
Abstract
Despite having serious clinical manifestations, Cellulosimicrobium cellulans remain under-reported with only three genome sequences available at the time of writing. Genome sequences of C. cellulans LMG16121, C. cellulans J36 and Cellulosimicrobium sp. strain MM were used to determine distribution of pathogenicity islands (PAIs) across C. cellulans, which revealed 49 potential marker genes with known association to human infections, e.g. Fic and VbhA toxin-antitoxin system. Oligonucleotide composition-based analysis of orthologous proteins (n = 791) across three genomes revealed significant negative correlation (P < 0.05) between frequency of optimal codons (Fopt) and gene G+C content, highlighting the G+C-biased gene conversion (gBGC) effect across Cellulosimicrobium strains. Bayesian molecular-clock analysis performed on three virulent PAI proteins (Fic; D-alanyl-D-alanine-carboxypeptidase; transposase) dated the divergence event at 300 million years ago from the most common recent ancestor. Synteny-based annotation of hypothetical proteins highlighted gene transfers from non-pathogenic bacteria as a key factor in the evolution of PAIs. Additonally, deciphering the metagenomic islands using strain MM’s genome with environmental data from the site of isolation (hot-spring biofilm) revealed (an)aerobic respiration as population segregation factor across the in situ cohorts. Using reference genomes and metagenomic data, our results highlight the emergence and evolution of PAIs in the genus Cellulosimicrobium.
Similar content being viewed by others

Introduction
With only three published species, C. cellulans1, C. funkie2, C. terreum3 and 31 16S rRNA gene sequences, the genus Cellulosimicrobium remains underrepresented in present NCBI reference databases. First proposed by Schumann et al.1, this genus has stayed taxonomically confounding with multiple reclassifications from the genera Cellulomonas, Oerskovia, Brevibacterium and Arthrobacter1. The ecological distribution of Cellulosimicrobium strains has been by and large limited to the mesophilic environments such as soil, marine sponges and clinical materials. However, only two instances of isolation from extreme environments have been reported, including hot springs4 and Antarctic snow5. Currently, there are four sequenced genomes for the genus Cellulosimicrobium, including Cellulosimicrobium sp. strain MM4, C. cellulans LMG 16121 (NZ_CAOI00000000.1), C. cellulans J36 (NZ_JAGJ00000000.1) and C. funkei U11 (NZ_JNBQ00000000.1), which were isolated from biofilms (surface temperature > 57 °C) at the Manikaran hot springs (surface water temperature > 95 °C), aluminium hydroxide gel antacid, compost and agricultural soil, respectively. The genus Cellulosimicrobium is associated with human infections such as meningitis, endocarditis, bacteremia, soft tissue infection, endophthalmitis, septic arthritis and prosthetic joint infections6. Here we perform a detailed genome wide investigation using two available C. cellulans genomes i.e. LMG 16121 and J36 along with Cellulosimicrobium sp. strain MM (for its > 97% whole genome proximity to C. cellulans strains) to determine evolutionary processes that have shaped pathogenicity across the species C. cellulans.
Previous genomic studies of emerging pathogenic bacteria have revealed that pathogenicity islands (PAIs) contribute significantly towards organismal evolution by expression of infection-related factors7. Using the three draft genome sequences and a metagenomically-derived minimal genome, we have deciphered the pathogenic gene complements of species C. cellulans, which is attributed to 80% incidences of the total human infections for this genus6. Highlighting the close proximity of the functionally coupled ORFs, Fic and VbhA, the results provide the evidence for the ‘selfish operon’ theory whereby their juxtaposition on a PAI results from a probable single horizontal gene transfer event8. PAIs ORF annotation revealed 49 virulence-encoding genes and suggested that horizontal transfer from non-pathogenic bacteria plays a signficiant role in the evolution of PAIs. Finally, these analyses provide a platform for using these 49 credible virulence markers to diagnose the presence of Cellulosimicrobium pathogens.
Results and Discussion
Phylogenomic analysis
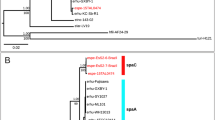
Very few genomes or 16S rRNA sequences exist for Cellulosimicrobium, therefore, phylogenetic reconstruction for the strains was performed using the Family Promicromonosporacae, which significantly increased the number of genomes and 16S rRNA sequences available. 16S rRNA gene (n = 31) based tree topology revealed genus-specific clustering for Promicromonospora (n = 11), Isoptericola (n = 6), Myceligenerans (n = 3), Xylanibacterium (n = 1), Xylanimonas (n = 1), Xylanimicrobium (n = 1) and Cellulosimicrobium (n = 7). Cellulosimicrobium sp. strain MM was clustered with C. cellulans LMG 16121 (Fig. 1a). Pairwise average nucleotide (ANI) values were also calculated generating a score of 98.23% (>95%) between Cellulosimicrobium needs to be italicized. sp. MM and C. cellulans LMG 16121, which suggested that the genome was a sub-species9. ANI values for C. cellulans J36, C. funkei U11, I. variabilis 225, P. kroppenstedtii DSM19349, P. sukumoe 327MFSha31 and X. cellulosilytica DSM 15894 with respect to strain MM, were 88.24%, 85.29%, 81.17%, 80.28%, 79.71% and 79.88%, respectively, distinctly demonstrating species/genus level delineation (see Supplementary Table S2). Further, DNA-DNA hybridization (DDH) values were also determined in order to resolve strain MM at species level. % DDH values were 75.6, 55.3, 56, 20.9, 15.7, 16, 17.2 for C. cellulans LMG16121, C. cellulans J36, C. funkei U11, I. variabilis 225, P. kroppenstedtii DSM19349, P. sukumoe 327MFSha31 and X. cellulosilytica DSM 15894, respectively (see Supplementary Table S3). Both ANI and DDH values assigned Cellulosimicrobium sp. strain MM under C. cellulans species with values greater than species delination cut-off i.e. 95% and 70%, respectively for each analysis9. Interestingly, C. cellulans J36 demonstrated values less than the species delineation cut-off for both ANI and DDH which here indicates that this strain needs further confirmation using biochemical and physiological tests, to be put under C. cellulans species. Overall, the 16S rRNA analysis identified that C. funkei and C. cellulans form a clade, with C. terreum as an outlier to this clade (Fig. 1a). Multiple strains of C. cellulans were scattered into two sub-clades with C. funkei possibly because of the low number of Cellulosimicrobium strains with an available 16S rRNA sequence (n = 6; Fig. 1a). Whole genome based (n = 8) phylogenetic reconstruction using both 31 single copy genes10 (Fig. 1b) and 400 conserved bacterial marker genes11 (see Supplementary Fig. S1) revealed similar tree topology whereby strain MM was grouped with C. cellulans LMG16121.

