
Abstract
We investigate the effect of Rh doping in Sr2IrO4 using X-ray absorption spectroscopy (XAS). We observed appearance of new electron-addition states with increasing Rh concentration (x in Sr2Ir1−xRhxO4) in accordance with the concept of hole doping. The intensity of the hole-induced state is however weak, suggesting weakness of charge transfer (CT) effect and Mott insulating ground states. Also, Ir Jeff = 1/2 upper Hubbard band shifts to lower energy as x increases up to x = 0.23. Combined with optical spectroscopy, these results suggest a hybridisation-related mechanism, in which Rh doping can weaken the (Ir Jeff = 1/2)–(O 2p) orbital hybridisation in the in-planar Rh-O-Ir bond networks.
Similar content being viewed by others
Introduction
Novel phenomena in 5d transition metal oxides (TMOs) have been a central subject in condensed matter physics. The large spin-orbit (SO) coupling transforms the 5d electron states from crystal-field-driven orbital states to certain SO-entangled Jeff states (Jeff : effective total angular momentum)1,2. Consequently, a variety of exotic phases, including a relativistic Mott insulator1,2,3,4,5,6, a quantum spin liquid7,8,9,10,11, and a topological insulator12,13,14, are shown and expected in 5d TMOs. In the case of the most well-known 5d TMO, Sr2IrO4, even a superconductivity is expected because its crystal structure15, low energy Hamiltonian16,17, and magnetic excitation18,19 are similar to those of the mother compound of high TC cuprates, La2CuO4. Indeed, recent angle-resolved photoemission spectroscopy studies showed the cuprate-like Fermi arc band and the d-wave gap in K doped Sr2IrO420,21.
One of the important issues to be resolved for 5d TMOs is how electron/hole doping affects their ground states. It is because the doping behaviours will provide us the clues for understanding emergent phenomena including superconductivity. Studies on La2−xSrxCuO4, for instance, manifested a charge transfer ground state where doped holes reside in the ligand O orbitals22,23. Such hole states in O 2p orbitals form the Zhang-Rice singlet state24, which has been known as the key component of high TC superconductivity. Therefore, studies on carrier doping effects on 5d TMOs are necessary.
This article focuses on the effect of Rh doping in Sr2IrO4. Our Rh L-edge X-ray absorption spectroscopy (XAS) study revealed that the valence of Rh at low Rh concentrations is mainly +3 rather than +4, consistent to recent studies25,26. Therefore, Sr2Ir1−xRhxO4 can be viewed as hole-doped iridates analogous to hole-doped cuprates. With O K-edge XAS, we particularly report two distinct features. One is that a new electron-addition state emerges near the chemical potential as a result of Rh doping. The intensity of the hole-induced states is, however, rather weak compared to the case of cuprates or nickelates. This suggests that the charge transfer (CT) effect is weak and doped holes mainly reside in the Ir 5d orbitals, being consistent with known Mott insulating ground state of Sr2IrO4. The other feature is that the 5d upper Hubbard band (UHB) hybridised with in-plane oxygen (Op) 2p orbitals undergoes a strong redshift whereas the same UHB but hybridised with apical oxygen (Oa) 2p orbitals hardly does. We propose that Rh doping can decrease the hybridisation between Ir and O via charge reconstruction in Op ions and/or via the less extended nature of Rh wave function. Optical spectroscopy study further showed that the onset energy of CT transition from Op 2p to Ir 5d indeed decreases with increasing Rh concentration, supporting the interpretation of the XAS results.
