
Abstract
The superconducting transition temperature (Tc) of tetragonal Fe1+δSe was enhanced from 8.5 K to 44 K by chemical structure modification. While insertion of large alkaline cations like K or solvated lithium and iron cations in the interlayer space, the [Fe2Se2] interlayer separation increases significantly from 5.5 Å in native Fe1+δSe to >7 Å in KxFe1−ySe and to >9 Å in Li1−xFex(OH)Fe1−ySe, we report on an electrochemical route to modify the superconducting properties of Fe1+δSe. In contrast to conventional chemical (solution) techniques, the electrochemical approach allows to insert non-solvated Li+ into the Fe1+δSe structure which preserves the native arrangement of [Fe2Se2] layers and their small separation. The amount of intercalated lithium is extremely small (about 0.07 Li+ per f.u.), however, its incorporation results in the enhancement of Tc up to ∼44 K. The quantum-mechanical calculations show that Li occupies the octahedrally coordinated position, while the [Fe2Se2] layers remain basically unmodified. The obtained enhancement of the electronic density of states at the Fermi level clearly exceeds the effect expected on basis of rigid band behavior.
Similar content being viewed by others

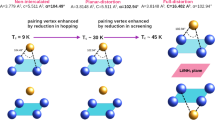
Introduction
Tetragonal iron selenide, Fe1+δSe (0.01 ≤ δ ≤ 0.04), is one of the most interesting representatives of iron-based superconductors discovered a few years ago1,2,3. Although the superconducting transition temperature is rather low (Tc = 8.5 K for δ = 0.01), Fe1+δSe offers unique opportunities for structure modification which significantly enhances Tc up to 40 K4,5,6,7,8,9. The increase of Tc is accompanied by changes of distances and interactions between the anti-fluorite layers [Fe2Se2]: adjacent [Fe2Se2] layers shift by (½, ½, 0) and the interlayer space is filled by heavy alkali metals4,5 or solvated metal cations (chemical intercalation using anoxic polar solvent6,7). Very interesting results were obtained in hydrothermal synthesis experiments which yielded the formation of Li1−xFex(OH)Fe1−ySe with Tc ≈ 40 K (refs 8,9). The Li1−xFex(OH)Fe1−ySe structure is a stacking arrangement of alternating [Fe2Se2] and [(Li,Fe)OH] layers, the latter acting as charge reservoirs9.
The literature information on electrochemical studies of iron chalcogenides is limited to just a few papers. So, the electrochemical Li-ion intercalation into FeSe1−xTex results in the formation of LiyFeSe1−xTex for 0.25 ≤ x ≤ 1 (ref. 10). However, the observed preservation of lattice parameters and Tc (even at high y values) are not consistent with the claimed formation of the Li-intercalated phases. Moreover, the structural characterization is insufficient. Carbon-coated FeSe-nanoparticles were investigated as an anode material for lithium-ion batteries with a sustainable reversible capacity of 340 mAh·g−1 (ref. 11). The reversible charge-discharge cycling implicates the amorphization of the initial iron selenide and probably involves the formation of metallic Fe-nanoparticles and Li2Se. Successful electrochemical intercalation of potassium and sodium into FeSe has been reported very recently12.
In this work we have successfully modified superconducting properties of Fe1+δSe for the first time by electrochemical lithium intercalation. This approach allows the preservation of the native structure and the enhancement of Tc up to 44 K due to changes of the carrier concentration. The obtained results provide the opportunity to shed new light on the mechanism of superconductivity in Fe-based superconductors and opens new ways for enhancing Tc. The detailed investigation of the electrochemical behavior of Fe1+δSe upon lithium intercalation conditions clarifies the existing contradictions and demonstrates the potential of the electrochemical approach to modify crystal structure and properties of Fe1+δSe.