Phylogenomic analysis and comparative functional potential of Cellulosimicrobium strains.
(a) Rooted Maximum likelihood tree based on Jukes-Cantor model for family Promicromonosporacae using 31 16S rRNA gene sequences with Cellulomonas aerilata 5420S-23 as outgroup, (b) Rooted tree based on 31 single copy genes from 7 whole genomes using Cellulosmonas flavigena DSM 20109 as outgroup. All the trees are drawn to scale, with branch lengths measured in the number of substitutions per site. The percentage (> 70%) of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) are shown next to the branches. (c) Heatmap with column dendrogram showing top 50 metabolic pathways reconstructed between three Cellulosimicrobium genomes i.e. strains MM, LMG16121 and J36. Three strains were clustered based on functional pathways using Manhattan distance metric, top 50 pathways with standard deviation 0.4 and having at least 0.8% of the total abundance were selected. Colour scale is representing the relative abundance of each functional pathway.
Comparative functional potential of Cellulosimicrobium cellulans strains
Metabolic pathway reconstruction for the three Cellulosimicrobium strains based on KAAS12, revealed a conserved set of central pathways like glycolysis/gluconeogenesis, TCA cycle, β -alanine metabolism, inositol phosphate metabolism, propanoate metabolism and two-component system (TCS). Cellulosimicrobium sp. strain MM exhibited the pathways for fatty acid metabolism, synthesis and degradation of ketone bodies and D-alanine metabolism, which were not present in the other two neighbors (Fig. 1c). D-alanine is proposed to be involved in biofilm production, adhesion and pathogenesis. In addition, synthesis and degradation of ketone bodies in strain MM appear to play a pivotal role in quorum sensing, which is also associated with biofilm formation13. LMG16121 uniquely encoded a Type-II secretion system (Fig. 1c) in contrast to the other strains. Out of 102 pathways reconstructed across all the strains, taurine and hypotaurine metabolism, which are involved in membrane stabilization, glycolysis and glycogenesis, were unique to C. cellulans J36 (Fig. 1c)14. Further, hierarchical clustering of the three genomes on the bases of top 50 enriched metabolic pathways, revealed closeness of strain MM and LMG16121, as also shown using phylogenetic analysis (Fig. 1b,c). In total, 791 orthologous genes were identified across the three genomes (see Supplementary Table S4), with the majority (n = 598) assigned to translation, metabolism and structure maintenance15.
Horizontal gene transfer (HGTs) candidates were determined across three Cellulosimicrobium strains. Strain LMG had the greatest number of potentially transferred genes (n = 367), followed by strain MM (n = 348) and J36 (n = 280) (see Supplementary Table S5). It is possible that the high number of horizontal transfer events in strains LMG16121 and MM is indicative of the extreme environment from which they were isolated, i.e. antacid and arsenic contaminated hot spring microbial mat, respectively. Another explaination for occurrence of frequent HGT events overall in all three genomes can be presence of mobile genetic elements and PAIs across C. cellulans genomes (as discussed in the section below), as mobile genetic elements facilitate HGTs16. Strain MM encoded 2-oxo-acid dehydrogenase, histidine kinase, FNR transcriptional regulator, FAD dependent oxidoreductase, Clp subunits and hemin transport proteins on HGT loci. Whereas LMG16121 HGT loci included cobyrinic acid ac-diamide synthase, TetR family transcription regulator, chitin binding protein, luxR, copper oxidase, bleomycin resistance protein and CheY proteins. The HGT loci for J36 revealed integrase, IS3/IS911 family transposase, daunorubicin resistance protein and CLG chitinase B (see Supplementary Table S5). Additionally, hierarchical clustering was performed on annotated HGT candidates across three genomes i.e. strain MM, LMG16121 and J36 along with heatmap showing relative abundance of HGT genes. Strain MM was clustered with J36 based on the annotation of the HGT genes which is interesting here as strain MM coordinated with LMG16121 in terms of frequency of HGT events (see Supplementary Fig. S2).
Evidence for G+C biased gene conversions across the genus Cellulosimicrobium
Pairwise correlation between Fopt and %G+ C across the Cellulosimicrobium orthologous gene complement (n = 791, see Supplementary Table S4) revealed a weak, but statistically significant (R2 < − 0.4, P-value ≤ 3.5e-15) negative correlation (Fig. 2a), which could be interpreted as a result of insufficient codon usage choices due to the 74%G+ C in this genus17. The weak-negative correlation between gene-based G+ C content and Fopt can be explained by both high genomic G+ C content (average %G+ C = 74) and G+ C biased gene conversion (gBGC) effect (Fig. 2a)17,18,19. Habitat specific variations were evident (P-value < 0.05) in the genome wide pairwise analysis of codon usage across Cellulosimicrobium ecotypes. Strain MM (a hot spring ecotype) showed a significantly different codon usage profile when compared with the mesophilic LMG16121 (P-value = 5.373e-06) and J36 (P-value = 4.722e-07) (see Supplementary Fig. S3) highlighting the differential impact of local environmental functional constraints.

Scatter plot analysis showing coupling between G+C content, Fopt and dN/dS across Cellulosimicrobium genomes.
(a) Pearson Product-Moment Correlation between %G+ C and Fopt with labeled R2 and P-value for Cellulosimicrobium sp. strain MM, Cellulosimicrobium cellulans LMG16121 and Cellulosimicrobium cellulans J36, based on 791 common orthologues between all three genomes. (b) dN/dS values for orthologous proteins in independent pairs of strains C. cellulans J36, C. cellulans LMG16121 and Cellulosimicrobium sp. strain MM. Black dotted line at dN/dS value of 1 represents the baseline criterion for positive natural selection. (c) Pairwise correlation analysis between Foptand dN/dS values for three genome pairs.
An average dN/dS of ≤ 0.8 for the 791 orthologous gene pairs revealed that the C. cellulans core genome was evolving under purifying selection (Fig. 2b), which is to be expected as these are all essential genes and the genome is G+ C rich18. A negative pairwise correlation was observed between Fopt and dN/dS values for all combinations (Fig. 2c), suggesting an association between the selection of protein sequences and the optimization of codon frequencies.
