
Abstract
WRKY transcription factors belong to one of the largest transcription factor families. These factors possess functions in plant growth and development, signal transduction and stress response. Here, we identified 95 DcWRKY genes in carrot based on the carrot genomic and transcriptomic data and divided them into three groups. Phylogenetic analysis of WRKY proteins from carrot and Arabidopsis divided these proteins into seven subgroups. To elucidate the evolution and distribution of WRKY transcription factors in different species, we constructed a schematic of the phylogenetic tree and compared the WRKY family factors among 22 species, which including plants, slime mold and protozoan. An in-depth study was performed to clarify the homologous factor groups of nine divergent taxa in lower and higher plants. Based on the orthologous factors between carrot and Arabidopsis, 38 DcWRKY proteins were calculated to interact with other proteins in the carrot genome. Yeast two-hybrid assay showed that DcWRKY20 can interact with DcMAPK1 and DcMAPK4. The expression patterns of the selected DcWRKY genes based on transcriptome data and qRT-PCR suggested that those selected DcWRKY genes are involved in root development, biotic and abiotic stress response. This comprehensive analysis provides a basis for investigating the evolution and function of WRKY genes.
Similar content being viewed by others

Introduction
Transcriptional regulation is the most important link for regulating gene expression in plants. Transcription factors are involved in controlling many important biological processes in the gene transcription regulatory network1,2. In the plant genome, a large proportion of genes belong to transcription factors and at least 58 transcription factor families have been determined3. In Arabidopsis and rice, approximately 8% and 4% of genes were identified as transcription factors, respectively4,5.
The WRKY family, whose name is derived from the highly conserved WRKY domain, is one of the largest transcription factor families6. The WRKY domain contains about 60 amino acids, comprising a highly conserved short peptide WRKYGQK and adjacent C2H2 or C2HC zinc finger structure. The conserved amino acid WRKYGQK also consists of various forms, such as WRKYGKK, WRKYDQK and WRKYDHK7,8. Based on the number of WRKY domains and the type of zinc finger, the WRKY family can be divided into three groups, namely, groups I, II and III. Group I contains two WRKY domains and a C2H2 zinc finger type (C-X4–5-C-X22–23-H-X1-H). Group II contains one WRKY domain and a C2H2 type zinc finger; this group can be further divided into five subgroups (IIa, IIb, IIc, IId and IIe). Group III also contains only one domain but has a C2HC zinc finger type (C-X7-C-X23-H-X1-C)6. Wu and his colleagues further analyzed the WRKY family in Arabidopsis and rice and revealed the distribution of group II into three subgroups (IIa+b, IIc and IId+e) based on sequence similarities9.
The first reported WRKY gene, SPF1, was cloned from sweet potato in 199410. Since then, several WRKY genes have been found in parsley11, Arabidopsis12, rice13 and even in green algae14. The completed genome sequencing of many plants has resulted in a more comprehensive identification of WRKY genes. The number of WRKY members in different species varies greatly. Thus far, 72 members have been found in Arabidopsis15, 100 in rice9, 41 in Physcomitrella patens3,16 and 19 in Selaginella moellendorffii3. Evolution analysis showed that group I was the oldest group and groups II and III originated from group I9,17. In the later study, WRKY factors were identified in the nonplant species Dictyostelium discoideum and Giardia lamblia, suggesting that WRKY factors have a very ancient origin14.
A large number of WRKY factors were found, but the functions of only a small number of these factors have been analyzed further. These WRKY factors are involved in seed germination, plant growth and development, signal transduction and metabolic regulation18,19,20. They also participate in biotic and abiotic stress responses, such as freezing, salinity, drought and pathogen infection21,22. A WRKY transcription factor, ABO3, mediates plant responses to ABA and drought stress in Arabidopsis23. Overexpression of two wheat WRKY genes, namely, TaWRKY2 and TaWRKY19, confer tolerance to salt, drought and freezing stresses in transgenic plants24. Similarly, Zhou et al. revealed that three GmWRKY genes from soybean play differential roles in abiotic stress tolerance in transgenic Arabidopsis plants25. Several WRKY genes are regulated by miRNAs26,27. In sunflower, miR396 regulates HaWRKY6 in response to high-temperature damage27.
Carrot is an important economic crop in the family Apiaceae and is rich in carotene and various nutrients. Studies on carrot transcription factors are rarely reported because of the lack of carrot genomic data28,29. The publication of the carrot genome draft database (CarrotDB) provides resources to make a bioinformatics identification and analysis on WRKY transcription factors. The genomic and transcriptomic database for carrot was built by our group (Lab of Apiaceae Plant Genetics and Germplasm Enhancement, http://apiaceae.njau.edu.cn/carrotdb/index.php)30. In this paper, we identified 95 DcWRKY genes in the genomic and transcriptomic carrot database and focused on the evolution and duplication of WRKY genes on different species. A total of 71 DcWRKY genes were detected to have expression on carrot root development based on transcriptome data. Moreover, the expression analysis of several DcWRKY genes under different stresses showed that DcWRKY genes participated in the abiotic stress response. Our results provide a basis for studying the evolution and function of WRKY transcription factors.
Results
Identification of DcWRKY transcription factors in the D. carota genome
To identify all the WRKY factors in the carrot genome, we employed the HMM profile of the WRKY domain (PF03106) as a query to search against the database using HMMER3.0 and BLAST. A total of 95 nonredundant genes were assigned as WRKY genes and termed DcWRKY1 to DcWRKY95 (Table 1 and S1). The lengths of the DcWRKY proteins ranged from 101 to 865 amino acids, with an average of 333 amino acids. The highly conserved domain WRKYGQK was present in 88 DcWRKY proteins, whereas the remaining seven proteins contained WRKYGKK, WRKYDHK, or WRKYDQK domain. Seventeen genes were identified to group I, which contained two WRKY domains and had a zinc finger motif of C2H2 type (C-X4-C-X22–23-H-X1-H). Sixty-seven DcWRKY members contained a zinc finger motif of C2H2 type (C-X4–5-C-X23-H-X1-H), which were classified as group II. Group III had 11 members, which contained a C2HC(C-X7-C-X23-H-X1-C) zinc finger.
Phylogenetic relationship and structure of the carrot DcWRKYs
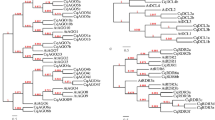
To investigate the classification and phylogenetic relationship of the WRKY proteins in carrot and Arabidopsis, we used the domain region of the WRKY proteins from carrot and Arabidopsis to construct a phylogenetic tree. Based on the phylogenetic tree (Fig. 1), all the DcWRKY factors could be divided into three groups. Group I is an independent branch, whereas groups II and III showed a relatively close relationship. Group II, in particular, could be further divided into three main groups with five subgroups. Subgroups IIa and IIb were separated from one clade and IIe and IId clustered to a branch.

