanding the phenotype of PPARgamma deficiency. J. Lipid Res. 53, 1968–1978 (2012).
Versini, M., Jeandel, P. Y., Rosenthal, E. & Shoenfeld, Y. Obesity in autoimmune diseases: not a passive bystander. Autoimmun. Rev. 13, 981–1000 (2014).
Klotz, L. et al. The nuclear receptor PPAR gamma selectively inhibits Th17 differentiation in a T cell-intrinsic fashion and suppresses CNS autoimmunity. J. Exp. Med. 206, 2079–2089 (2009).
Roszer, T. et al. Autoimmune kidney disease and impaired engulfment of apoptotic cells in mice with macrophage peroxisome proliferator-activated receptor gamma or retinoid X receptor alpha deficiency. J. Immunol. 186, 621–631 (2011).
Setoguchi, K. et al. Peroxisome proliferator-activated receptor-gamma haploinsufficiency enhances B cell proliferative responses and exacerbates experimentally induced arthritis. J. Clin. Invest. 108, 1667–1675 (2001).
Natarajan, C., Muthian, G., Barak, Y., Evans, R. M. & Bright, J. J. Peroxisome proliferator-activated receptor-gamma-deficient heterozygous mice develop an exacerbated neural antigen-induced Th1 response and experimental allergic encephalomyelitis. J. Immunol. 171, 5743–5750 (2003).
Sato, S., Hasegawa, M., Fujimoto, M., Tedder, T. F. & Takehara, K. Quantitative genetic variation in CD19 expression correlates with autoimmunity. J. Immunol. 165, 6635–6643 (2000).
Chen, Z., Stockton, J., Mathis, D. & Benoist, C. Modeling CTLA4-linked autoimmunity with RNA interference in mice. Proc. Natl. Acad. Sci. USA 103, 16400–16405 (2006).
Kawasaki, A. et al. Association of IRF5 polymorphisms with systemic lupus erythematosus in a Japanese population: support for a crucial role of intron 1 polymorphisms. Arthritis. Rheum. 58, 826–834 (2008).
Myhr, C. B. et al. The autoimmune disease-associated SNP rs917997 of IL18RAP controls IFNgamma production by PBMC. J. Autoimmun. 44, 8–12 (2013).
Tsai, Y. S., Xu, L., Smithies, O. & Maeda, N. Genetic variations in peroxisome proliferator-activated receptor gamma expression affect blood pressure. Proc. Natl. Acad. Sci. USA 106, 19084–19089 (2009).
Tsai, Y. S. et al. Decreased PPAR gamma expression compromises perigonadal-specific fat deposition and insulin sensitivity. Mol. Endocrinol. 23, 1787–1798 (2009).
Kakoki, M. et al. Altering the expression in mice of genes by modifying their 3′ regions. Dev. Cell 6, 597–606 (2004).
Choi, J. H. et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature 466, 451–456 (2010).
Moser, B. & Loetscher, P. Lymphocyte traffic control by chemokines. Nat. Immunol. 2, 123–128 (2001).
Matloubian, M. et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 427, 355–360 (2004).
Allende, M. L., Dreier, J. L., Mandala, S. & Proia, R. L. Expression of the sphingosine 1-phosphate receptor, S1P1, on T-cells controls thymic emigration. J. Biol. Chem. 279, 15396–15401 (2004).
Shaffer, A. L. et al. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity 17, 51–62 (2002).
Basso, K. & Dalla-Favera, R. BCL6: master regulator of the germinal center reaction and key oncogene in B cell lymphomagenesis. Adv. Immunol. 105, 193–210 (2010).
Park, H. J. et al. PPARgamma negatively regulates T cell activation to prevent follicular helper T cells and germinal center formation. PloS One 9, e99127 (2014).
Cock, T. A. et al. Enhanced bone formation in lipodystrophic PPARgamma(hyp/hyp) mice relocates haematopoiesis to the spleen. EMBO Rep. 5, 1007–1012 (2004).
Wan, Y., Chong, L. W. & Evans, R. M. PPAR-gamma regulates osteoclastogenesis in mice. Nat. Med. 13, 1496–1503 (2007).
Kluk, M. J. & Hla, T. Signaling of sphingosine-1-phosphate via the S1P/EDG-family of G-protein-coupled receptors. Biochim. Biophys. Acta 1582, 72–80 (2002).
Mandala, S. et al. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science 296, 346–349 (2002).
Koch, A. et al. Thiazolidinedione-dependent activation of sphingosine kinase 1 causes an anti-fibrotic effect in renal mesangial cells. Br. J. Pharmacol. 166, 1018–1032 (2012).
Koch, A. et al. PPARgamma agonists upregulate sphingosine 1-phosphate (S1P) receptor 1 expression, which in turn reduces S1P-induced [Ca] increases in renal mesangial cells. Biochim. Biophys. Acta 1831, 1634–1643 (2013).
Reif, K. et al. Balanced responsiveness to chemoattractants from adjacent zones determines B-cell position. Nature 416, 94–99 (2002).
Mitsdoerffer, M. et al. Proinflammatory T helper type 17 cells are effective B-cell helpers. Proc. Natl. Acad. Sci. USA 107, 14292–14297 (2010).
Berger, H. et al. SOCS3 transactivation by PPARgamma prevents IL-17-driven cancer growth. Cancer Res. 73, 3578–3590 (2013).
Li, B., Reynolds, J. M., Stout, R. D., Bernlohr, D. A. & Suttles, J. Regulation of Th17 differentiation by epidermal fatty acid-binding protein. J. Immunol. 182, 7625–7633 (2009).