Anaerobic respiration leads to Cellulosimicrobium population segregation for hot spring ecotypes
Metagenomic reads from microbial mat at Manikaran hot spring were recruited on the genome of strain MM, whereby the regions of the MM genome that were underrepresented in the metagenome (Metagenomic Islands; MGIs) highlighted the accessory genome and environment-specific genetic repertoire. MGIs maintained 33 ORFs encoding for multiple tRNA synthetases, such as lysyl, aspartyl, isoleucyl, cysteinyl, etc. (Fig. 3a, see Supplementary Tabe S5). tRNAs were also annotated within MGIs, which supports their horizontal gene transfer potential20. Besides the greater abundance of ABC transporters and DNA associated proteins, genes encoding quorum sensing, including oxygen sensor proteins, were enriched in the MGIs (see Supplementary Table S6 Fig. 3a). The oxygen sensing machinery included proteins such as FAD linked oxidoreductase (n = 16), histidine kinase (n = 18), NADH-ubiquinone and quinone oxidoreductase (n = 12), ubiquinone and menaquinone biosynthesis (n = 4) and luciferase family oxidoreductase (n = 4) (see Supplementary Table S6). Additionally, pyridine nucleotide-disulfide oxidoreductase dimerization protein (n = 4), LuxR (n = 13), arsenic resistance protein transcriptional regulator (ArsR) (n = 10), glycerol dehydrogenase like oxidoreductase (n = 6), succinate dehydrogenase (n = 11) and fumarate hydratase (n = 5) were also annotated on the MGIs (see Supplementary Table S6). The Manikaran hot spring microbial mats are characterized by both oxic and anoxic micro-niches21, hence Cellulosimicrobium hot spring ecotypes may use oxygen sensing for niche adaptation. Strain MM maintained the genetic potential for arsenic mediated respiration (Ars operon, 27.11% identity) and detoxification (Arr operon, 27.95%) with respect to E. coli K12. The Ars operon (arsA, arsB, arsC, arsD, arsR) can support respiration across oxic and anoxic conditions, whereas, Arr is only aerobic. Hence, we conclude that respiration might be a splitting factor for the Cellulosimicrobium hot spring ecotypes, given that these microbial mats possess oxic and anoxic micro-niches and occurrence of an(aerobic) respiration related genes on the accessory genome of strain MM21.

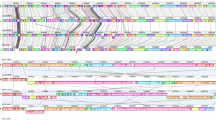
Illustration of pathogenicity islands and metagenomic islands across Cellulosimicrobium sp. strain MM.
(a) Twelve MGIs depicted across the genome of Cellulosimicrobium sp. strain MM after mapping of metagenomic reads from biofilm at Manikaran hot springs. (b) Whole genome alignments. Rings from inside to outside: 1, Whole genome synteny plot of three Cellulosimicrobium genomes using 5 kb window size. Black, blue and red rings represent whole genome sequences for strain MM, LMG16121 and J36. BLASTN comparisons of strain MM with two reference genomes i.e. strains LMG16121 and J36: 2, Black solid represents the genome sequence of strain MM. 3, Green colored rings represent the location of 5 PAIs deciphered across strain MM. 4, Circular black line graph shows %G+ C content of strain MM with regions highlighted for sudden variability (P-value < 0.05) across the extent of 5 PAIs. 5, Blue ring represents genome sequence of strain LMG16121. 6, Red ring represents genome sequence of strain J36. (c) The schematic representation for the annotation of 5 PAIs deciphered in the genome of Cellulosimicrobium sp. strain MM. The direction of the ORFs shows the gene orientation. A standard nomenclature was followed for each PAI belonging to strain MM as MM_CPAI1, MM_CPAI2, MM_CPAI3, MM_CPAI4, MM_CPAI5 where “MM” stands for the strain and “C” stands for the genus Cellulosimicrobium. Blue and red colored blocks represent non-virulent and virulent ORFs, respectively as predicted by VirulentPred. Grey colored blocks represent hypothetical proteins. On MM_CPAI2, ORFs for Fic and VbhA following ‘selfish operon’ theory are highlighted.
Identification and characterization of PAIs across Cellulosimicrobium cellulans ecotypes
Putative PAIs were identified across the 3 genomes by analyzing variations in %G+ C, codon usage patterns (Fig. 3b) and ‘true’ PAIs were assigned using gene content annotation, e.g. tRNA and virulence genes (Fig. 3b,c, Table 1). VirulentPred22 supplemented with MP323 predicted 80 virulent ORFs for the PAIs (MM = 19; LMG16121 = 36; J36 = 25; Fig. 3, see Supplementary Figs S4 and S5). Among these virulent proteins, 61% (49/80) are well known to cause human infections and have been associated with other human pathogens such as Mycobacterium tuberculosis24, Staphylococcus aureus25 and Pseudomonas aeruginosa26 (Table S1).
The whole genome virulence profile showed that 32% (942/3082) of the total protein sequences of Cellulosimicrobium sp. strain MM were pathogenic with a threshold score above 0.2 (using MP3). Similarly, LMG16121 had 31% (998/3217) and J36 had 28% (784/2770) pathogenic proteins. It has already been reported that pathogens including, Mycobacterium tuberculosis H37Rv, Pseudomonas aeruginosa B136–33, Vibrio cholera IEC224 and Neisseria menigitides 053422, maintain 30.28, 26.2, 18 and 16.3%, respectively. This suggests that Cellulosimicrobium carries a high pathogenic potential. A total of 25–28% of the Cellulosimicrobium pathogenic genes could be annotated using the KEGG GENES database (see Supplementary Table S7), with 155 genes shared across all three genomes (see Supplementary Table S8). These core pathogenic genes included UDP-N-acetylmuramate dehydrogenase (murB), chitinase, penicillin amidase, ABC transporters (n = 41), multidrug resistance proteins (emrB, n = 4) and drug exporter proteins (n = 3) (see Supplementary Table S8). These common pathogenic genes were also assigned COG classes, whereby 20% were unknown function, 18% were involved in carbohydrate metabolism and transport, 9% in defense mechanisms, 5% in cell wall/membrane/envelope biogenesis and 2.5% in cell motility (see Supplementary Fig. S6). Cell motility especially was variably distributed across the 3 strains, being nearly absent in J36 (see Supplementary Fig. S6). Interestingly, all the above pathways have recently been proposed as drug targets against Brucella melitensis 16M27 and therefore may present possible drug targets for treating Cellulosimicrobium infections.
The PAI gene content specific to each strain is outlined in the Supplemental information (see Supplementary Text S1) and Fig. 1 (also see Supplementary Figs S4 and S5). Strain MM had some interesting examples, including locus MM_CPAI1 which maintained mobile genetic elements along side multidrug efflux proteins and inositol synthesis and MM_CPAI2 which maintained genes encoding for anti-toxin VbhA and Fic (filamentation induced by cyclic AMP) proteins (Fig. 3c). Fic are effector proteins which work in a complex with VbhT (toxin) and VbhA (anti-toxin) system28. MM_CPAI2 was marked by the presence of juxtaposing Fic and VbhA proteins, speculating that conjugative systems are transferred together via HGT on the pathogenicity or genomic islands loci following the ‘selfish operon model’ (SOM) (Fig. 3c)8. As an alternative to SOM, occurrence of these functionally coupled ORFs can also be justified by the co-regulation model whereby genes are clustered in an operon by mere rearrangements followed by selection for co-regulation8. The presence of fluoroquinolone resistance and DEAD-box helicase, pemK, comEC coding regions supports the annotation of these regions as essential for pathogenicity. Similarly, LMG16121 PAIs included genes encoding for UDP-glucose pyrophosphorylase, phage-associated proteins, laminarinase, rhamnolipids and β -glucosidase-related glycosidases (see Supplementary Fig. S4). Finally, J36 PAIs encoded for an abundance of mobile genetic elements, hemin ABC transporter protein, tetracyclin/bleomycin/doxorubicin/methyl viologen resistance proteins and dihydrofolate reductase (see Supplementary Fig. S5).