Results and Discussion
Electrochemical intercalation of Li-ions into tetragonal Fe1+δSe was performed using a single-phase polycrystalline sample and large aggregates of single crystals. The polycrystalline sample consisted of agglomerates of particles with linear sizes between 1 and 10 μm (Fig. SI1). The lattice parameters a = 3.7720(2) Å and c = 5.5231(4) Å obtained from powder X-ray diffraction data (PXRD, Fig. SI2) are in good agreement with the literature13,14. According to the SEM study, crystal aggregate of tetragonal Fe1+δSe with a linear size of approximately 1 × 2 mm contains oriented single-crystal blocks (Fig. SI3).
Cyclic voltammetry (CVA) studies of polycrystalline Fe1+δSe electrodes between 1.3 V and 2 V vs. Li/Li+ (Fig. 1a) revealed the occurrence of a redox process with a significant reversible capacity (ca. 300 mAh·g−1). In the 1st cycle the reduction peak is at ca. 1.3 V, the oxidation peak is at ca. 1.85 V. Upon cycling, the potential of the reduction peak shifts towards more positive potential values (1.4–1.5 V), while the position of the corresponding oxidation peak does not change considerably, although a significant broadening of the peak occurs. Ex-situ PXRD data for the electrode material charged or discharged at different potential values show a drastic decrease of the reflections intensities of Fe1+δSe without noticeable lattice parameters variation. No evidence for new crystalline phases is observed (Fig. 1b). These findings are compatible with an irreversible phase transformation.

(a) CVAs curves (first three cycles) obtained for the polycrystalline Fe1+δSe electrode in the potential range 1.3−2.0 V vs. Li/Li+ (scan rate of 0.05 mV·s−1); (b) a part of ex-situ PXRD patterns for polycrystalline Fe1+δSe electrodes at different potentials during 1st cycle. Reflections of tetragonal Fe1+δSe are indexed. The diffraction maximum at 2θ of approx. 23° is unidentified. Electrochemical investigations at the same conditions (potential region, scan rates) revealed the appearance of this maximum also in the patterns of electrodes based on other active materials and even of the ‘idle’ electrode. Thus, the maximum is assigned to products of the electrolyte reduction occurring at the effective electrode surface.
The observed capacity value suggests irreversible decomposition of iron selenide in the 1st cycle according to reaction equation (1). This assignment is also in agreement with earlier conclusions11. We assume that this reaction results in the formation of iron and lithium selenide nanoparticles which could not be detected by our PXRD measurements.

The reduction process was studied in more detail by means of slow scan-rate linear voltammetry (LVS) and potentiostatic intermittent-titration technique (PITT) combined with chronoamperometry (CA). This study reveals the complex character of the 1st reduction curve including additional peaks at ca. 2.1, 1.8 and 1.6 V vs Li/Li+ (Fig. 2, Fig. SI4) preceding the main conversion peak (starting at ca. 1.5 V), similarly to previous observations11. The results of analogous investigation of carbon-free electrodes allowed attributing the rather intense peak at ca. 2.1 V to the electrolyte reduction at the carbon surface (Fig. SI4). The charge passed through the cell in the potential region of 3.0−1.55 V (before the major reduction peak) corresponds to ca. 11 mAh·g−1 (0.055 Li+ per f.u.). Further application of CA at 1.7 V for a few days did not result in significant growth of the current. This voltage is between the peaks of the assumed Li+ ion intercalation and the main conversion process in the differential capacity plot (Fig. 2). We suppose that the amount of the intercalated Li in LixFe1+δSe at this voltage corresponds to x ≈ 0.06. According to PXRD, the Fe1+δSe phase is preserved in the PITT procedure. Even after long CA treatment at 1.7 V (several days up to a week) all samples exhibit the same behavior: after turning-off the electrochemical cell, the potential increases rather rapidly from 1.7 V to approx. 2−2.5 V vs. Li/Li+ as in case of initial Fe1+δSe. We regard this effect as the result of the spontaneous decomposition of the intercalated phase LixFe1+δSe. Indeed, magnetic measurements of the electrode treated at 1.7 V vs. Li/Li+ and kept inside the glovebox at room temperature for several days showed the presence of the initial Fe1+δSe phase only.

