
Abstract
Microbes play key roles in shaping the physiology of insects and can influence behavior, reproduction and susceptibility to pathogens. In Sub-Saharan Africa, two major malaria vectors, Anopheles gambiae and An. coluzzii, breed in distinct larval habitats characterized by different microorganisms that might affect their adult physiology and possibly Plasmodium transmission. We analyzed the reproductive microbiomes of male and female An. gambiae and An. coluzzii couples collected from natural mating swarms in Burkina Faso. 16S rRNA sequencing on dissected tissues revealed that the reproductive tracts harbor a complex microbiome characterized by a large core group of bacteria shared by both species and all reproductive tissues. Interestingly, we detected a significant enrichment of several gender-associated microbial biomarkers in specific tissues, and surprisingly, similar classes of bacteria in males captured from one mating swarm, suggesting that these males originated from the same larval breeding site. Finally, we identified several endosymbiotic bacteria, including Spiroplasma, which have the ability to manipulate insect reproductive success. Our study provides a comprehensive analysis of the reproductive microbiome of important human disease vectors, and identifies a panel of core and endosymbiotic bacteria that can be potentially exploited to interfere with the transmission of malaria parasites by the Anopheles mosquito.
Similar content being viewed by others

Introduction
The interplay between hosts and their endogenous microbiota is crucial for the physiology of a vast number of organisms ranging from mammals to plants1. In insects, the resident microbiome regulates nutrition, digestion, metabolism, reproduction, longevity and immunity2,3. The insect microbiota participate in the synthesis of essential nutrients that are scarce or unavailable in the host diet4, the production of enzymes increasing digestion efficiency5, and the provision of vitamins6. Microorganisms can also detoxify harmful chemicals such as flavonoids, tannins, and alkaloids present in plants, enabling the survival of many insects7.
In insect vectors of human diseases, the study of the microbiome holds tremendous potential for the development of microbial-based strategies to control vector-borne pathogens8,9. Prior microbiome studies of the Anopheles species that transmit malaria have focused on the analysis of the female midgut, the first tissue where Plasmodium parasites establish infection. Anopheles gut symbiotic bacteria dramatically influence sporogonic development of the most deadly malaria parasite, Plasmodium falciparum10,11,12,13,14, by either stimulating the host immune response13 or via the generation of damaging free radicals14. Interactions between Anopheles and the gut microbiota are complex as some bacteria interfere with parasite development12, while antibiotic-mediated perturbation of gut bacteria can increase P. falciparum infection rates15. Resistance to Plasmodium has also been genetically engineered using bacterial symbionts secreting anti-parasite molecules in the gut16, an approach known as paratransgenesis (reviewed in17).
In contrast with the evolving literature on Anopheles gut microbiota, the microbiome associated with insect reproductive organs has been largely unexplored, aside from studies focusing on Asaia and Wolbachia. Wolbachia, a maternally transmitted intracelullar bacterium residing in the ovaries and testes of nearly 60% of insect species, influences host reproductive success, immunity and lifespan, and can limit the development of vector-borne pathogens such as dengue virus and Plasmodium18,19. Recently, maternally transmitted Wolbachia infections were found in natural populations of An. gambiae and An. coluzzii, opening novel possibilities for malaria control20. Asaia, a bacterial genus populating both the male and female reproductive tracts of Anopheles as well as the gut21,22,23, has been proposed for paratransgenesis strategies given its ability to be paternally and maternally transmitted21,22,23.
Besides their role in pathogen resistance, reproductive tissue microbes may affect reproductive fitness and mating preferences. In Drosophila melanogaster, assortative mating was associated with symbiotic bacteria via the microbiota’s effects on cuticle mating pheromone synthesis24. Assortative mating behavior has been observed between An. gambiae and An. coluzzii, two closely related species (previously named S and M molecular forms, respectively) that live in sympatry over large areas in Sub-Saharan Africa but are only occasionally found to swarm together and form hybrids at very low frequencies25,26,27. These species are adapted to largely diverse larval habitats, with An. coluzzii using permanent breeding grounds like rice fields and An. gambiae predominantly found in temporary breeding sites28. Besides possible genetically-determined differences29 in cuticular hydrocarbon profiles30 and other mechanisms for reproductive isolation such as divergence in wing beat frequencies31, distinctive adult reproductive tract microbiomes fostered by different larval habitats may have favored the emergence of pre-mating barriers between these species, explaining the low prevalence of hybrids found in natural populations.
Such largely dissimilar habitats occupied during larval development may foster distinctive adult reproductive tract microbiomes, possibly creating pre-mating barriers and explaining the low prevalence of hybrids found in natural populations.
Microbiome surveys of the anopheline reproductive tracts have not been previously reported, despite the potential relevance for both reproductive success and mating ecology. Here we performed 16S rRNA amplicon sequencing and DNAseq investigations of the microbiomes populating male and female reproductive tissues of An. gambiae and An. coluzzii mosquitoes collected from natural mating swarms. Our results reveal that these two species share a highly abundant core microbiome present in both male and female tissues that could be exploited for paratransgenesis-based control strategies. Moreover, identification of swarm-specific microbial biomarkers suggests a spatial connection between larval breeding sites and swarm locations and provides unexpected insights into the mating biology of these important malaria vectors.