Molecular clock analysis of pathogenicity island proteins
A Bayesian approach was used to calculate the evolutionary protein clock for three PAI proteins, namely Fic, D-alanyl-D-alanine carboxypeptidase and transposase (for selection criterion please see “Methods”)29. Molecular clock analysis for the Fic protein (dN/dS = 1.128) across the multiple bacterial lineages encoding this protein on PAIs (including Cellulosimicrobium), predicted the most recent common ancestor (MRCA) to have occurred 27 million years ago (mya) (Fig. 4a). The maximum clade credibility tree revealed Clavibacter michiganensis subsp. michiganensis NCPPB382 (r = 0.009 to 4.874) and Rhodococcus equi (r = 0.011 to 5.533) with highest substitution rates (r at 95% Highest Posterior Density (HPD) interval) in comparison to rest of the strains (shown by branch thickness and black color, Fig. 4a). While Clavibacter michiganensis is well established to be evolving at higher rates using evolutionary dating methodology30, Rhodococcus equi is also known as an emerging pathogen31. The topology of the tree placed Cellulosimicrobium strains MM and J36 together and exhibited significant homology (Posterior Probability = 0.97) to that of Mycobacterium tuberculosis (Fig. 4a), in which Fic protein has been established to be involved in pathogenicity32.

Maximum clade credibility tree summarizing the results of the Bayesian molecular clock analysis of (a) Fic protein, (b) D-alanyl-D-alanine carboxypeptidase and (c) transposase. The protein sequences of these genes harbored by PAIs of different bacterial lineages were aligned by CLUSTALW and evolutionary rate estimation was performed using BEAST. The timeline indicates the age (mya, million years ago) of nodes. Values above the branches indicate posterior probability values and blue horizontal node bars show the length of the 95% highest posterior density (HPD) interval of node ages. The Cellulosimicrobium strains are labeled in red. The branch color gradient (red to black) and width is set according to the increasing substitution rate (r at 95% HPD interval) with black and increased thickness representing the higher substitution rate.
D-alanyl-D-alanine carboxypeptidase found on a PAI in strain LMG16121, has a widespread phylogenetic distribution and a significant role in pathogenesis33. The summary tree showed relatively high substitution rates (95% HPD interval) across all the nodes (Fig. 4b). LMG16121 (r = 0.001 to 2.9596) was grouped with Streptomyces turgidiscabies (r = 0.004 to 2.8708) and Clavibacter michiganensis (r = 0.0001 to 2.6795) (Posterior probability = 0.89). When observed closely (branch thickness), these 3 strains were characterized with relatively lower substitution rates as compared to Enterococcus faecium E980 (r = 0.005 to 3.4292), Escherichia coli 536 (r = 0.0003 to 3.3744) and Staphylococcus aureus (r = 0.005 to 3.4292) (Fig. 4b). The MRCA for D-alanyl-D-alanine carboxypeptidase was calculated at ~40 mya. A similar analysis was also performed across transposase protein sequences from multiple bacterial lineages, including all Cellulosimicrobium strains for its frequent presence on PAIs (Fig. 4c). Consistent with previous reports of molecular clock studies of transposable elements34, the MRCA for transposase (dN/dS = 1.06) across the Cellulosimicrobium dataset was calculated at ~250 mya, indicating a relative age >100 mya. As expected the majority (n = 11) of the Cellulosimicrobium transposases were grouped together, highlighting the species specific nature of this gene family (Fig. 4).
HGT drives the evolution of PAIs via hypothetical proteins from avirulent isolates
The hypothetical proteins occurring on the PAIs were annotated using ACLAME35 which were functionally assigned broadly to mobilization proteins, phage-related proteins, recombinase and DNA-related proteins (Table 2). All the annotated phage proteins were mapped to prophages or viral peptides in hosts such as Rhodobacter sphaeroides, Paracoccus denitrificans PD1222 and Burkholderia pseudomallei. However, one specific “phage lambda-related host specificity protein J” found on J36_PAI2 was found with origin in plasmid pMT1 of Yersinia pestis biovar Microtus str. 91001 (Table 2). Strikingly, only 37% (17/46) of the hypothetical proteins annotated across the 13 PAIs in the Cellulosimicrobium pan-genome were predicted to have originated from pathogenic bacteria (Table 2). The remaining 63% were predicted to have originated from non-pathogenic bacterial hosts, which highlights that HGT interactions between pathogens and other bacteria can play a significiant role in the evolution of PAIs. High number of hypothetical proteins has also been found on PAIs of other pathogenic bacteria such as Pseudomonas aeruginosa PAO116. PAIs themselves are mobile genetic elements and are well known to have been acquired during speciation of pathogens from non-pathogenic or environmental ancestors. Hence, PAIs harbor both virulent and avirulent ORFs and thus origin of hypothetical proteins can be mapped to avirulent bacterial isolates as well.
Metagenomic recruitment of PAIs across Manikaran hot springs
We mapped environmental metagenomic reads from Manikaran hot-spring microbial mats to the 13 PAIs. Only 38% of the PAIs showed significant recruitment, with the majority in strain MM, which was isolated from these microbial mats (Fig. 5). Metagenomic reads mapped to the PAIs were enriched for ORFs encoding for integrase, transposase, secY, inositol-3-phosphate synthase and Clp subunits (Fig. 5). Inositol-3-phosphate synthase is known to be laterally transferred from Archaea to thermophillic bacteria36 and also plays a role in Mycobacterium tuberculosis pathogenicity37. The mapping of metagenomic reads to the secY protein (Fig. 5), suggests that the hot spring community is experiencing an elevated stress response (with respect to both temperature i.e. 57 °C and arsenic) across this environment21. Clp subunits (ClpX and ClpP), which are virulence markers, were also enriched in the microbial mats (Fig. 5)38. This suggest that the microbial community is under considerable environmental stress and maintains significant virulence potential.

Recruitment plot showing binning of metagenomic reads from biofilm at Manikaran hot springs on pathogenicity islands (PAIs).