LVS curve obtained for the polycrystalline Fe1+δSe electrode (scan rate 0.02 mV·s−1, 2.5–1.2 V vs. Li/Li+).
The inset shows the LVS curve and the differential capacity obtained by PITT in the potential range 2.5–1.7 V vs. Li/Li+ in detail.
Magnetic measurements of the polycrystalline Fe1+δSe electrode treated with LVS and CA at 1.7 V vs. Li/Li+ unveiled drastic changes in comparison with native Fe1+δSe (Fig. 3). The temperature dependence of the magnetic susceptibility χ(T) shows a strong diamagnetic signal at low temperatures with a kink at about 8 K and a broad maximum at about 70 K. The pronounced, negative slope of the magnetic susceptibility at low temperature indicates the presence of initial superconducting tetragonal Fe1+δSe3. The broad maximum at high temperatures signals the presence of several percent of elemental iron in the system, presumably in nano-structured form15. This attribution is strongly supported by magnetization curves measured at 2 and 12 K, see inset of Fig. 3. Both curves evidence hysteresis loops which are typical for iron nanoparticles below the blocking temperature15,16. Besides, the M(H) dependence measured at 2 K is decorated with a weak diamagnetic response seen in low fields due to the presence of the superconducting phase. Thus, the redox process between 1.8 V and 1.6 V vs Li/Li+ is characterized by a very small capacity value and accompanied by the conservation of the Fe1+δSe structure. The observed capacity value corresponds to a small amount of Li reacted with Fe1+δSe and being intercalated as Li+. The spontaneous potential increase after cell turning-off together with the magnetic measurements data can be understood taking into account the instability of the product of the electrochemical redox process if the potential is not applied.

Temperature dependence of the magnetic susceptibility of the initial polycrystalline Fe1+δSe electrode (blue open curve) and of the polycrystalline Fe1+δSe electrode treated at 1.7 V vs Li/Li+ (black solid spheres) measured in the ZFC regime at 0.01 T.
The inset represents magnetization curves measured at 2 K and 12 K.
To clarify the behavior of Fe1+δSe in the applied electrochemical potentials, in-situ PXRD studies in galvanostatic mode (GCPL) were performed. In the potential region of 2−1.5 V vs. Li/Li+ (corresponding to approximately 0−10 mAh·g−1 in the E−C curve), the original Fe1+δSe structure exhibits no measurable alterations (Fig. 4a,b). The intensity and position of the 101 reflection remains apparently stable (taking into account experimental errors), but the 001 reflection broadens and its center of gravity shifts to slightly lower angles (Fig. 4c,d). Concordantly, the shape of the E−C curve is typical for a solid solution intercalating process involving a single phase. Further decrease of the potential (1.5−0.7 V vs. Li/Li+, which corresponds to 10−400 mAh·g−1 on the E−C curve) results in the rapid decomposition of Fe1+δSe monitored by the drastic decrease in the intensities of both the 001 and 101 reflections (Fig. 4a). No new diffraction maxima are observed. The E−C curve reveals an almost flat plateau in this potential region, which unambiguously indicates the two-phase state of the system in accordance with reaction (1).

In-situ X-ray powder diffraction experiment performed for the polycrystalline Fe1+δSe electrode during galvanostatic charge with potential limitation (approx. C/20 rate, 2.0−0.7 V vs. Li/Li+).
(a) obtained PXRD patterns (reflections of the tetragonal Fe1+δSe are indexed, additional observed maxima correspond to the components of the in-situ electrochemical cell); (b) E−C curve corresponding to the PXRD patterns; (c) dependence of 001 reflection position on the passed charge; (d) transformation of the 001 reflection at different potentials under potentiostatic conditions.