Results
16S rRNA amplicon sequencing of reproductive tissues from Anopheles gambiae and An. coluzzii mating couples
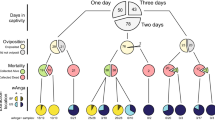
To characterize the reproductive tract microbiomes of An. gambiae and An. coluzzii adults, mating couples were collected in natural swarms in three villages near Bobo-Dioulasso, Burkina Faso: Vallée du Kou 5 (VK5) and Vallée du Kou 7 (VK7), highly populated by An. coluzzii, and Soumousso, where An. gambiae are predominant (see Fig. 1 and Supplementary Table S1). Four reproductive tissues were isolated: the testes and male accessory glands (MAGs) from males, and the ovaries and lower reproductive tract (LRT, which comprises the atrium, the spermatheca and the parovarium) from females. We collected mating couples rather than resting males and females as we wanted to specifically study sexually active individuals. While the vast majority of collections (26 out of 30 mating couples) were composed of conspecific couples, we found 4 mixed An. gambiae/An. coluzzii couples, providing a 13.3% frequency of interspecific matings similar to previously reported hybridization frequencies in this geographical area25.

Samples were subjected to 16S rRNA amplicon sequencing (variable region V4, see Methods), and sequences were processed with diversity, taxonomic, and functional profiling pipelines32,33 to characterize the microbiome structure, composition, and variability. A number of samples (18) failed the analysis, providing a final number of 102 tissues successfully sequenced (Supplementary Table S2).
The male and female reproductive tracts share a large core microbiome
We detected a high intra-sample diversity (i.e. alpha-diversity, defined as the number of different organisms, Supplementary Fig. S1) in the microbiome of the four reproductive tissues analyzed, with no clear differences driven by gender or species. At a subsampling rate of 3,500 sequences, we estimated a number of species-level sequence clusters (Operational Taxonomic Units, OTUs) in each sample ranging from 240 to 763 (average 500 s.d. 96). This overall high diversity was driven by a large number of OTUs present at low prevalence (i.e. fraction of positive samples) but high relative abundance (i.e. fraction of reads belonging to a given OTU in a sample, Supplementary Fig. S2).
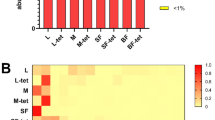
Despite this large intra-sample variability, we identified a highly conserved core microbiome present in all samples and comprising OTUs spanning seven different bacterial genera with variable levels of abundance (Fig. 2). Acinetobacter, and specifically OTU 4482598, was the quantitatively dominant microorganism in the majority of the samples (avg. 16.3% s.d. 17.8%, min 0.4%, max 73.3%). This OTU matched the corresponding fragment of several distinct Acinetobacter lwoffii sequences stored in the public repositories, although some identical matches were also detected for other closely related species and unnamed organisms in the same genus. Other OTUs present in the core microbiome included Pseudomonas and Staphylococcus, genera containing species ubiquitous to a large number of environmental and host-associated habitats. Acinetobacter, Pseudomonas and Staphylococcus have also been detected at lower percentages in the midgut of An. gambiae females11 and other anophelines34. Two OTUs assigned to the Enterobacteriaceae were also present in all samples; the 16S rRNA sequence for bacteria in this family can be inconclusive at the genus level, but several organisms in this clade have been reported to colonize the Anopheles gut11,35 with potential interaction with Plasmodium11,14,36. Corynebacterium was also identified in all samples. As in previous sequencing studies with similar detection sensitivity11 this genus was only occasionally found in the Anopheles midgut, our finding possibly reflects specificity for reproductive tissues or for our geographical cohort.

(A) Genera from both Gram-positive (Staphylococcus, Corynebacterium, Geobacillus, Micrococcus) and Gram-negative (Acinetobacter, Pseudomonas) bacteria constitute the core microbiome of the A. gambiae (Agam) and A. coluzzii (Acol) male and female reproductive tissues collected from three villages (VK5, VK7 and Soumousso). (B) Different core operational taxonomic units (OTUs) are present within the same genus, suggesting that a consistent species-level microbial diversity characterizes the reproductive tract microbiome. Gender, tissues (Ovaries and Lower Reproductive Tract for females, Testes and Male Accessory Glands for males), villages (Soumousso, VK5 and VK7), species (An. gambiae: Agam; and An. coluzzii: Acol) and swarm types (individual species or mixed) are color-coded as described in the legend on the left of the figure.
We found little evidence of qualitative or quantitative differences in microbial diversity between An. gambiae and An. coluzzii. Some potentially discriminatory (p-value < 0.01) microorganisms including members of Geobacillus, Bacillus, Desemzia, Oscillospira and Burkholderia were more abundant in An. gambiae, while some OTUs in the Corynebacterium, Phycicoccus, Proteiniclasticum, Nesterenkonia, Macrococcus were more associated with An. coluzzii (0.01 ≤ p-value < 0.02). None of these OTUs, however, were significantly differentiated between species (p-value > 0.05) following correction for multiple hypotheses testing.
Whole-metagenome shotgun sequencing (WGS)37 of three An. coluzzii reproductive tissues confirmed our results and identified Acinetobacter lwoffii as the species in the Acinetobacter genus previously detected by 16S rRNA sequencing (Fig. 3A,B). Our A. lwoffii reads provided almost complete genome coverage and placed the Anopheles strain close to the previously published NIPH 478 (Fig. 3B). Escherichia coli was the prevalent microorganism in the Enterobacteriaceae family, and Propionibacterium acnes were identified in all three samples. We observed lower intra-sample diversity than in the 16S rRNA dataset likely due to the reduced sensitivity achievable by WGS, especially given the high fraction of host DNA contamination. This may explain the absence of Acinetobacter in one of three samples sequenced (Fig. 3A).