(a) One dot represents each read aligned onto the PAIs of Cellulosimicrobium sp. strain MM namely MM_CPAI1, (b) MM_CPAI2, (c) MM_CPAI3, (d) MM_CPAI5 and (e) J36_CPAI2 from Cellulosimicrobium cellulans J36. x-y axes represent the sequence co-ordinates and sequence identity, respectively. Blue, red and gray blocks represent non-virulent, virulent (as predicted by VirulentPred) and hypothetical proteins, respectively.
Conclusions
C. cellulans has been associated with human pathogenicity, which is likely acquired and conferred through mobile genetic elements including pathogenicity islands. Using 3 reference genomes, C. cellulans LMG16121, C. cellulans J36 and Cellulosimicrobium sp. strain MM, we annotated 13 PAIs, encoding 49 potential virulence factors well-established to cause human infections. However, 32% (63/200) of the annotated PAI ORFs encoded for unknown proteins, of which 63% mapped to non-pathogenic bacteria, supporting the role of HGT in the evolution of PAIs16. Characterized with a high G+ C content (average 74%), genus Cellulosimicrobium was predicted to experience high selection pressure, with an average dN/dS value of 0.8 (< 1) based on 791 orthologous genes. This study provides first insights into the evolution of PAIs across the genus Cellulosimicrobium and reveals 49 virulence genes such as, Fic, VbhA toxin/antitoxin system, FtsK, ClpX, etc. which can be used as diagnostic markers for pathogenic Cellulosimicrobium strains.
Methods
Phylogenomic analysis
Phylogenomic analysis was performed in order to assign phylogenetic status to the uncharacterized (at species level) Cellulosimicrobium strain MM. Given a limited number of genomes sequenced for the genus Cellulosimicrobium (n = 4), we used dataset from the complete family Promicromonosporacae (n = 8). 16S rRNA gene sequence, 31 single copy genes10 and 400 conserved marker protein sequence based methods11 were used to perform the phylogenetic analysis for the family. The 16S rRNA gene sequences (n = 31) were retrieved from the NCBI database for all seven genera included in family Promicromonosporacae namely: Cellulosimicrobium, Isoptericola, Myceligenerans, Promicromonospora, Xylanibacterium, Xylanimicrobium and Xylanimonas including Cellulomonas aerilata 5420S-23 as the outgroup. Similarly, phylogenomic reconstruction was performed based on amino acid sequences of 400 conserved bacterial markers and 31 single copy genes belonging to eight genome sequences of the family Promicromonosporacae from NCBI database viz. Cellulosimicrobium cellulans J36, Cellulosimicrobium cellulans LMG16121, Cellulosimicrobium sp. strain MM, Cellulosimicrobium funkei U11, Isoptericola variabilis strain 225, Promicromonospora kroppenstedtii DSM 19349 and Promicromonospora sukumoe 327MFSha31 and Xylanimonas cellulosilytica DSM 15894 (http://www.ncbi.nlm.nih.gov/genome/browse/representative/). The concatenated sequences were aligned using CLUSTALW39 and a maximum likelihood40 tree was constructed at bootstrap value of 1000, implemented in software MEGA 6.041. Further, in order to support 16S rRNA and marker gene based phylogenetic analysis, ANI and DDH values9 were also calculated for the 8 whole genomes from Promicromonosporacae. ANI was calculated at minimum alignment length cut-off of 700 bp, minimum identity cut-off of 70% and window size of 1000 bp.
Functional annotations and metagenomic recruitment of Cellulosimicrobium strains
To determine the functional variability across three Cellulosimicrobium strains, i.e. MM, LMG16121 and J36, comparative genomic analysis was performed based on proteins and metabolic pathways. ORFs were predicted from the three genome sequences using FragGeneScan 1.1842 followed by gene finding using KAAS (KEGG Automatic Annotation Server) by BLASTp against the KEGG (Kyoto Encyclopedia of Genes and Genomes) GENES database at E-value of 1e-5 and identity cut-off of 70%. Metabolic pathways were reconstructed and filtered using MinPath (Minimal set of Pathways)43 for all the Cellulosimicrobium genomes, i.e. strains MM, LMG16121 and J36. Further, one-way hirerachical clustering was performed on the top 50 variables (i.e. metabolic pathways) and were plotted at relative abundance of 0.8% and standard deviation cut-off of 0.4%. Protein orthologues were determined using pairwise reciprocal smallest distance (RSD) algorithm initially, followed by retrieving the common set in all three genomes44 at E-value of 1e-15 and divergence cut-off of 0.5. Whole genome alignments were created in Mauve 2.4.045 and visualized in Circos46 at 10 Kb minimum cut-off. In order to elucidate the effect of arsenic contamination across microbial mats, arsenic related gene clusters were reconstructed from strain MM’s genome using PSI BLAST and identity cut-off of 25%.
HGT events were determined for three Cellulosimicrobium genomes based on codon usage deviation using Hidden Markov models (HMM) implemented in program SIGI-HMM47. Hierarchical clustering was performed on the annotated HGT loci using Euclidean distance matrix across three Cellulosimicrobium genomes. Putative alien (pA) genes were hence determined on the stretch of genomic islands (GIs) using Viterbi algorithm based on codon usage variations of HGTs from the rest of the genome47. Paired-end metagenomic reads (n = 78,891,278) from the biofilms at Manikaran hot springs21 were mapped over the genome of strain MM using GASSST (Global Alignment Short Sequence Search Tool)48 at sequence similarity cut-off of 85% to allow for Cellulosimicrobium strain MM specific recruitments49. Abundance-weighted average coverage analysis using Nonpareil50 revealed that the metagenome dataset had a genome coverage of 93% against strain MM. Therefore, we believe that our sequencing depth was good enough to represent the in situ microbial diversity.MGIs were annotated as continuous stretches of gaps in the metagenome recruitment plot by subjecting them to ORF prediction and further BLASTp against the NCBI nr database at E-value of 1e-5, minimum bit-score cut-off of 100 and identity cut-off of 70%49.
Determination of pathogenicity islands (PAIs)
Three genomes representing C. cellulans i.e. strains LMG16121, MM and J36 were used for deciphering PAIs following a segregative approach discriminating islands from the core genome on the basis of %G+ C content, dinucleotide frequency and codon usage implemented in program PAI-DA20,51 with window size of 5 Kb per genome. Both %codon usage bias and %G+ C content were plotted against the individual genome to identify skewed “island-like” regions. Simultaneously, PAI-DB v2.052 was used to validate occurrence of these PAI like regions against databases for both pathogenicity islands (PAIs) and resistance islands (REIs) at E-value of 0.01. Given that highly expressed genes (HEGs) such as genes encoding for ribosomal subunits, transcription and terminator genes and repair genes might also exhibit a skewed codon usage and G+ C content20, we used sequence composition information (manual curation) to avoid the false negative predictions. Further, tRNA scan was used to check whether PAIs are flanked by tRNAs to validate the PAIs53.