The results above can be summarized in a scenario for the electrochemical processes of a polycrystalline Fe1+δSe electrode. In agreement with earlier results11, the major process favored by thermodynamics is the decomposition and complete reduction of Fe1+δSe to metallic Fe. Electrochemical data together with in-situ PXRD measurements unveil that at higher potentials (ca. 1.7 V vs. Li/Li+) a subtle redox process takes place. This process conserves the initial structure topology and leads to the formation of a ternary phase LixFe1+δSe with slightly larger lattice parameter c compared with the one of Fe1+δSe. The potential increase which is observed after turning-off the electrochemical cell can be interpreted as spontaneous decomposition of the ternary phase due to its instability (cf. below). The small lattice parameters variation together with the small charge transfer indicates the insertion of a tiny amount of Li+ which leads to the formation of the phase LixFe1+δSe with preserved structure topology. However, studies of bulk LixFe1+δSe are hampered by its extreme instability and the overlap of intercalation and decomposition processes in the narrow potential region.
We assumed that the use of Fe1+δSe crystal aggregates instead of a polycrystalline electrode can kinetically inhibit the decomposition process. Polycrystalline electrodes consist of a large amount of relatively small crystallites (1–10 μm in size), crystal aggregates contain several single crystals of much larger size (100–1000 μm). Surface effects (including the decomposition of LixFe1+δSe and the formation of metallic Fe) in the latter case should be much less pronounced. Furthermore, the application of crystals allows avoid the use of carbon black and polyvinylidenfluorid in the measurement cell that makes the corresponding electrochemical side reactions negligible. The electrochemical behavior of Fe1+δSe crystal agglomerates differs only slightly from that of the polycrystalline electrodes (Fig. SI5). The differences mainly concern a shift of the redox peaks to higher potential values. The first reduction peak, which corresponds to the suggested intercalation process, appears at ca. 1.8 V vs. Li/Li+. It is followed by the major peak (attributed to the conversion reaction) with an onset at ca. 1.7 V. This major peak is distorted in the CVA curve because the drastic volume change causes the destruction of the crystal. I–t curves obtained by long-time potentiostatic experiment at 1.7 V are close to those of the polycrystalline material. The current (in module) increases in the course of the 30 hours experiment. The amount of charge passed through the cell before the current increases corresponds to ca. 0.06 Li+ per f.u.
Magnetic measurements of the Fe1+δSe crystal aggregates after treatment at 1.7 V indicate the presence of inhomogeneous superconducting phases. The temperature dependence of the magnetic susceptibility (Fig. 5) demonstrates a strong diamagnetic response (Meissner effect) at low temperatures corresponding to 100% volume fraction of the superconducting phases. At higher temperatures the χ(T) curve shows three kinks at T1 ~ 9 K, T2 ~ 30 K and T3 ~ 44 K. It becomes positive in the range T2 < T < T3. The T1 anomaly can be attributed to the superconducting phase transition in binary (tetragonal) Fe1+δSe which predominates in the studied sample. The magnetization curve measured at 2 K (lower inset to Fig. 5) is typical for Fe1+δSe single crystals17. Two other anomalies can be attributed to two new superconducting phases. Some confirmation of the superconducting nature of the phases in the region T1 < T < T3 can be found in magnetization curves measured at 12 K and 35 K (upper inset to Fig. 5) indicating a small diamagnetic response. From the results of the chronopotentiometric experiment (Fig. SI6) we assume that in the products of the electrochemical treatment of the Fe1+δSe crystal aggregates there are single-phase domains with slightly different content of Li.

Temperature dependence of the magnetic susceptibility of initial Fe1+δSe crystal aggregates (open blue triangles) and that after treatment at 1.7 V vs. Li/Li+ (solid black circles) measured in the ZFC regime at 0.01 T.
The insets show the field dependence of magnetization at temperatures below the superconducting transition measured at 12 K and 35 K (top) and 2 K (bottom).
The enhancement of Tc observed for the Fe1+δSe crystal after electrochemical treatment confirms the suggested formation of LixFe1+δSe. As additional technique to determine the amount of Li+, ICP-MS analysis was performed for polycrystalline electrodes (Table SI1). The highest Li content was detected in the electrodes subjected to the LVS followed by rather long CA at 1.55 V vs. Li/Li+. The relative Li-content in the electrode subjected to the intercalation reaction conditions (PITT with CA at 1.7 V) can be estimated as 0.06 Li per f.u. Fe1+δSe. The result is in perfect agreement with the electrochemical data but this agreement merely reflects the tendencies for the different redox processes since capacity values are strongly sensitive to experimental conditions like the distribution of Fe1+δSe particles in the electrode active layer and its thickness.