(A) Species level abundances as estimated by MetaPhlAn highlight the presence of Acinetobacter lwoffi, Propionibacterium acnes, Kocuria, Pseudomonas, Kocuria rhizophila and Micrococcus luteus in at least two of the three samples. The other organisms present in one sample only are Streptococcus mitis, Stenotrophomonas maltophilia and Aereococcus vididans. (B) Mapping of the metagenomic sample with highest A. lwoffii abundance against the genetically closest genome in the species (NIPH 478) highlights a consistent coverage of the large majority of the genes.
Female and male reproductive tracts harbor quantitatively distinct microbial populations
While core bacteria were common to female and male reproductive organs, their quantitative distribution differed between genders. Acinetobacter (OTU 4482598 and several other OTUs) drove the sample clustering and were consistently more abundant in the female reproductive tract using the linear discriminant effect size tool, LEfSe38 (uncorrected p < 1e-5, Fig. 4A). In contrast, Enterobacteriaceae and Aerococcaceae OTUs were significantly higher in male reproductive tissues (p < 0.01). When expanding this analysis to non-core OTUs, LEfSe detected additional microbial biomarkers associated with female (Desemzia and Granulicatella) or male (Agrobacterium, Pseudomonas, Bacteroides, and Cloacibacterium) reproductive tissues (Fig. 4A).

Tissue-enriched microbial clades were found in each of the four reproductive organs (Fig. 4B). The LRT, in particular, was enriched in bacteria from the phylum Proteobacteria (order Gammaproteobacteria) and in Acinetobacter and environmental uncultivable bacteria from the SC3 and TM7 candidate phyla clades. We also identified organisms with evidence of niche adaptation to the MAGs (Ruminococcus and Trabulsiella, an Enterobacteraceae that resembles Salmonella), testes (Neisseria, a colonizers of mucosal surfaces of many organisms, as well as members of the Intrasporangiaceae family), and ovaries (order Deinococcales). The finding of tissue-specific tropism is in line with results from other hosts39 including humans40. Future studies will be needed to determine whether these microorganisms exert a functional role in modulating the biochemical properties of mosquito reproductive tissues.
An average of 46 (s.d. 10) distinct OTUs with a relative abundance higher than 0.1% were shared between the reproductive tissues of each sex (Supplementary Fig. 3).
The microbiome diversity in the cohort is enriched by highly variable and sample-specific taxa
Even in the presence of a large core microbiome, we observed high alpha diversity (i.e. intra-sample diversity) due to the presence of many bacteria with partial prevalence but occasional high abundance. Many OTUs showed a large coefficient of variation across samples (Fig. 5), including members of Enterobacteriaceae, Pseudomonadaceae, Achromobacter, and Deftia. These highly variable OTUs were not statistically correlated with any of the metadata available (gender, species, tissue, village), which may suggest they are not strictly required for normal micro-ecology and host-microbiome homeostasis, and may result from local environmental acquisition events, specific amplification in favorable conditions, or horizontal transmission.

The 30 OTUs with highest score are reported here after computing the coefficient of variation for all the OTUs in our dataset. Each value in the heatmap represents the relative abundance of an OTU in a sample.
Some highly abundant microorganisms were found only in a very small fraction of samples. This group included endosymbionts (see below) and other genera such as Leuconostoc, a lactic acid-producing coccus, Arcobacter, Ruminococcus and Lautropia, that may be considered opportunistic colonizers (Supplementary Table 4). Given their ability to occasionally invade the reproductive tracts at high relative abundances, these microorganisms might be potentially used for microbiome perturbation strategies aimed at interfering with the physiological host-microbiome homeostasis.
We found an enrichment in specific bacteria in some swarm locations. Three males collected in the VK7 village from the same mating swarm (swarm 2.3, adjacent to a rice paddy, Fig. 1) showed a highly significant enrichment in Shewanella, Rhodocyclacea, Pseudomonas and Azospira in both testes and MAGs (Fig. 6). These bacteria were detected at much lower abundance in females mated to these males, as well as in males and females from swarm locations at the other end of the VK7 village or in the other villages. Shewanella, Rhodocyclacea and Pseudomonas share some genetic similarities with each other and are often found in water, including rice fields, while some Azospira genera are root bacteria with metal bioactivity. As the reproductive microbiome of adults is largely shaped during larval development, it is likely that local environmental factors have favored the proliferation of these microbes in the rice fields surrounding the swarm location. These findings suggest the hypothesis that males from the same larval breeding sites may tend to swarm together. Genetic analyses will be needed to test the hypothesis of a possible degree of kinship within male swarms.