Results
Rh L3-edge XAS was performed to estimate the hole concentration. Figure 1(a) shows the spectra of the Sr2Ir1−xRhxO4 crystals for x = 0.10, 0.23 and 0.41. The peak near 3006 eV corresponds to the L3-edge white line, which represents the transitions from Rh 2p3/2 to 4d states. We fitted our spectra to linear combinations of known spectra of Rh3+ in Rh2O3 (the red lines) and Rh4+ in Sr2RhO4 (the blue lines)26. The valence of Rh is almost +3 rather than +4 in low-x samples (x ≤ 0.23) while the portion of Rh4+ ions starts to be prominent in x = 0.41 sample, consistent with a previous result26. Figure 1(b) shows the concentration ratio (y) of dopants to host metal ions (y = [Rh3+]/[Ir]), where [Rh3+] is the Rh3+ concentration according to our fitting results. It is clearly shown y increases almost linear to x, manifesting the hole doping effect.

(a) Rh L3-edge XAS spectra of Sr2Ir1−xRhxO4 crystals of x = 0.10, 0.23 and 0.41. The open circles show the experimental spectra and the black solid curves show the fitting results. The spectra of Rh3+ in Rh2O3 (red curve) and Rh4+ in Sr2RhO4 (blue curve) were taken from ref. 26 for the fitting. (b) The deduced values of Rh3+ concentration per one Ir site as a function of Rh doping ratio (x). Almost linear relation manifests the hole doping scheme at least for x ≤ 0.23. The error bars account for the uncertainty in determining the peak areas.
The O K-edge XAS spectra show that the Rh doping considerably changes the electronic structure of Sr2IrO4 near the chemical potential. Figure 2 shows the O K-edge XAS spectra of Sr2Ir1−xRhxO4 (x = 0, 0.10, 0.23, 0.41, and 0.71). The spectra near the threshold reflect transitions from O 1s core levels to unoccupied O 2p states hybridised with Ir/Rh d bands. The solid and dashed curves are the XAS spectra taken with σ- and π-polarised light, respectively. The experimental geometry is shown in the inset of Fig. 2. We fixed the angle of the incident beam to be 60°, so the polarisation of σ-polarised light was parallel to the sample surface, while the polarisation of π-polarised light was almost perpendicular to the surface. The huge polarisation dependence in the spectra suggests a quasi-2-dimensional nature in all the samples. With increasing Rh concentration, we observe large and consistent changes in the spectra, especially below the photon energy of 531 eV.

The solid and dashed curves show the spectra taken with σ- and π-polarised light, respectively. The inset shows the measurement geometry.
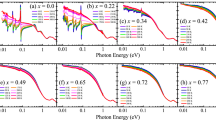
We can assign the peaks below 531 eV as in-plane and apical (Op and Oa) 2p states hybridised with Ir or Rh d orbitals, consistent with the previous studies1,27. Figure 3 shows the O K-edge XAS spectra of Sr2Ir1−xRhxO4 near the threshold with the polarisation vectors ( E ) (a) parallel and (b) perpendicular to the normal axis of the surface ( c in the inset of Fig. 2), respectively. In the case of pure Sr2IrO4, there exist a single peak near 530.2 eV in the E // c spectrum and a doublet near 529.6 eV and 530.2 eV in the E ⊥ c spectrum. It is well known that the unoccupied state at lowest energy in Sr2IrO4 is close to a singlet of Ir 5d Jeff = 1/2 UHB,  . Thus, we attribute the split (of roughly 0.6 eV) as a chemical shift due to different chemistry between Op and Oa1,27. The peaks highlighted by filled triangles in Fig. 3 are the Ir 5d UHB hybridised with Op 2p orbitals (UHBp). Likewise, the peaks marked as open triangles correspond to the Ir 5d UHB hybridised with Oa 2p orbitals (UHBa).
. Thus, we attribute the split (of roughly 0.6 eV) as a chemical shift due to different chemistry between Op and Oa1,27. The peaks highlighted by filled triangles in Fig. 3 are the Ir 5d UHB hybridised with Op 2p orbitals (UHBp). Likewise, the peaks marked as open triangles correspond to the Ir 5d UHB hybridised with Oa 2p orbitals (UHBa).