In-situ PXRD data together with the magnetic measurements indicate bulk (i.e. not surface) formation of the superconducting phase which is structurally similar to tetragonal Fe1+δSe. In case of tetragonal Fe1+δSe, such a Tc increase is mainly achieved by electron doping4,5,6,7. The electrochemical insertion of Li into the Fe1+δSe structure can be considered as one possible way of electron doping. Due to the close-packed features of the Fe1+δSe structure (defect anti-fluorite blocks), there are tetrahedral and distorted octahedral voids (Fig. 6). For tetrahedral coordination, the calculated Li−Se distances (ca. 2.28 Å) are significantly shorter than those in the Li2Se structure (2.6 Å)18. For octahedral voids, the estimated Li−Se distances (ca. 2.57–2.95 Å) are comparable to the ones in Li2Se, while the Li−Fe distances (ca. 2.39 Å) are too short. However, shift of the Li in [001] direction results in a square-pyramidal coordination of Li with longer Li−Fe contacts.

The optimized 3 × 3 supercell of the tetragonal structure of Li1/18FeSe (left top; the 4-fold symmetry around Li (red) is essentially preserved, relaxation of Fe (purple) and Se (green) is taking place basically for nearest neighbors, only) with Li filling tetrahedral voids (right top) and octahedral ones (ideal–left bottom, optimized–right bottom).
Relaxing all atoms with respect to the total energy shifts Li off-center and reduces the undulation of the Fe-Se layers (right bottom).
A similar effect is observed in LiFeAs (PbClF type)19, where the anti-PbO-like topology is combined with a square-pyramidal Li+ coordination. Here, Li cations stabilize the whole structure because of Fe3+/Fe2+ reduction. It should be noted that the size of the anti-fluorite [Fe2As2] layer in LiFeAs is very close to that of the [Fe2Se2] layer in Fe1+δSe. Obviously, complete Li insertion into Fe1+δSe would correspond to the complete reduction Fe2+/Fe1+, thereby limiting the amount of inserted Li.
The total energy for different model structures and Li concentration x was calculated to investigate the stability of LixFeSe with Li atoms occupying different sites. From chemical experience, it is expected that Li occupies tetrahedral or octahedral voids between the Se atoms (Fig. 6). Without relaxation of Se coordinates, we find a binding energy of slightly more than 1 eV per Li atom occupying the octahedral void, essentially irrespective of the Li concentration (Table SI2). For the tetrahedral position, the binding energy is reduced considerably to less than 0.5 eV per Li. Due to this large energy difference and the large Li mobility, lithium occupation of the tetrahedral void can be safely discarded near the equilibrium. Relaxing all atoms without symmetry constraint (space group P1) in a Li1/18FeSe super cell (Fig. 6) and with a Li concentration close to the experimental value, the binding energy increases (Table SI2) by about 0.2 eV per Li. The predominant structural feature is an off-center Li position (Fig. 6) and a smoothing of the FeSe layers. The fourfold symmetry of the Li position and its environment however, is essentially preserved.
We also calculated the stability of LixFeSe (x = 1/18 = 0.056, near the experimental Li content) with respect to its decomposition into Li2Se, Fe and FeSe. Li1/18FeSe is not stable and thus should, under equilibrium conditions, decompose towards FeSe, Li2Se and Fe with an energy gain between 0.20 eV (for non-magnetic Fe clusters) and 0.42 eV (for magnetic Fe clusters) per Li (Table SI2). This is in agreement with the available experimental data. The calculated decomposition energy is only slightly dependent on the magnetic state of Fe in the clusters formed during the decomposition. For the experimentally observed superparamagnetic iron clusters the energy should be in-between the two limiting cases.