Schrieber, L. et al. Aberrant lymphocyte trafficking in murine systemic lupus erythematosus. Rheumatol. Int. 6, 215–219 (1986).
Dorner, T., Giesecke, C. & Lipsky, P. E. Mechanisms of B cell autoimmunity in SLE. Arthritis Res. Ther. 13, 243 (2011).
Shao, W. H. & Cohen, P. L. Disturbances of apoptotic cell clearance in systemic lupus erythematosus. Arthritis Res. Ther. 13, 202 (2011).
Katsiari, C. G., Liossis, S. N. & Sfikakis, P. P. The pathophysiologic role of monocytes and macrophages in systemic lupus erythematosus: a reappraisal. Semin. Arthritis Rheum. 39, 491–503 (2010).
Weening, J. J. et al. The classification of glomerulonephritis in systemic lupus erythematosus revisited. J. Am. Soc. Nephrol. 15, 241–250 (2004).
Carlucci, F. et al. Genetic dissection of spontaneous autoimmunity driven by 129-derived chromosome 1 Loci when expressed on C57BL/6 mice. J. Immunol. 178, 2352–2360 (2007).
Heidari, Y. et al. A lupus-susceptibility C57BL/6 locus on chromosome 3 (Sle18) contributes to autoantibody production in 129 mice. Genes Immun. 10, 47–55 (2009).
Deeb, S. S. et al. A Pro12Ala substitution in PPARgamma2 associated with decreased receptor activity, lower body mass index and improved insulin sensitivity. Nat. Genet. 20, 284–287 (1998).
Butt, C., Gladman, D. & Rahman, P. PPAR-gamma gene polymorphisms and psoriatic arthritis. J. Rheumatol. 33, 1631–1633 (2006).
Jalil, S. F. et al. Association of Pro12Ala (rs1801282) variant of PPAR gamma with Rheumatoid Arthritis in a Pakistani population. Rheumatol. Int. 34, 699–703 (2014).
Klotz, L., Schmidt, S., Heun, R., Klockgether, T. & Kolsch, H. Association of the PPARgamma gene polymorphism Pro12Ala with delayed onset of multiple sclerosis. Neurosci. Lett. 449, 81–83 (2009).
Pawlak-Adamska, E. et al. PPARg2 Ala(1)(2) variant protects against Graves’ orbitopathy and modulates the course of the disease. Immunogenetics 65, 493–500 (2013).
Song, J. et al. Peroxisome proliferator-activated receptor gamma C161T polymorphisms and survival of Japanese patients with immunoglobulin A nephropathy. Clin. Genet. 64, 398–403 (2003).
Zhao, W. et al. The peroxisome-proliferator activated receptor-gamma agonist pioglitazone modulates aberrant T cell responses in systemic lupus erythematosus. Clin. Immunol. 149, 119–132 (2013).
Okunuki, Y. et al. Peroxisome proliferator-activated receptor-gamma agonist pioglitazone suppresses experimental autoimmune uveitis. Exp. Eye Res. 116, 291–297 (2013).
Settipane, G. A., Pudupakkam, R. K. & McGowan, J. H. Corticosteroid effect on immunoglobulins. J. Allergy Clin. Immunol. 62, 162–166 (1978).
Cupps, T. R., Gerrard, T. L., Falkoff, R. J., Whalen, G. & Fauci, A. S. Effects of in vitro corticosteroids on B cell activation, proliferation and differentiation. J. Clin. Invest. 75, 754–761 (1985).
McKinley, L. et al. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J Immunol. 181, 4089–4097 (2008).
Schewitz-Bowers, L. P. et al. Glucocorticoid-resistant Th17 cells are selectively attenuated by cyclosporine A. Proc. Natl. Acad. Sci. USA 112, 4080–4085 (2015).
Barak, Y. et al. PPAR gamma is required for placental, cardiac and adipose tissue development. Mol. Cell 4, 585–595 (1999).
Cartharius, K. et al. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics 21, 2933–2942 (2005).
Lin, S. C. et al. Suppression of dual-specificity phosphatase-2 by hypoxia increases chemoresistance and malignancy in human cancer cells. J. Clin. Invest. 121, 1905–1916 (2011).
Acknowledgements
We thank for Dr. Ronald M. Evans at Salk Institute for kindly providing us with Pparg+/− mice and Dr. Iain. C. Bruce at Zhejiang University and Dr. Yohan Kendrick at Wallace Academic Editing for discussions. This work was supported by grants from Ministry of Science and Technology (104-2321-B-006-022 and 102-2321-B-006-007), National Health Research Institutes (EX104-10231SI and EX105-10511SI), National Cheng Kung University Aim for the Top University Project and National Institute of Health (HL42630 and HL77145).
Author information
Authors and Affiliations
Contributions
Y.H.L. researched data, contributed to discussion, wrote manuscript. Y.S.T. contributed to discussion, wrote manuscript, reviewed/edited manuscript. S.C.L researched data, contributed to discussion. N.S.L. contributed to discussion, reviewed/edited manuscript. M.S.J. contributed to discussion, reviewed/edited manuscript. C.T.L. and S.W.H. researched data. W.C.C. and J.M.S. researched data, contributed to discussion. N.M. contributed to discussion, reviewed/edited manuscript. P.J.T. contributed to discussion, wrote manuscript, reviewed/edited manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Liu, YH., Tsai, YS., Lin, SC. et al. Quantitative PPARγ expression affects the balance between tolerance and immunity. Sci Rep 6, 26646 (2016). https://doi.org/10.1038/srep26646
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep26646
This article is cited by
-
Peroxisome proliferator-activator receptor γ and psoriasis, molecular and cellular biochemistry
Molecular and Cellular Biochemistry (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