Bar plots represent the relative abundance (indicated on the Y-axis) of Shewanella, Rhodocyclaceae, Pseudomonas, and Azospira in mosquito samples from three villages (VK5, VK7 and Soumousso, indicated by different color codes. VK5: green; VK7: blue; Soumousso: pink). Each swarm is identified by a numerical code (top bar). Swarm location is provided in Fig. 1. Reproductive tissues collected from male and female individuals from each swarm are represented by color-coded bars. Ovaries: pink; Lower Reproductive Tract (LRT): yellow; Male Accessory Glands (MAGs): green; Testes: blue. Species are indicated by a different bar outline color (An. gambiae: red; An. coluzzii: black). Shewanella, Rhodocyclaceae, Pseudomonas, and Azospira were highly enriched in male tissues (both MAGs and testes) from a specific swarm (swarm 2.3) from the VK7 village. Please note that some tissues collected from females from swarm 2.3 failed the 16S rRNA sequencing. A detailed list of tissues sequenced for each swarm is provided in Supplementary Table 2.
Endosymbionts with potential relevance for malaria control are present at partial prevalence
Among the organisms present at high relative abundance in a small fraction of the samples were the endosymbionts Asaia23, Rickettsia, Spiroplasma41, Thorsellia anophelis42 and the previously characterized Wolbachia20 (Table 1). Bacterial endosymbionts which live within the tissues or cells of their host are widespread across arthropods species43 and generally persist by maternal transmission. Asaia were detected in only 5% of the collected samples (Table 1), in contrast with prior reports on laboratory colonies and field populations39. We did, however, detect members of the Acetobacteraceae family in 72% of the samples, with OTUs assigned to the genera Acetobacter, Gluconobacter, Roseococcus, and Roseomonas, all acetic acid-producing bacteria known to colonize a wide range of insects39. Spiroplasma, an endosymbiont that can colonize germline cells and manipulate host reproductive output44, was identified in the LRT of an An. gambiae female and, at lower abundances, in the testes of an An. coluzzii male (Supplementary Fig. 4). To our knowledge, this is the first time that Spiroplasma has been identified in the Anopheles reproductive tract. In addition, the gammaproteobacterium Thorsellia anopheles was detected in ovaries and testes from both species (Table 1). T. anophelis was first identified in the midgut of An. arabiensis in central Kenya45 and appears to have adapted to the female anopheline midgut by utilizing blood and tolerating the alkaline conditions present in this tissue42. Thorsellia is closely related to the genus Arsenophonus42 which comprises endosymbionts of arthropod species46.
Discussion
Our analysis of the reproductive tract microbiomes of two major malaria vectors reveals the presence of a large core microbiome spanning seven bacterial genera (12 OTUs) shared by all tissues (Fig. 2). When relaxing the definition of core microbiome by considering organisms present in at least 90% of the individuals, the number of common reproductive tract colonizers expands to 54 OTUs. Given the number of samples and their diversity in terms of host species (An. gambiae and An. coluzzii), tissues (MAGs, testes, LRT, ovaries), gender, and geographical origin (three villages and several distinct swarms), our findings suggest that the core reproductive microbiome identified here may be shared by other anopheline populations, with broad implications for using these bacteria to stop malaria transmission by the mosquito vector.
Our original hypothesis of different reproductive microbiomes populating An. gambiae and An. coluzzii was not supported by our data. Although surprising given the vastly diverse larval habitats occupied by these two species, this result may be due to the depth of 16S rRNA analysis, and therefore we cannot conclusively rule out a role of the reproductive microbiome in mediating adaptation to different ecological niches or in shaping specific adult behaviors including mating. Our analyses, however, unexpectedly revealed a number of bacteria (Shewanella, Rhodocyclacea, Pseudomonas and Azospira) selectively enriched in males from the same swarm, a mixed swarm containing both An. gambiae and An. coluzzii males. These two mosquito species mate in swarms that are formed at dusk in specific locations which are conserved during the same season and even across different years47. How mosquitoes recognize these specific locations is not known but choice is thought to depend on how sites are attractive to females, for instance for blood feeding opportunities27. Swarm 2.3 from which those males were captured is next to a rice field (Fig. 1), presumably representing the nearest breeding sites available for larval development. Apart from swarm 2.2, which is situated in close proximity to swarm 2.3 but from which only one male tissue could be sequenced (Fig. 6, Supplementary Table 2), all other swarms from VK7 are located in the opposite end of the village and may be associated with different breeding sites (Fig. 1). The four bacterial genera enriched in males from swarm 2.3 can all grow in rice paddies, and it is plausible that the specific microenvironment of neighboring rice paddies specifically favored their proliferation. As bacteria are likely to colonize reproductive tissues during larval development, our observation of males (but not females) from the same swarm sharing specific bacteria suggests these males originated from the same breeding site, indicating a possible degree of kinship between males in swarms. Females are thought to fly over longer distances than males48, potentially explaining why the same bacteria were not detected in the reproductive tract of the single female that we analyzed from that swarm. An alternative explanation is bacterial tropism for male rather than female reproductive tissues. Further studies are needed to test this hypothesis and to further unravel the mating ecology of these mosquitoes.
A number of endosymbionts were identified that may have reproductive interactions with their mosquito hosts. We identified Spiroplasma bacteria in the testes and ovaries of two individuals. Endosymbiotic Spiroplasma infect approximately 5–15% of all insect species43,49, living primarily in the hemolymph and gut41 from where they can be horizontally transmitted50. Spiroplasma infections have been detected in An. gambiae45, An. funestus45 and several species of the Aedes and Culex genera51,52,53,54, but this may be the first time they have been identified in the reproductive tract of malaria mosquitoes. These bacteria can persist in their female host through two main strategies: either providing an indirect fitness advantage to females by inducing male killing55 or by directly protecting the host against natural pathogens44,56. Therefore, Spiroplasma infections possess two key characteristics that might be exploited for disease control, namely an ability to spread through mosquito populations and a protective function against pathogens.