For a better understanding of the superconducting properties of LixFe1+δSe, the change of the electronic density of states near the Fermi level EF upon Li insertion is a crucial parameter. Comparison of the calculated DOS (Fig. 7 and Table SI2) for Li1/18FeSe and the parent compound FeSe yields three main results: (i) Surprisingly, the insertion of Li leads to a sizeable re-structuring of the DOS which goes beyond the changes expected on basis of a rigid band picture and results in an increase of the DOS at EF. (ii) The total band width is basically unchanged. (iii) The main increase of the DOS at EF originates from more distant Se and Fe neighbors and not from the closer Li environment. This demonstrates that the additional electron donated by the Li ion is rather delocalized. However, a possible formation of polarons (beyond the DFT calculations) might modify this picture.

Total and partial electronic density of states (DOS) for Li1/18FeSe.
The Fermi level EF is at zero energy. Upper panel: Insertion of Li leads to a significant change of the electronic structure beyond a rigid band behavior (shift of EF, only) compared to the parent compound FeSe (with optimized Se position, see text), in particular to a sizeable increase of the DOS at EF (insert). Lower panel: As in the idealized parent compound FeSe, the states near EF originate almost exclusively from Fe with a small Se admixture; the Li-contribution is very small owing to the small Li concentration.
The observed changes in the electronic structure evidence the formation of a ternary compound which is chemically different from Fe1+δSe. The appearance of a DOS spike at the Fermi level is in line with the observed higher critical temperature of the superconductivity.
In summary, our results are in line with earlier findings that the Fe1+δSe structure pattern has a strong tendency to preserve the electron count of the [Fe2Se2] layers. Thus, the homogeneity range of these structure units with respect to heterovalent substitution or the insertion of electropositive metals is rather limited. Even in case of structure alterations which result in substantial increases of the Tc values, the charge of the anti-fluorite layer changes only marginally7,8. Our finding suggests that the significant Tc-enhancement of Fe1+δSe can be achieved by a merely minute reduction of the Fe cations without increasing the [Fe2Se2]-interlayer separation. Therefore, it appears promising to investigate if the carrier concentration (or the iron valence) influences the Tc-value of layered iron-based selenides to a much larger extent than the interlayer separation.
Methods
Preparation
Polycrystalline sample of Fe1+δSe was prepared under argon atmosphere using iron pieces (99,995%) and selenium shots (99,999%). Glassy carbon crucibles with a lid were filled with Fe/Se mixture with molar ratio close to 1 : 1 (Fe : Se = z : 1 and 0.98 ≤ z ≤ 1.02), placed into vacuumed quartz ampoule and subjected to high-temperature annealing (for details see ref. 13). Large crystals of Fe1+δSe were prepared using the KCl/AlCl3 flux at 427 °C in evacuated quartz ampoules in constant temperature gradient20.
Basic characterization
Microstructure investigation of polycrystalline samples and crystals was performed with JEOL JSM 5510 (LaB6 cathode, 30 kV) scanning microscope. Phase identification and lattice parameters determination (for polycrystalline sample and crystals) were performed using room-temperature X-ray powder diffraction data obtained by image plate Guinier camera Huber G670, (CoKα1 radiation, λ = 1.78892 Å). Crystalline Ge (a = 5.6576 Å, ref. 21) was used as internal standard. Ex-situ X-ray diffraction data for polycrystalline electrodes were collected in air using Bruker D8-Advance diffractometer (CuKα1 radiation, λ = 1.540598 Å, LynxEye PSD) in reflection mode. The electrodes previously were covered by air-protective one side sticky tape.