Low energy parts in the O K-edge XAS spectra with (a) E // c and (b) E ⊥ c , where c is the surface normal axis. The spectra of Sr2RhO4 were taken from ref. 27. The E ⊥ c spectra are reproduced from the σ-polarisation data, and the E//c spectra are deduced from the relation:  . Solid (open) triangles highlight the peak positions of the Ir Jeff = 1/2 UHB hybridised with the in-plane (apical) O 2p.
. Solid (open) triangles highlight the peak positions of the Ir Jeff = 1/2 UHB hybridised with the in-plane (apical) O 2p.
With increasing x, we observe appearance of lower-energy peaks, A and B, as highlighted in Fig. 3. The energies of peaks A and B are clearly distinct from those of the Sr2IrO4 and Sr2RhO4 peaks. Such lower-energy peaks are generally shown in hole-doped systems including nickelates28 or cuprates22,23, because new electron-addition states appear due to hole doping. Since the energy of such states should be close to that of original occupied states, the difference in energy between UHB and the hole-induced states should roughly reflects the charge gap. We see that the energy difference between peak A and Ir UHBp (also, between peak B and Ir UHBa) is roughly 0.5 eV, similar to the known value of the charge gap in Sr2IrO4. Therefore, peaks A and B correspond to the electron-addition states hybridised with Op and Oa orbitals, respectively. The energy difference between peaks A and B (~0.6 eV) in Fig. 3(b) reflects the difference in chemistry between Op and Oa, similar to the cases of UHBp and UHBa.
Apart from the low-energy peaks (A and B), it is clearly shown in both Fig. 3(a,b) that the UHBp peaks shift to lower energy with increasing x. The amount of the UHBp redshift with respect to the case of undoped iridate is approximately 0.25 eV at x = 0.23. Interestingly, the redshift is much less predominant in the case of UHBa. Such an anisotropic peak shift in the two peaks is not easy to understand because they share identical Ir 5d orbitals (Jeff = 1/2 UHB). As will be discussed in the next section, the redshifts are the signatures of reduced O 2p–Ir 5d hybridisation strength upon the hole doping.
To conclude this section, we observe two distinct features due to Rh doping in the O K-edge spectra: One is the emergence of new electron-addition states (A and B) at low energy and the other is a concomitant redshift of Ir UHBp. These will be further analysed in Discussion.
Discussion
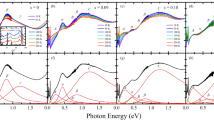
To describe the spectral evolution upon Rh doping quantitatively, we fitted the O K-edge spectra with E // c using Lorentz-Gaussian models. For x = 0.04–0.23 samples, only Rh3+ (d6) prevails so that there could exist three unoccupied states near the chemical potential: the hole-induced states (peak A), Ir UHBp, Rh eg band with increasing order of energy. Figure 4(a) shows the best fitting result of x = 0.23 spectra using the three peaks with a higher energy background for Ir e_g band (dashed line). Meanwhile, for highly doped samples (x ≥ 0.41), it was difficult to obtain reliable fitting results because of the mixed contributions of Rh3+ and Rh4+ states.

(a) The E // c spectra of x = 0.23 sample. The open circles are the experimental data and the black solid line is the fitting result. The red, blue, purple, and dashed curves are for Ir UHBp, peak A, Rh eg, and the Ir eg background, respectively. (b) IA/IUHB as functions of y (concentration ratio of dopants to host cations). The data of La2CuO4 is taken from ref. 23 for the comparison.
We note that the intensity of peak A in Rh doped Sr2IrO4 is much smaller than those in cuprates and nickelates. Figure 4(b) shows the areal intensity of peak A normalised by that of UHBp, namely IA/IUHB of the Rh-doped iridates as a function of y. Small error bars account for the uncertainty in determining the peak intensity. For comparison, we appended the IA/IUHB values in (La1−xSrx)2CuO423 as a function of the number of holes per Cu as well. For the case of the cuprates, IA/IUHB exceeds 1 even at y = 0.1, representatively showing the strong nature of CT insulator. In contrast, IA/IUHB in Rh doped Sr2IrO4 at y = 0.11 (or x = 0.10), is only approximately 0.14. This indicates that the CT effect in iridates is much weaker compared to that in the cuprates. This observation is consistent with the Mott insulating ground state in Sr2IrO4, in which doped holes should reside mainly in metal d orbitals rather than ligand orbitals.