Contrary to higher estimates previously obtained using targeted PCR investigations39, we detected Asaia only in 5% of samples. This apparent inconsistency could be explained by a potential positive bias of the PCR primers used in the 16S rRNA sequencing for the entire Acetobacteraceae family that comprises Asaia. Many OTUs could only be assigned at the family level, which suggests either the presence of unknown genera in this family or the existence of organisms belonging to subclades of known genera (including Asaia) that are still uncharacterized in the 16S rRNA databases. We hypothesize that bacteria from Asaia and very closely related organisms are indeed common colonizers of the reproductive tracts of An. gambiae and An. coluzzii, and that the current characterization and sampling of Asaia is not capturing the overall in-field diversity of this genus. As Asaia is also identified in the mosquito gut and has been proposed for paratransgenesis strategies for its ability to be paternally and maternally transmitted21,22,23, it will be important to accurately determine its prevalence in natural Anopheles populations.
The finding of a core reproductive microbiome is highly relevant for blocking Plasmodium infections. Paratransgenesis approaches have been proposed to control the transmission of malaria parasites through the use of bacteria producing anti-Plasmodium agents17. The strength of these approaches depends on factors including the ability to re-engineer bacterial genomes, the infectivity of the bacteria for mosquitoes, and the fitness costs associated with infection that affect their ability to spread through insect populations. Our study identified several candidates in the core microbiome that could be used for this purpose. Of particular interest is the widespread presence of A. lwoffii, a relatively well-characterized, cultivable and non-pathogenic bacterium commonly found in the rhizosphere that has already been proposed as a biocontrol agent for plant protection and has been found in the midgut of An. gambiae11 and other anophelines34. The strain we have identified in field Anopheles populations is highly abundant in the reproductive organs and may have potential for paratransgenic approaches. At a time when widespread resistance to all classes of insecticides currently used for mosquito control is threatening the success of our best weapons against malaria57, these and other core bacteria may furnish novel and desperately needed tools for the control of Plasmodium transmission in mosquito populations.
Materials and Methods
Mosquito collections and rRNA sequencing
Mosquito samples were collected during August-September 2011 in three villages near Bobo-Dioulasso, Burkina Faso. Two villages in Vallée du Kou, VK5 (11°23′N; 4°24′W), which is delimited by rice fields, and VK7 (11°24′N; 04°24′W) surrounded by savannah to the North and rice fields to the South, where An. coluzzii (M form) is more abundant58. The village of Soumousso (11°00′N; 4°02′W) is characterized by savannah and by temporary breeding sites that are more favorable to An. gambiae (S form)58. We collected 30 mating couples in copula from different swarms and we dissected the male and female reproductive tracts 1 to 3 h later. Reproductive tissues included the testes and male accessory glands (MAGs) for males, and the ovaries and lower reproductive tract (LRT, which comprises the atrium, the spermatheca and the parovarium) for females.
For An. gambiae and An. coluzzii species determination, DNA from legs was extracted by incubating an individual leg in 40 μl of grinding buffer (10 mM Tris-HCl pH 8.2, 1 mM EDTA, 25 mM NaCl) with 0.2 mg/ml proteinase K for 45 min at 37 °C, then 5 min at 95 °C. One μl of each DNA extract was then used for the locus S200 × 6.1 PCR amplification. DNA extraction and 16S rRNA sequencing were performed as described in20.
Genomic DNA was extracted using DNeasy (Qiagen) and subjected to 16S rRNA amplifications similar to59. Briefly, primers comprising a sample barcode sequence and the Illumina adapters were used to allow directional sequencing covering of the variable region V4 (15F: 5′GTGCCAGCMGCCGCGGTAA-3′; and 806R: 5′GGACTACHVGGGTWTCTAAT-3′). PCR reactions included 10 μl of DNA template diluted 1:50, 10 μl of HotMasterMix with the HotMaster Taq DNA Polymerase (5 Prime), and 5 μl of primer mix (2 μM of each primer). The cycling conditions were: 94 °C for 3 min, 30 cycles at 94 °C for 45 sec, 50 °C for 60 sec, 72 °C for 5 min, and a final 72 °C for 10 min. DNA amplification was quantified and pooled in equimolar concentrations on the Pippin Prep (Sage Sciences, Beverly, MA) with size selected (375–425 bp) to minimize non-specific amplification products from host DNA. A final library size and quantification was done on an Agilent Bioanalyzer 2100 DNA 1000 chips (Agilent Technologies, Santa Clara, CA). Sequencing was performed on the Illumina MiSeq v2 platform, and paired-end reads of 175b in length in each direction were generated. The overlapping paired-end reads were stitched together (approximately 97 bp overlap), and size selected to reduce non-specific amplification products from host DNA (225–275 bp).
16S rRNA and whole-genome shotgun sequencing and analysis
MiSeq sequencing of the V4 variable region of the 16S rRNA gene generated a total of 2.32 M reads (average 22,560, s.d. 14,471 reads per sample) from 102 high-quality samples (18 samples were removed because of technical failures). This dataset was pre-processed and analyzed with the Qiime pipeline32 for taxonomic composition, alpha diversity, and beta diversity analysis32,33, as previously described20, to characterize the microbiome structure, composition, and variability.