Assigning functions to PAIs
Protein coding sequences were obtained for the PAIs of all three genomes by FragGeneScan 1.1842. Functions were assigned to the ORFs using BLASTp (E-value = 1e-5) against non-redundant protein database, KEGG54 and Gene Ontology (GO) (Gene Ontology Consortium, 2008) database. Protein sequences were also searched against the Pfam library of hidden Markov models (HMMs) using HMMER55 for family level prediction. Further, the virulent content of the PAIs was determined using multiple databases such as Virulent Factor Database (VFDB)56, VirulentPred22 and MP323. MP3 was used at a threshold value of 0.2 and minimum protein length of 30.The hypothetical proteins which were abundant on PAIs were then checked for their origin using the ACLAME database35.
Molecular clock analysis of PAI gene content
After functional assignment of ORFs predicted on PAIs of the three genomes, we selected three proteins, i.e. Fic, D-alanyl-D-alanine carboxypeptidase and transposase from strain MM, LMG16121 and J36, respectively, to infer the divergence time of most recent common ancestor (MRCA) among multiple bacterial lineages harboring these genes on PAIs. In case where multiple strains of one bacterial species were found to be carrying the gene of interest on PAIs, only single strain was taken into account for tree construction. Fic protein from strain MM was selected for its significant association with pathogenesis57 as well as its frequent occurrence on PAIs in other pathogenic bacterial lineages (n = 9). PAIs from strain LMG16121 were characterized by the repeated presence (n = 9) of ORFs encoding for proteins involved in cell-wall biogenesis out of which D-alanyl-D-alanine carboxypeptidase was chosen for its association with PAIs of other pathogenic bacteria (n = 5). Transposase was also selected for this analysis, for its repeated presence on PAIs of strain J36 (n = 7) as well as forming a significant part of PAIs in other bacterial genera (n = 19). The protein coding sequences for each of these genes (n = 3) was retrieved from Cellulosimicrobium genomes (n = 3) as well as from other bacteria (NCBI) reported for the presence of these genes on PAIs followed by multiple sequence alignment using CLUSTALW. The alignment was then used in BEAST version 1.8.229 to perform Bayesian molecular clock analysis using the following parameters: Clock = Random Local Clock, Substitution model = WAG (for amino acids), Site heterogeneity = Gamma, Tree Prior = Coalescent Constant Size, Length of Monte Carlo Markov Chain (MCMC) = 1000000 and Burnin = 100. Random Local Clock and Gamma distribution (relaxed model) was used to account for maximum heterogeneity in terms of substitutions given inter-genera, diverse nature of the dataset. TreeAnnotator (http://beast.bio.ed.ac.uk/TreeAnnotator) was further used to summarize the information from multiple sample trees generated from BEAST into a single target tree, i.e. “Maximum clade credibility” tree with values for the rate of substitution at 95% Highest Posterior Density (HPD) interval, posterior probability, length and height of 95% HPD interval of the node ages. The final annotated tree was then visualized using FigTree version 1.4.0 (http://tree.bio.ed.ac.uk/software/figtree/).
Statistical analysis
For Cellulosimicrobium genomes undertaken in this study, pairwise correlation was computed between gene centric optimal codon frequencies Fopt (a measure of codon usage bias) and %G+ C content using Pearson Product-Moment Correlation Coefficient (R2) at 95% confidence level based on 791 common orthologues found between three genomes17. Fopt values were calculated using CodonW (version 1.4.4, http://codonw.sourceforge.net) for the 791 orthologous proteins from each of three Cellulosimicrobium genomes. Further, pairwise comparisons between the codon usage bias (measured as Fopt values) were performed between Cellulosimicrobium sp. MM, C. cellulans LMG16121 and C. cellulans J36, using Wilcoxon-Mann-Whitney test with continuity correction to elucidate the significance of variable codon bias patterns across different genomes inhabiting different environments58. To further estimate coupling between selection on codon usage and selection of amino acids, Pearson correlation was computed between Fopt and the dN/dS for orthologous gene pairs (n = 791) between all three genome pairs. For this the mean Fopt value of two orthologous genes was taken as the Fopt value for that gene pair. dN/dS values for each orthologous gene pair was calculated by pairwise aligning protein sequences by CLUSTALW followed by codon to codon alignment of corresponding nucleotide sequences using PAL2NAL59. Further substitution rates were estimated using yn00 module implemented in PAML60. All the above statistical analyses and scatter plotting were performed in R (R Core Team, http://www.R-project.org/).
Accession numbers
Sequence data were obtained for the Cellulosimicrobium genomes from NCBI Genome database: Cellulosimicrobium sp. strain MM [GenBank:NZ_JPQW00000000.1], Cellulosimicrobium cellulans LMG16121 [NZ_CAOI00000000.1], Cellulosimicrobium cellulans J36 [NZ_JAGJ00000000.1], Cellulosimicrobium funkei U11 [NZ_JNBQ00000000.1], Isoptericola variabilis 225 [NC_015588.1], Promicromonospora kroppenstedtii DSM 19349 [NZ_AZXR00000000.1], Promicromonospora sukumoe 327MFSha31 [NZ_ARQM00000000.1] and Xylanimonas cellulosilytica DSM 15894 [NC_013530.1]. 16S rRNA gene sequence data was used from the family Promicromonosporacae under accession numbers: Promicromonospora citrea [X83808.1], Promicromonospora endophytica EUM 273 [GU434253.2], Promicromonospora enterophila [X83807.1], Promicromonospora flava [AM992980.1], Promicromonospora sp. UTMC 792 [JN038073.1], Promicromonospora sukumoe [AB023375.1], Promicromonospora thailandica [AB560974.1], Promicromonospora kroppenstedtii RS16 [AM709608.1], Promicromonospora aerolata [AJ487303.1], Promicromonospora umidemergens [FN293378.1], Promicromonospora vindobonesis [AJ487302.1], Promicromonospora xylanilytica strain YIM61515 [FJ214352.1], Cellulosimicrobium cellulans [X83809.1], Cellulosimicrobium funkei strain W6122 [AY501364.1], Cellulosimicrobium terreum strain DS-61 [EF076760.1], Isoptericola chiayiensis strain 06182M-1 [FJ469988.1], Isoptericola dokdonensis strain DS-3 [DQ387860.1], Isoptericola halotolerans strain YIM 70177 [AY789835.1], Isoptericola hypogeus [AJ854061.1], Isoptericola jiangsuensis strain CLG [EU852101.1], Isoptericola nanjingensis strain H17 [HQ222356.1], Myceligenerans crystallogenes [FR733716.1], Myceligenerans sp. XJEEM 11063 [EU910872.1], Myceligeneris xiligouensis strain XLG9A10.2 [AY354285.1], Xylanibacterium ulmi [AY273185.2], Xylanimicrobium pachnodae VPCX2 [AF105422.1], Xylanimonas cellulosilytica DSM 15894 [CP001821.1] and Cellulomonas aerilata strain 5420S-23 [EU560979.1]. Metagenome sequence (NGS) data were obtained from DDBJ/EMBL/GenBank under the accession number of PRJEB4614 (http://www.ebi.ac.uk/ena/data/view/PRJEB4614).