Another important finding in the O K-edge XAS spectra is that only UHBp undergoes a redshift whereas UHBa does not despite they represent the same UHB. Figure 5(a) shows the amount of redshifts (ΔE) of UHBp and UHBa with respect to those of undoped Sr2IrO4 as a function of y. Only the data for x ≤ 0.23, which exhibit little secondary phase (Rh4+), are collected. The error bars in the figure account for the uncertainty in determining the peak position through the fitting processes. In order to understand the origin of the redshifts, we first suspect the consistency with the structural evolution. It has been reported that with increasing x, the Ir-O bond is slightly shortened in average and the Ir-O-Ir bonding angle increases in average29. This moderate structural distortion would tend to increase the hybridisation strength of the Ir Jeff = 1/2 UHB–O 2p orbitals, which should have resulted in a blueshift of UHBp or UHBa. Therefore, structural change upon Rh doping cannot explain the redshifts of UHB.

(a) Relative peak shifts (ΔE) of UHBp (squares) and UHBa (triangles) as functions of y with error bars. The solid lines are guides to the eye. (b) Schematic diagram of hybridisation change in Rh doped Sr2IrO4. (c) σ1(ω) obtained by optical spectroscopy. The extrapolations toward the abscissa roughly visualize the onsets of the charge transfer excitations from in-plane O 2p to Ir 5d UHB.
The plausible mechanism is the decrease of orbital hybridisation in the Ir-O bonds. The energy of UHB can be significantly influenced by the orbital hybridisation of bare Ir Jeff = 1/2 state and O 2p state. The schematic for the orbital hybridisation is illustrated in Fig. 5(b). As a result of the hybridisation, formed are an antibonding state π*, which is commonly called “Ir UHB”, above the chemical potential and a bonding state π, commonly called “O 2p”, below the chemical potential. What is observed in the O K-edge XAS data is the lowering of π*(UHB) hybridised with Op (UHBp). Therefore, the redshift of UHBp can be easily understood if Rh doping can effectively reduce the orbital hybridisation. This mechanism is more effective in the in-planar directions because the influence of Rh can reach Ir only via the in-plane O, namely, through Rh-Op-Ir bond chains due to quasi-2-dimensional crystal structure of Sr2IrO4. This explains why UHBp suffers much more intense redshift compared to UHBa (see Fig. 5(a)).
We propose two possible microscopic origins how Rh doping can decrease the orbital hybridisation between Ir d and O 2p orbitals. One is related to the difference of valence of ions. When the iridate is doped with Rh3+ ions, charge redistribution in Op ions occurs: Since Rh can supply only 3 electrons to the Op ions, Op tend to attract more electrons from neighbouring Ir ions. It effectively increases the electronegativity of Op in Ir-Op bond, thereby making the Ir-Op bond ionic. Similar mechanism but opposite trend has been discussed in H doped VO230. The enhanced ionic character of the Ir-Op bond can decrease hybridisation between two ions. The other is related to the less extended nature of Rh wave functions. According to the results of previous dynamical mean-field theory calculations, hybridisation between Ir and O ions is much stronger than that between Rh and O ions31. Because the valence d orbitals comprise the admixture of Ir and Rh d orbitals, Rh doping can effectively decrease the hybridisation between Ir 5d and O 2p orbitals.