Three samples were also subject to deep shotgun metagenomic sequencing (Illumina HiSeq 2000 with NexteraXT Library preparation) generating a total of 1,29 M reads (avg 431 M, s.d. 22 M, Supplementary Table S3), that were processed for host DNA removal with BowTie260 against the Anopheles genomes, species-level taxonomic profiling with MetaPhlAn33,61, coverage analysis with SAMTools62, and biomarker discovery with LEfSe38. Cladograms and phylogenetic trees were displayed using GraPhlAn63.
Additional Information
Accession codes: Sequence reads for the 16S-rRNA amplicon samples have been deposited in the NCBI Sequence Read Archive (SRA) under accession code SRR610826. The three shotgun metagenomic samples are available in the NCBI Sequence Read Archive (SRA) under accession codes PRJNA244534 andSRX1631720 .
How to cite this article: Segata, N. et al. The reproductive tracts of two malaria vectors are populated by a core microbiome and by gender- and swarm-enriched microbial biomarkers. Sci. Rep. 6, 24207; doi: 10.1038/srep24207 (2016).
Accession codes
References
Cabreiro, F. & Gems, D. Worms need microbes too: microbiota, health and aging in Caenorhabditis elegans. EMBO molecular medicine 5, 1300–1310, 10.1002/emmm.201100972 (2013).
Dillon, R. J. & Dillon, V. M. The gut bacteria of insects: nonpathogenic interactions. Annual review of entomology 49, 71–92, 10.1146/annurev.ento.49.061802.123416 (2004).
Dale, C. & Moran, N. A. Molecular Interactions between Bacterial Symbionts and Their Hosts. Cell 126, 453–465 (2006).
Douglas, A. E. Nutritional interactions in insect-microbial symbioses: aphids and their symbiotic bacteria Buchnera. Annual review of entomology 43, 17–37, 10.1146/annurev.ento.43.1.17 (1998).
Terra, W., Ferreira, C., Jordao, B. & Dillon, R. In Biology of the insect midgut 153–194 (Springer, 1996).
Salem, H. et al. Vitamin supplementation by gut symbionts ensures metabolic homeostasis in an insect host Vol. 281 (2014).
Douglas, A. E. Microbial brokers of insect-plant interactions revisited. Journal of chemical ecology 39, 952–961, 10.1007/s10886-013-0308-x (2013).
Weiss, B. & Aksoy, S. Microbiome influences on insect host vector competence. Trends Parasitol. 27, 514–522, 10.1016/j.pt.2011.05.001 (2011).
Gendrin, M. & Christophides, G. K. The Anopheles mosquito microbiota and their impact on pathogen transmission. Anopheles mosquitoes-New insights into malaria vectors 10, 55107 (2013).
Dong, Y., Manfredini, F. & Dimopoulos, G. Implication of the mosquito midgut microbiota in the defense against malaria parasites. Plos pathogens 5, e1000423, 10.1371/journal.ppat.1000423 (2009).
Boissiere, A. et al. Midgut microbiota of the malaria mosquito vector Anopheles gambiae and interactions with Plasmodium falciparum infection. Plos pathogens 8, e1002742, 10.1371/journal.ppat.1002742 (2012).
Tchioffo, M. T. et al. Modulation of malaria infection in Anopheles gambiae mosquitoes exposed to natural midgut bacteria. Plos one 8, e81663, 10.1371/journal.pone.0081663 (2013).
Meister, S. et al. Anopheles gambiae PGRPLC-mediated defense against bacteria modulates infections with malaria parasites. Plos pathogens 5, e1000542, 10.1371/journal.ppat.1000542 (2009).
Cirimotich, C. M. et al. Natural microbe-mediated refractoriness to Plasmodium infection in Anopheles gambiae. Science (New York, N.Y.) 332, 855–858, 10.1126/science.1201618 (2011).
Gendrin, M. et al. Antibiotics in ingested human blood affect the mosquito microbiota and capacity to transmit malaria. Nature communications 6, 5921, 10.1038/ncomms6921 (2015).
Wang, S. et al. Fighting malaria with engineered symbiotic bacteria from vector mosquitoes. Proceedings of the National Academy of Sciences of the United States of America 109, 12734–12739, 10.1073/pnas.1204158109 (2012).
Wang, S. & Jacobs-Lorena, M. Genetic approaches to interfere with malaria transmission by vector mosquitoes. Trends in biotechnology 31, 185–193, 10.1016/j.tibtech.2013.01.001 (2013).
Hoffmann, A. A., Hercus, M. & Dagher, H. Population dynamics of the Wolbachia infection causing cytoplasmic incompatibility in Drosophila melanogaster. Genetics 148, 221–231 (1998).
Bian, G. et al. Wolbachia invades Anopheles stephensi populations and induces refractoriness to Plasmodium infection. Science (New York, N.Y.) 340, 748–751, 10.1126/science.1236192 (2013).
Baldini, F. et al. Evidence of natural Wolbachia infections in field populations of Anopheles gambiae. Nature communications 5, 3985, 10.1038/ncomms4985 (2014).
Capone, A. et al. Interactions between Asaia, Plasmodium and Anopheles: new insights into mosquito symbiosis and implications in malaria symbiotic control. Parasit Vectors 6, 182 (2013).