Additional Information
How to cite this article: Sharma, A. et al. (Meta)genomic insights into the pathogenome of Cellulosimicrobium cellulans. Sci. Rep. 6, 25527; doi: 10.1038/srep25527 (2016).
References
Schumann, P., Weiss, N. & Stackebrandt, E. Reclassification of Cellulomonas cellulans (Stackebrandt and Keddie 1986) as Cellulosimicrobium cellulans gen. nov., comb. nov. Int. J. Syst. Evol. Microbiol. 51, 1007–1010 (2001).
Brown, J. M. et al. Characterization of clinical isolates previously identified as Oerskovia turbata: proposal of Cellulosimicrobium funkei sp. nov. and emended description of the genus Cellulosimicrobium. Int. J. Syst. Evol. Microbiol. 56, 801–804 (2006).
Yoon, J.-H. et al. Identification of Saccharomonospora strains by the use of genomic DNA fragments and rRNA gene probes. Int. J. Syst. Bacteriol. 46, 502–505 (1996).
Sharma, A., Hira, P., Shakarad, M. & Lal, R. Draft genome sequence of Cellulosimicrobium sp. strain MM, isolated from arrsenic-rich microbial mats of a Himalayan hot spring. Genome Announc. 2, e01020–14; doi: 10.1128/genomeA.01020-14 (2014).
Antony, R., Krishnan, K. P., Thomas, S., Abraham, W. P. & Thamban, M. Phenotypic and molecular identification of Cellulosimicrobium cellulans isolated from Antarctic snow. Antonie Van Leeuwenhoek 96, 627–634 (2009).
Petkar, H. et al. Cellulosimicrobium funkei: First report of infection in a nonimmunocompromised patient and useful phenotypic tests for differentiation from Cellulosimicrobium cellulans and Cellulosimicrobium terreum. J. Clin. Microbiol. 49, 1175–1178 (2011).
Dobrindt, U., Hochhut, B., Hentschel, U. & Hacker, J. Genomic islands in pathogenic and environmental microorganisms. Nat. Rev. Microbiol. 2, 414–424 (2004).
Price, M. N., Huang, K. H., Arkin, A. P. & Alm, E. J. Operon formation is driven by co-regulation and not by horizontal gene transfer. Genome Res. 15, 809–819 (2005).
Konstantinidis, K. T. & Tiedje, J. M. Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. USA 102, 2567–2572 (2005).
Wu, M. & Scott, A. J. Phylogenomic analysis of bacterial and archaeal sequences with AMPHORA2. Bioinformatics 28, 1033–1034 (2012).
Segata, N., Börnigen, D., Morgan, X. C. & Huttenhower, C. PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat. Commun. 4, 2304; doi: 10.1038/ncomms3304 (2013).
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C. & Kanehisa, M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–185 (2007).
Kim, S. et al. RNAseq-based transcriptome analysis of Burkholderia glumae quorum sensing. Plant Pathol. J. 29, 249–259 (2013).
Mosier, A. C. et al. Metabolites associated with adaptation of microorganisms to an acidophilic, metal-rich environment identified by stable-isotope-enabled metabolomics. MBio 4, e00484–00412; doi: 10.1128/mBio.00484-12 (2013).
Jelsbak, L. et al. Identification of metabolic pathways essential for fitness of Salmonella Typhimurium in vivo. PLos One 9, e101869; doi: 10.1371/journal.pone.0101869 (2014).
Schmidt, H. & Hensel, M. Pathogenicity islands in bacterial pathogenesis. Clin. Microbiol. Rev. 17, 14–56 (2004).
Ran, W., Kristensen, D. M. & Koonin, E. V. Coupling between protein level selection and codon usage optimization in the evolution of bacteria and archaea. MBio 5, e00956–14; doi: 10.1128/mBio.00956-14 (2014).
Bohlin, J., Brynildsrud, O., Vesth, T., Skjerve, E. & Ussery, D. W. Amino acid usage is asymmetrically biased in AT- and GC-rich microbial genomes. PLos One 8, e69878; doi: 10.1371/journal.pone.0069878 (2013).
Lassalle, F. et al. GC-Content evolution in bacterial genomes: the biased gene conversion hypothesis expands. PLos Genet. 11, e1004941; doi: 10.1371/journal.pgen.1004941 (2015).
Che, D., Hasan, M. S. & Chen, B. Identifying Pathogenicity Islands in bacterial pathogenomics using computational approaches. Pathogens 3, 36–56 (2014).
Sangwan, N. et al. Arsenic rich Himalayan hot spring metagenomics reveal genetically novel predator-prey genotypes. Environ. Microbiol. Rep. 7, 812–823 (2015).
Garg, A. & Gupta, D. VirulentPred: a SVM based prediction method for virulent proteins in bacterial pathogens. BMC Bioinformatics 9, 62; doi: 10.1186/1471-2105-9-62 (2008).
Gupta, A., Kapil, R., Dhakan, D. B. & Sharma, V. K. MP3: a software tool for the prediction of pathogenic proteins in genomic and metagenomic data. PLos One 9, e93907; doi: 10.1371/journal.pone.0093907 (2014).
Schmid, M. C. et al. A translocated bacterial protein protects vascular endothelial cells from apoptosis. PLos Pathog. 2, e115; doi: 10.1371/journal.ppat.0020115 (2006).
Michel, A. et al. Global regulatory impact of ClpP protease of Staphylococcus aureus on regulons involved in virulence, oxidative stress response, autolysis and DNA repair. J. Bacteriol. 188, 5783–5796 (2006).
Kovačić, F. et al. Structural and functional characterisation of TesA - a novel lysophospholipase A from Pseudomonas aeruginosa. PLos One 8, e69125; doi: 10.1371/journal.pone.0069125 (2013).
Pradeepkiran, J. A., Kumar, K. K., Kumar, Y. N. & Bhaskar, M. Modeling, molecular dynamics and docking assessment of transcription factor rho: a potential drug target in Brucella melitensis 16M. Drug Des. Devel. Ther. 9, 1897–1912 (2015).