To support the hybridisation mechanism discussed above, we measured the difference in energy between π and π* using optical spectroscopy. Figure 5(c) shows the in-plane real part of optical conductivity σ1(ω) of the x = 0, 0.10, and 0.23 samples. According to a previous study32, the broad features above photon energy of 2.5 eV can be attributed to a charge excitation from π (O 2p) to π* (Ir UHB). Meanwhile, the broad peak near 1 eV and the bump near 2.3 eV can be attributed to Ir d-d transition, which is less relevant to the main interest in this work. It is clearly observed that the onset energy of the high energy bump decreases with increasing x. This suggests that the Ir UHB–O 2p hybridisation becomes weaker as the hole doping is promoted, consistent with our XAS observations and interpretations.
The energy differences between π and π* bands can be roughly estimated from the extrapolations toward the abscissa. Compared to the case of undoped Sr2IrO4, the onset energy of x = 0.23 (x = 0.10) decreased by approximately 0.5 eV (0.3 eV), which is about two times larger than the amount of the UHBp redshift (shown in Fig. 5(a)). This is consistent with the hybridisation mechanism for weakened hybridisation not only lowers the π* band but also lifts up the π band by similar magnitude. Therefore, σ1(ω) data supports the hybridisation mechanism for the UHB redshifts.
It is worthwhile to consider the consequence of reduced Ir UHB–O 2p hybridisation due to Rh doping on the low-energy physics in Sr2IrO4. It is well known that the low energy physics in strongly correlated electron systems can be largely determined by the ratio of on-site Coulomb interaction (U) to the single electron bandwidth (W). According to the results of a recent ab initio calculation, the value of U would decrease due to enhanced ligand screening31 so that U/W should decrease altering the electronic structure near the bandgap. However, the results of recent angle-resolved photoemission experiment show that Rh doping induces only a rigid shift of bands without an appreciable change in band dispersions33. This controversy can be resolved by noting that the value of W should decrease as well, because the decrease in hybridisation strength (by Rh doping) will reduce the bandwidth of the UHB. Therefore, the value of U/W should not change substantially.
In conclusion, Rh doped Sr2IrO4 exhibit peculiar evolution in electronic structure: i) a charge transfer state emerges near the chemical potential but the feature is not that strong as in the hole-doped cuprate, consistent to the Mott insulating ground state, and ii) the Ir UHB energy is lowered by a few tenths eV due to reduced Ir Jeff = 1/2 UHB–O 2p hybridisation strength upon the hole doping.
Methods
Materials
High-quality single crystals Sr2Ir1−xRhxO4 (0 ≤ x ≤ 0.71) were synthesized from off-stoichiometric quantities of SrCl2, SrCO3, IrO2, and RhO2 using self-flux method. Detailed methods are described elsewhere29.
X-ray absorption spectroscopy
Polarization-dependent O K-edge X-ray absorption spectroscopy (XAS) was performed at the 2A beamline of the Pohang Light Source in total electron yield mode. To obtain a clean surface, we cleaved samples in situ in an ultra-high vacuum (~6 × 10−8 Pa). We fixed the angle of the incident beam as 60 degrees, and changed the direction of polarisation to resolve the in- and out-of-plane O responses. Rh L-edge XAS was performed at the 16A1 beamline of the National Synchrotron Radiation Research Center in Taiwan in fluorescence yield mode. All the XAS spectra were collected at room temperature.
Optical spectroscopy
We performed ellipsometry measurement to obtain the real part of optical conductivity between 0.74 and 4 eV at room temperature using a V-VASE ellipsometer (J. A. Woollam Co.).
Additional Information
How to cite this article: Sohn, C. H. et al. X-ray Absorption Spectroscopy Study of the Effect of Rh doping in Sr2IrO4. Sci. Rep. 6, 23856; doi: 10.1038/srep23856 (2016).
References
Kim, B. J. et al. Novel J eff = 1/2 Mott State Induced by Relativistic Spin-Orbit Coupling in Sr2IrO4 . Phys. Rev. Lett. 101, 076402 (2008).