Shane, J. L., Bongio, N. J., Favia, G. & Lampe, D. J. Draft Genome Sequence of Asaia sp. Strain SF2. 1, an Important Member of the Microbiome of Anopheles Mosquitoes. Genome announcements 2, e01202–01213 (2014).
Favia, G. et al. Bacteria of the genus Asaia stably associate with Anopheles stephensi, an Asian malarial mosquito vector. Proceedings of the National Academy of Sciences 104, 9047–9051 (2007).
Sharon, G. et al. Commensal bacteria play a role in mating preference of Drosophila melanogaster. Proceedings of the National Academy of Sciences of the United States of America 107, 20051–20056, 10.1073/pnas.1009906107 (2010).
Dabire, K. R. et al. Assortative mating in mixed swarms of the mosquito Anopheles gambiae s.s. M and S molecular forms, in Burkina Faso, West Africa. Medical and veterinary entomology 27, 298–312, 10.1111/j.1365-2915.2012.01049.x (2013).
Diabate, A. et al. Mixed swarms of the molecular M and S forms of Anopheles gambiae (Diptera: Culicidae) in sympatric area from Burkina Faso. Journal of medical entomology 43, 480–483 (2006).
Diabate, A. et al. Spatial swarm segregation and reproductive isolation between the molecular forms of Anopheles gambiae. Proceedings. Biological sciences/The Royal Society 276, 4215–4222, 10.1098/rspb.2009.1167 (2009).
Lehmann, T. & Diabate, A. The molecular forms of Anopheles gambiae: a phenotypic perspective. Infection, genetics and evolution : journal of molecular epidemiology and evolutionary genetics in infectious diseases 8, 737–746, 10.1016/j.meegid.2008.06.003 (2008).
Lawniczak, M. K. et al. Widespread divergence between incipient Anopheles gambiae species revealed by whole genome sequences. Science 330, 512–514, 10.1126/science.1195755 (2010).
Caputo, B. et al. Comparative analysis of epicuticular lipid profiles of sympatric and allopatric field populations of Anopheles gambiae s.s. molecular forms and An. arabiensis from Burkina Faso (West Africa). Insect biochemistry and molecular biology 37, 389–398, 10.1016/j.ibmb.2007.01.002 (2007).
Pennetier, C., Warren, B., Dabire, K. R., Russell, I. J. & Gibson, G. “Singing on the wing” as a mechanism for species recognition in the malarial mosquito Anopheles gambiae. Current biology : CB 20, 131–136, 10.1016/j.cub.2009.11.040 (2010).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nature methods 7, 335–336, 10.1038/nmeth.f.303 (2010).
Segata, N. et al. Metagenomic microbial community profiling using unique clade-specific marker genes. Nature methods 9, 811–814, 10.1038/nmeth.2066 (2012).
Osei-Poku, J., Mbogo, C. M., Palmer, W. J. & Jiggins, F. M. Deep sequencing reveals extensive variation in the gut microbiota of wild mosquitoes from Kenya. Molecular ecology 21, 5138–5150, 10.1111/j.1365-294X.2012.05759.x (2012).
Jiang, J., Alvarez, C., Kukutla, P., Yu, W. & Xu, J. Draft genome sequences of Enterobacter sp. isolate Ag1 from the midgut of the malaria mosquito Anopheles gambiae. Journal of bacteriology 194, 5481, 10.1128/jb.01275-12 (2012).
Eappen, A. G., Smith, R. C. & Jacobs-Lorena, M. Enterobacter-activated mosquito immune responses to Plasmodium involve activation of SRPN6 in Anopheles stephensi. Plos one 8, e62937, 10.1371/journal.pone.0062937 (2013).
Segata, N. et al. Computational meta’omics for microbial community studies. Mol Syst Biol 9, 666 (2013).
Segata, N. et al. Metagenomic biomarker discovery and explanation. Genome biology 12, R60, 10.1186/gb-2011-12-6-r60 (2011).
Crotti, E. et al. Acetic acid bacteria, newly emerging symbionts of insects. Applied and Environmental Microbiology 76, 6963–6970 (2010).
Segata, N. et al. Composition of the adult digestive tract bacterial microbiome based on seven mouth surfaces, tonsils, throat and stool samples. Genome Biol. 13, R42 (2012).
Gasparich, G. E. Spiroplasmas: evolution, adaptation and diversity. Frontiers in bioscience : a journal and virtual library 7, d619–640 (2002).
Briones, A. M., Shililu, J., Githure, J., Novak, R. & Raskin, L. Thorsellia anophelis is the dominant bacterium in a Kenyan population of adult Anopheles gambiae mosquitoes. ISME J 2, 74–82, 10.1038/ismej.2007.95 (2008).
Duron, O. et al. The diversity of reproductive parasites among arthropods: Wolbachia do not walk alone. BMC biology 6, 27, 10.1186/1741-7007-6-27 (2008).
Jaenike, J., Unckless, R., Cockburn, S. N., Boelio, L. M. & Perlman, S. J. Adaptation via symbiosis: recent spread of a Drosophila defensive symbiont. Science (New York, N.Y.) 329, 212–215, 10.1126/science.1188235 (2010).