Electrochemical processing
Electrochemical treatments were performed in two-electrode setup with metallic Li as counter and reference electrodes. The commercial electrolyte (1 mol L−1 LiPF6 in a mixture of dimethyl carbonate and ethylene carbonate (1: 1 by volume, Merck) was used. The glass fiber was applied as separator. Working electrode (in case of polycrystalline samples) was prepared by screen printing the slurry containing the active material, carbon black and PVDF (mass ratio 85 : 5 : 10) in N-methylpyrrolidone onto Al-substrate. Load of active material was approx. 1 mg·cm−2. In a case of Fe1+δSe crystals, one crystal was placed by its flat face between current collector and separator directly. Electrochemical cells were assembled and disassembled in Ar atmosphere. Potentiostat/galvanostat Biologic VMP-3 was used for data collecting. Several electrochemical techniques were applied: linear and cyclic voltammetry (LVS and CVA) between 1.0 V and 2.5 V vs. Li/Li+ with scan rate of 0.02 mV·s−1 and 0.05 mV·s−1; potentiostatic intermittent titration technique (PITT) with 0.01 V step in combination with chronoamperometry (CA) at selected potentials; and galvanostatic charge with potential limitation (GCPL) at current density of approx. 20 mA·g−1.
In-situ powder X-ray diffraction
The experiment was carried out on polycrystalline electrodes. The two-electrode electrochemical cell was analogous to the mentioned above but with Be window at the anode side and porous polypropylene separator (“UFIM”, Russia) instead of glass fiber. The diffraction data were collected using Bruker D8-Advance diffractometer (CuKα1 radiation, λ = 1.540598 Å, LynxEye PSD) in reflection mode. During the experiment, a constant current of −15 μA (approx. 20 mA·g−1) was applied at the working electrode; diffraction patterns were collected every 30 min until the potential decreased to 0.7 V vs. Li/Li+ and the electrochemical cell was switched off.
Magnetic measurements
Magnetisation was measured by vibration magnetometer (PPMS-9T, Quantum Design) applying fields of 10–1000 Oe. The specimens (polycrystalline samples, polycrystalline electrodes, crystals aggregates) were put inside plastic holders and then placed into the magnetometer in Ar atmosphere.
Chemical analysis
The lithium/iron ratio in electrochemically treated electrodes was analyzed by ICP-MS by means of ICP-MS spectrometer ELAN DRC II.
Band structure calculations
Relativistic density functional (DFT) electronic structure calculations were performed using the full-potential local-orbital FPLO code22,23 (version fplo14.00-47). For the exchange-correlation potential, within the local density approximation (LDA) and the general gradient approximation (GGA), the parametrization of Perdew-Wang24 and Perdew-Burke-Ernzerhof25 were chosen, respectively. To obtain precise total energy and band structure information, the calculations were carried out on well converged meshes of up to 3888 k points depending on the cell volume. For all calculations, the respective experimental lattice parameters have been used.
Additional Information
How to cite this article: Alekseeva, A. M. et al. New superconductor LixFe1+δSe (x ≤ 0.07, Tc up to 44 K) by an electrochemical route. Sci. Rep. 6, 25624; doi: 10.1038/srep25624 (2016).
References
Hsu, F.-C. et al. Superconductivity in PbO-type structure α-FeSe, PNAS, 105(38), 14262–14264 (2008).
Williams, A. J., McQueen, T. M. & Cava, R. J. The stoichiometry of FeSe, Solid State Commun., 149, 1507–1509 (2009).
McQueen, T. M. et al. Extreme sensitivity of superconductivity to stoichiometry in Fe1+δSe, Phys. Rev. B, 79, 014522 (2009).
Guo, G. et al. Superconductivity in iron selenide KxFe2Se2 (0 ≤ x ≤ 1.0), Phys. Rev. B, 82, 180520(R) (2010).
Ying, J. J. et al. Superconductivity and magnetic properties of single crystals of K0.75Fe1.66Se2 and Cs0.81Fe1.61Se2, Phys. Rev. B, 83, 212502 (2011).
Ying, T. P. et al. Observation of superconductivity at 30 ∼ 46 K in AxFe2Se2 (A = Li, Na, Ba, Sr, Ca, Yb and Eu), Sci. Rep. 2, 426 (2012).
Burrard-Lucas, M. et al. Enhancement of the superconducting transition temperature of FeSe by intercalation of a molecular spacer layer, Nature Materials, 12, 15–19 (2013).