Kim, B. J. et al. Phase-Sensitive Observation of a Spin-Orbital Mott State in Sr2IrO4 . Science 323, 1329–1332 (2009).
Moon, S. J. et al. Dimensionality-Controlled Insulator-Metal Transition and Correlated Metallic State in 5d Transition Metal Oxides Srn+1IrnO3n+1 (n = 1, 2, and ∞). Phys. Rev. Lett. 101, 226402 (2008).
Okabe, H. et al. Ba2IrO4: A spin-orbit Mott insulating quasi-two-dimensional antiferromagnet. Phys. Rev. B 83, 155118 (2011).
Comin, R. et al. Na2IrO3 as a Novel Relativistic Mott Insulator with a 340-meV Gap. Phys. Rev. Lett. 109, 266406 (2012).
Ohgushi, K. et al. Resonant X-ray Diffraction Study of the Strongly Spin-Orbit-Coupled Mott Insulator CaIrO3 . Phys. Rev. Lett. 110, 217212 (2013).
Chaloupka, J., Jackeli, G. & Khaliullin, G. Kitaev-Heisenberg Model on a Honeycomb Lattice: Possible Exotic Phases in Iridium Oxides A2IrO3 . Phys. Rev. Lett. 105, 027204 (2010).
Singh, Y. et al. Relevance of the Heisenberg-Kitaev Model for the Honeycomb Lattice Iridates A2IrO3 . Phys. Rev. Lett. 108, 127203 (2012).
Chaloupka, J., Jackeli, G. & Khaliullin, G. Zigzag Magnetic Order in the Iridium Oxide Na2IrO3 . Phys. Rev. Lett. 110, 097204 (2013).
Cao, G. et al. Evolution of magnetism in the single-crystal honeycomb iridates (Na1−xLix)2IrO3 . Phys. Rev. B 88, 220414(R) (2013).
Takayama, T. et al. Spin-orbit coupling induced semi-metallic state in the 1/3 hole doped hyper-kagome Na3Ir3O8 . Sci. Rep. 4, 6818 (2014).
Shitade, A. et al. Quantum Spin Hall Effect in a Transition Metal Oxide Na2IrO3 . Phys. Rev. Lett. 102, 256403 (2009).
Kim, C. H., Kim, H.-S., Jeong, H., Jin, H. & Yu, J. Topological Quantum Phase Transition in 5d Transition Metal Oxide Na2IrO3 . Phys. Rev. Lett. 108, 106401 (2012).
Kim, H.-S., Kim, C. H., Jeong, H., Jin, H. & Yu, J. Strain-induced topological insulator phase and effective magnetic interactions in Li2IrO3 . Phys. Rev. B 87, 165117 (2013).
Crawford, M. K. et al. Structural and magnetic studies of Sr2IrO4 . Phys. Rev. B 49, 9198–9201 (1994).
Wang, F. & Senthil, T. Twisted Hubbard Model for Sr2IrO4: Magnetism and Possible High Temperature Superconductivity. Phys. Rev. Lett. 106, 136402 (2011).
Watanabe, H., Shirakawa, T. & Yunoki, S. Monte Carlo Study of an Unconventional Superconducting Phase in Iridium Oxide J eff = 1/2 Mott Insulators Induced by Carrier Doping. Phys. Rev. Lett. 110, 027002 (2013).
Kim, J. et al. Magnetic Excitation Spectra of Sr2IrO4 Probed by Resonant Inelastic X-Ray Scattering: Establishing Links to Cuprate Superconductors. Phys. Rev. Lett. 108, 177003 (2012).
Kim, J. et al. Excitonic quasiparticles in a spin–orbit Mott insulator. Nat. Commun. 5, 4453 (2014).
Kim, Y. K. et al. Fermi arcs in a doped pseudospin-1/2 Heisenberg antiferromagnet. Science 345, 187–190 (2014).