Lindh, J. M., Terenius, O. & Faye, I. 16S rRNA gene-based identification of midgut bacteria from field-caught Anopheles gambiae sensu lato and A. funestus mosquitoes reveals new species related to known insect symbionts. Appl Environ Microbiol 71, 7217–7223, 10.1128/aem.71.11.7217-7223.2005 (2005).
Gherna, R. L. et al. NOTES: Arsenophonus nasoniae gen. nov., sp. nov., the Causative Agent of the Son-Killer Trait in the Parasitic Wasp Nasonia vitripennis. International Journal of Systematic Bacteriology 41, 563–565 (1991).
Marchand, R. P. Field Observations On Swarming and Mating in Anopheles Gambiae Mosquitoes in Tanzania1. 34, 367–387, 10.1163/002829684X00209 (1983).
Charlwood, J. D. Studies on the bionomics of male Anopheles gambiae Giles and male Anopheles funestus Giles from southern Mozambique. Journal of vector ecology : journal of the Society for Vector Ecology 36, 382–394, 10.1111/j.1948-7134.2011.00179.x (2011).
Hackett, K. J. & Clark, T. B. Ecology of spiroplasmas. The mycoplasmas 5, 113–200 (1989).
Clark, T. Spiroplasma sp., a new pathogen in honey bees. Journal of Invertebrate Pathology 29, 112–113 (1977).
Hung, S. H., Chen, T., Whitcomb, R., Tully, J. & Chen, Y. Spiroplasma culicicola sp. nov. from the Salt Marsh Mosquito Aedes sollicitans†. International journal of systematic bacteriology 37, 365–370 (1987).
Williamson, D. L. et al. Spiroplasma diminutum sp. nov., from Culex annulus mosquitoes collected in Taiwan. Int J Syst Bacteriol 46, 229–233 (1996).
Abalain-Colloc, M. et al. Spiroplasma taiwanense sp. nov. from Culex tritaeniorhynchus mosquitoes collected in Taiwan. International journal of systematic bacteriology 38, 103–107 (1988).
Abalain-Colloc, M. et al. Spiroplasma sabaudiense sp. nov. from mosquitoes collected in France. International journal of systematic bacteriology 37, 260–265 (1987).
Poulson, D. F. & Sakaguchi, B. Nature of “sex-ratio” agent in Drosophila. Science (New York, N.Y.) 133, 1489–1490 (1961).
Xie, J., Vilchez, I. & Mateos, M. Spiroplasma bacteria enhance survival of Drosophila hydei attacked by the parasitic wasp Leptopilina heterotoma. Plos one 5, e12149, 10.1371/journal.pone.0012149 (2010).
Liu, N. Insecticide resistance in mosquitoes: impact, mechanisms, and research directions. Annual review of entomology 60, 537–559 (2015).
della Torre, A. et al. Speciation within Anopheles gambiae–the glass is half full. Science (New York, N.Y.) 298, 115–117, 10.1126/science.1078170 (2002).
Caporaso, J. G. et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. The ISME journal 6, 1621–1624 (2012).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature methods 9, 357–359, 10.1038/nmeth.1923 (2012).
Truong, D. T. et al. MetaPhlAn2 for enhanced metagenomic taxonomic profiling Nature methods 12, 902–903 (2015).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics (Oxford, England) 25, 2078–2079, 10.1093/bioinformatics/btp352 (2009).
Asnicar, F., Weingart, G., Tickle, T. L., Huttenhower, C. & Segata, N. Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ 3, e1029, 10.7717/peerj.1029 (2015).
Acknowledgements
We thank S. Ilboudo, H. Guel, D.D. Soma for help with sample collection and dissection, V. De Sanctis and R. Bertorelli (NGS Facility at the Centre for Integrative Biology, University of Trento) for WGS sequencing, and D.E. Neafsey for helpful discussions. We thank the Broad Institute sequencing platform for 16S sequencing.
Author information
Authors and Affiliations
Contributions
F.C., E.A.L., F.B., J.P., N.S. and A.D. designed the experiments. F.B. and J.P. performed the experiments. N.S., F.C., F.B., E.A.L. and W.S.G. analyzed the data. N.S. and D.T.T. performed the sequencing analysis. R.K.D. and A.D. provided samples for the analysis. N.S., F.B. and F.C. wrote the manuscript. All authors reviewed the manuscript. N.S. and F.B. contributed equally to this study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Segata, N., Baldini, F., Pompon, J. et al. The reproductive tracts of two malaria vectors are populated by a core microbiome and by gender- and swarm-enriched microbial biomarkers. Sci Rep 6, 24207 (2016). https://doi.org/10.1038/srep24207
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep24207
This article is cited by
-
Overview of paratransgenesis as a strategy to control pathogen transmission by insect vectors
Parasites & Vectors (2022)
-
The dynamic gut microbiota of zoophilic members of the Anopheles gambiae complex (Diptera: Culicidae)
Scientific Reports (2022)
-
Re-Analysis of 16S rRNA Gene Sequence Data Sets Uncovers Disparate Laboratory-Specific Microbiomes Associated with the Yellow Fever Mosquito (Aedes aegypti)
Microbial Ecology (2022)
-
Microbial Diversity of Adult Aedes aegypti and Water Collected from Different Mosquito Aquatic Habitats in Puerto Rico
Microbial Ecology (2022)
-
Characterization of the reproductive tract bacterial microbiota of virgin, mated, and blood-fed Aedes aegypti and Aedes albopictus females
Parasites & Vectors (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