Engel, P. et al. Adenylylation control by intra- or intermolecular active-site obstruction in Fic proteins. Nature 482, 107–110 (2012).
Drummond, A. J. & Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214; doi: 10.1186/1471-2148-7-214 (2007).
Bentley, S. D. et al. Genome of the actinomycete plant pathogen Clavibacter michiganensis subsp. sepedonicus suggests recent niche adaptation. J. Bacteriol. 190, 2150–2160 (2008).
Weinstock, D. M. & Brown, A. E. Rhodococcus equi: an emerging pathogen. Clin. Infect. Dis. 34, 1379–1385 (2002).
Mishra, S. et al. Cloning, expression, purification and biochemical characterisation of the FIC motif containing protein of Mycobacterium tuberculosis. Protein Expr. Purif. 86, 58–67 (2012).
Hung, W., Jane, W.-N. & Wong, H. Association of a D-alanyl-D-alanine carboxypeptidase gene with the formation of aberrantly shaped cells during the induction of viable but nonculturable Vibrio parahaemolyticus. Appl. Environ. Microbiol. 79, 7305–7312 (2013).
Giordano, J. et al. Evolutionary History of Mammalian Transposons Determined by Genome-Wide Defragmentation. PLos Comput. Biol. 3, e137; doi: 10.1371/journal.pcbi.0030137 (2007).
Leplae, R., Hebrant, A., Wodak, S. J. & Toussaint, A. ACLAME: A CLAssification of Mobile genetic Elements. Nucleic Acids Res. 32, D45–D49 (2004).
Michell, R. H. Inositol lipids: from an archaeal origin to phosphatidylinositol 3,5-bisphosphate faults in human disease. FEBS J. 280, 6281–6294 (2013).
Reynolds, T. B. Strategies for acquiring the phospholipid metabolite inositol in pathogenic bacteria, fungi and protozoa: making it and taking it. Microbiology 155, 1386–1396 (2009).
Ollinger, J., O’Malley, T., Kesicki, E. A., Odingo, J. & Parish, T. Validation of the essential ClpP protease in Mycobacterium tuberculosis as a novel drug target. J. Bacteriol. 194, 663–668 (2012).
Thompson, J. D., Higgins, D. G. & Gibson, T. J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680 (1994).
Felsenstein, J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol. 17, 368–376 (1981).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013).
Rho, M., Tang, H. & Ye, Y. FragGeneScan: predicting genes in short and error-prone reads. Nucleic Acids Res. 38, e191; doi: 10.1093/nar/gkq747 (2010).
Ye, Y. & Doak, T. G. A Parsimony Approach to Biological Pathway Reconstruction/Inference for Genomes and Metagenomes. PLos Comput. Biol. 5, e1000465; doi: 10.1371/journal.pcbi.1000465 (2009).
Wall, D. P. & Deluca, T. Ortholog detection using the reciprocal smallest distance algorithm. Methods Mol. Biol. 396, 95–110 (2007).
Darling, A. C. E., Mau, B., Blattner, F. R. & Perna, N. T. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403 (2004).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
Waack, S. et al. Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinformatics 7, 142; doi: 10.1186/1471-2105-7-142 (2006).
Rizk, G. & Lavenier, D. GASSST: global alignment short sequence search tool. Bioinformatics 26, 2534–2540 (2010).
Sharma, A. et al. Pan-genome dynamics of Pseudomonas gene complements across hexachlorocyclohexane dumpsite. BMC Genomics 16, 313; doi: 10.1186/s12864-015-1488-2 (2015).
Rodriguez-R, L. M. & Konstantinidis, K. T. Nonpareil: a redundancy-based approach to assess the level of coverage in metagenomic datasets. Bioinformatics 30, 629–635 (2014).
Tu, Q. & Ding, D. Detecting pathogenicity islands and anomalous gene clusters by iterative discriminant analysis. FEMS Microbiol. Lett. 221, 269–275 (2003).
Yoon, S. H., Park, Y.-K. & Kim, J. F. PAIDB v2.0: exploration and analysis of pathogenicity and resistance islands. Nucleic Acids Res. 43, D624–630 (2015).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Kanehisa, M. The KEGG database. Novartis Found. Symp. 247, 91–101; Discussion 101–103, 119–128, 244–252 (2002).
Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37 (2011).
Chen, L. et al. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 33, D325–328 (2005).
Ma, Z. et al. Insight into the specific virulence related genes and toxin-antitoxin virulent pathogenicity islands in swine streptococcosis pathogen Streptococcus equi ssp. zooepidemicus strain ATCC35246. BMC Genomics 14, 377; doi: 10.1186/1471-2164-14-377 (2013).
Torre, A. R. D. L., Lin, Y.-C., Peer, Y. V. de & Ingvarsson, P. K. Genome-wide analysis reveals diverged patterns of codon bias, gene expression and rates of sequence evolution in Picea gene families. Genome Biol. Evol. 7, 1002–1015 (2015).
Suyama, M., Torrents, D. & Bork, P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34, W609–612 (2006).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Acknowledgements
The authors acknowledge funds from Department of Biotechnology (DBT) and National Bureau of Agriculturally Important Microorganisms (NBAIM). AS gratefully acknowledge National Bureau of Agriculturally Important Microorganisms (NBAIM) for providing research fellowship. This paper was partly written during the visit by R.L. under DST-DAAD project to Germany (Helmholtz Zentrum für Umweltzforschung-UFZ, Leipzig).
Author information
Authors and Affiliations
Contributions
A.S. and R.L. performed the data analysis and data interpretation. A.S. prepared the figures. R.L., A.S. and J.A.G. wrote the manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Sharma, A., Gilbert, J. & Lal, R. (Meta)genomic insights into the pathogenome of Cellulosimicrobium cellulans. Sci Rep 6, 25527 (2016). https://doi.org/10.1038/srep25527
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep25527
This article is cited by
-
Bioinformatic Approach to Investigate Larvae Gut Microbiota Cellulosimicrobium protaetiae via Whole-Genome Analysis
Molecular Biotechnology (2024)
-
Genome sequence analysis and bioactivity profiling of marine-derived actinobacteria, Brevibacterium luteolum, and Cellulosimicrobium funkei
Archives of Microbiology (2021)
-
Cellulosimicrobium fucosivorans sp. nov., isolated from San Elijo Lagoon, contains a fucose metabolic pathway linked to carotenoid production
Archives of Microbiology (2021)
-
Taxonomically Characterized and Validated Bacterial Species Based on 16S rRNA Gene Sequences from India During the Last Decade
Indian Journal of Microbiology (2020)
-
Comparative metagenomic analyses of a high-altitude Himalayan geothermal spring revealed temperature-constrained habitat-specific microbial community and metabolic dynamics
Archives of Microbiology (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