Kim, Y. K., Sung, N. H., Denlinger., J. D. & Kim, B. J. Observation of a d-wave gap in electron-doped Sr2IrO4 . Nat. Phys. 12, 37–41 (2016).
Nücker, N., Fink, J., Fuggle, J. C., Durham, P. J. & Temmerman, W. M. Evidence for holes on oxygen sites in the high-T c superconductors La2−xSrxCuO4 and YBa2Cu3O7−y . Phys. Rev. B 37, 5158–5163 (1988).
Chen, C. T. et al. Electronic states in La2−xSrxCuO4+δ probed by soft-x-ray absorption. Phys. Rev. Lett. 66, 104–107 (1991).
Zhang, F. C. & Rice, T. M. Effective Hamiltonian for the superconducting Cu oxides. Phys. Rev. B 37, 3759–3761 (1988).
Klein, Y. & Terasaki, I. Insight on the electronic state of Sr2IrO4 revealed by cationic substitutions. J. Phys.: Condens. Matter 20, 295201 (2008).
Clancy, J. P. et al. Dilute magnetism and spin-orbital percolation effects in Sr2Ir1−xRhxO4 . Phys. Rev. B 89, 054409 (2014).
Moon, S. J. et al. Electronic structures of layered perovskite Sr2MO4 (M = Ru, Rh, and Ir). Phys. Rev. B 74, 113104 (2006).
Kuiper, P., Kruisinga, G., Ghijsen, J., Sawatzky, G. A. & Verweij, H. Character of holes in LixNi1−xO and their magnetic behavior. Phys. Rev. Lett. 62, 221–224 (1989).
Qi, T. F. et al. Spin-orbit tuned metal-insulator transitions in single-crystal Sr2Ir1−xRhxO4 (0 ≤ x ≤ 1). Phys. Rev. B 86, 125105 (2012).
Filinchuk, Y. et al. In situ diffraction study of catalytic hydrogenation of VO2: Stable phases and origins of metallicity. J. Am. Chem. Soc. 136, 8100–8109 (2014).
Sohn, C. H. et al. Orbital-Dependent Polaron Formation in the Relativistic Mott Insulator Sr2IrO4 . Phys. Rev. B 90, 041105(R) (2014).
Martins, C., Aichhorn, M., Vaugier, L. & Biermann., S. Reduced effective spin-orbital degeneracy and spin-orbital ordering in paramagnetic transition-metal oxides: Sr2IrO4 versus Sr2RhO4 . Phys. Rev. Lett. 107, 266404 (2011).
Cao, Y. et al. Hallmarks of the Mott-Metal Crossover in the Hole Doped J = 1/2 Mott insulator Sr2IrO4 . arXiv :1406.4978.
Acknowledgements
We thank L.-Y. Jang and B.-J. Su for assisting Rh L-edge x-ray absorption spectroscopy and H.D. Kim and C.H. Kim for fruitful discussion. This work was supported by IBS-R009-D1. D.-Y.C. acknowledges support from Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Science, ICT and Future Planning (NRF-2015R1C1A1A02037514). G.C. acknowledges support by the U.S. National Science Foundation via Grant No. DMR-1265162.
Author information
Authors and Affiliations
Contributions
C.H.S. and D.-Y.C. conceived the experiments, and C.H.S., D.-Y.C. and C.-T.K. performed the XAS measurements. L.J.S. performed the optical measurement. T.F.Q. and G.C. fabricated the crystals. D.-Y.C. and T.W.N. supervised the research. All authors contributed to the writing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Sohn, C., Cho, DY., Kuo, CT. et al. X-ray Absorption Spectroscopy Study of the Effect of Rh doping in Sr2IrO4. Sci Rep 6, 23856 (2016). https://doi.org/10.1038/srep23856
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep23856
This article is cited by
-
Quantum criticality in a layered iridate
Communications Physics (2021)
-
Comparative study of catalytic activities among transition metal-doped IrO2 nanoparticles
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