Sun, H. et al. Soft chemical control of superconductivity in lithium iron selenide hydroxides Li1−xFex(OH)Fe1−ySe, Inorg. Chem., 54, 1958–1964 (2015).
Lu, X. F. et al. Coexistence of superconductivity and antiferromagnetism in (Li0.8Fe0.2)OHFeSe, Nature Materials, 14, 325–329 (2015).
Abe H., Noji T., Kato M. & Koike Y. Electrochemical Li-intercalation into the Fe-based superconductor FeSe1−xTex, Physica C, 470, S487–S488 (2010).
Wei, D. et al. Layer structured α-FeSe: A potential anode material for lithium storage, Electrochem. Comm., 38, 124–127 (2014).
Shen S.-J. et al. Electro-chemical synthesis of alkali-intercalated iron selenide Superconductors. Chin. Phys. B 24, 117406 (2015).
Koz, C. et al. Synthesis and crystal growth of tetragonal β-Fe1.01Se, ZAAC, 640, 1600–1606 (2014).
Pomjakushina, E., Conder, K., Pomjakushin, V., Bendele, M. & Khasanov, R. Synthesis, crystal structure and chemical stability of the superconductor FeSe1−x, Phys. Rev. B, 80, 024517 (2009).
Huber, D. L. Synthesis, Properties and Applications of Iron Nanoparticles, Small 1, 482–501 (2005).
M. W. Grinstaff, M. B. Salamon & K. S. Suslik, Magnetic properties of amorphous iron. Phys. Rev. B, 48, 269 (1993).
Abdel-Hafiez, M. et al. Temperature dependence of lower critical field Hc1(T) shows nodeless superconductivity in FeSe, Phys. Rev. B, 88, 174512 (2013).
Inorganic Crystal Structure Database (ICSD). Database issued by Fachinformationszentrum Karlsruhe (FIZ), Germany, Version 2014-1.
Morozov, I. et al. Single crystal growth and characterization of superconducting LiFeAs, Crystal Growth and Design, 10, 4428–4432 (2010).
Chareev, D. et al. Single crystal growth and characterization of tetragonal FeSe1−x superconductors. Cryst. Eng. Comm., 15, 1989–1993 (2013).
Powder Diffraction File (PDF-2). Database issued by International Center for Diffraction Data, Newton Square, USA, 1998.
Koepernik, K. & Eschrig, H. Full-potentional nonorthogonal local orbital-minimum basis band-structure scheme, Phys. Rev. B, 59, 1743–1757 (1999).
Opahle, I., Koepernik, K. & Eschrig, H. Full-potentional band-structure calculation of iron pyrite, Phys. Rev. B, 60, 14035 (1999).
Perdew J. P. & Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy, Phys. Rev. B 45, 13244 (1992).
Perdew, J. P., Burke K. & Ernzerhof, M. Generalized gradient approximation made simple, Phys. Rev. Lett. 77, 3865–3867 (1996).
Acknowledgements
This work was partially supported by Russian Scientific Foundation grant No. 14-13-00738.
Author information
Authors and Affiliations
Contributions
A.M.A., E.V.A. and Y.G. designed the research, O.A.D., K.A.D. and A.M.A. performed electrochemical studies and characterizations, C.K. and U.S. prepared polycrystalline sample of Fe1+δSe, D.A.C. prepared crystal aggregates of Fe1+δSe, K.V.Z., O.S.V. and A.N.V. performed physical measurements, H.R. made electronic structure calculations, all authors contributed to the text of the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Alekseeva, A., Drozhzhin, O., Dosaev, K. et al. New superconductor LixFe1+δSe (x ≤ 0.07, Tc up to 44 K) by an electrochemical route. Sci Rep 6, 25624 (2016). https://doi.org/10.1038/srep25624
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep25624
This article is cited by
-
Research Progress of FeSe-based Superconductors Containing Ammonia/Organic Molecules Intercalation
Topics in Current Chemistry (2022)
-
Solid state chemistry for developing better metal-ion batteries
Nature Communications (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