Phylogenetic tree of all WRKY proteins from carrot and Arabidopsis.
Group I proteins with the suffix ‘N’ or ‘C’ indicates the N-terminal WRKY domains or the C-terminal WRKY domains.
The exon–intron structure analysis was performed to gain more insight into the DcWRKY genes. High variation was observed in numbers of exons and introns among DcWRKY genes (Fig. 2A,B). Forty-two DcWRKYs had two introns and accounted for the largest proportion, followed by eighteen DcWRKYs possessed only one intron and twelve DcWRKYs had three introns. DcWRKY genes belonging to the same group seemed to have similar exon–intron structures. For example, eight genes (DcWRKY59, 67, 15, 28, 41, 82, 91, 95) did not contain any intron and four of them (DcWRKY59, 67, 15 and 28) were classified into group I, three of them (DcWRKY82, 91 and 95) were classified into group IIc. Group III contained eleven members, nine genes had two introns. For subgroups IIb, most genes contain four introns, while most IId and IIe genes had two introns. These results showed a strong correlation between exon–intron structure and phylogenetic relationship, which providing an additional foundation to support the classification.

Structure analysis of carrot WRKY transcription factors.
(A) NJ phylogenetic tree of DcWRKYs. (B) Exon–intron composition of DcWRKY genes. (C) Distribution of conserved motifs of DcWRKYs. (D) Logo of each motif. Different motifs are shown by different colors numbered 1 to 10. See legend for detailed color annotation.
The conserved motifs were predicted by MEME program to explore the diversity in each group. As illustrated in Fig. 2C,D, motifs 1, 3 and 5 contained a WRKYGQK sequence. The DcWRKY proteins that share a similar motif composition were clustered into the same group. For example, most members of groups IIa and IIb contained motifs 1, 2, 6 and 8, whereas group IId and IIe shared motifs 1 and 2. Motifs 1 and 2 were also present in group IIc. Motifs 2, 3, 4, 5 and 7 were present in group I, which contained two WRKY domains, whereas group III possessed motifs 3 and 4.
Physicochemical analysis of deduced DcWRKY proteins
To analyze the physical and chemical characterizations of the carrot DcWRKYs, we calculated all 95 DcWRKY proteins using the Protparam tool (Table S1). The values of theoretical pI ranged from 4.61 to 10.04 and the average for all proteins was 7.24. No distinct difference was found between the percentage of positive and negative amino acids in all groups except for group IId, in which the percentage of positive amino acids was two-fold higher compared with negative amino acids. The content of aliphatic amino acids in all the proteins was very high, which accounted for an average of 16%, whereas the percentage of aromatic amino acids was only 7%. The average value of the aliphatic index reached 59.57, which suggested that the DcWRKY proteins contained rich aliphatic amino acids. Almost all DcWRKY proteins were calculated to be unstable proteins, only seven DcWRKY proteins were considered to be stable with aliphatic index values of less than 40. The grand average of hydropathicity of all DcWRKYs was less than zero, indicating that DcWRKY proteins were hydrophilic. Most of the physical/chemical properties of DcWRKY proteins were quite similar, but several differences were still observed, which may be due to the nonconserved regions in the protein sequences.
The cis-regulatory elements in all DcWRKY genes promoters were analyzed using the online software PlantCARE based the carrot genome data. A number of different kinds of cis elements were found and the 10 most common elements were represented in Fig. 3. These elements included a fungal elicitor responsive element W-box, two light responsive elements (G-box and Sp1 elements), three hormone responsive elements (CGTCA-motif, ERF and ABRE elements), a motif named Skn-1 associated with endosperm expression, a stress induction responding site TC-rich, a drought responsive element MBS and a heat stress responsive site HSE. Most DcWRKY genes contained more than one cis element in their promoter regions. WRKY proteins usually functioned as transcriptional regulators by binding to W-box ((C/T)TGAC(T/C)) to regulate defense-related genes11. We found that several DcWRKY genes also contained W-box element in their promoter regions. The same findings were identified in Chinese cabbage and Arabidopsis31,32, suggesting that these DcWRKY genes may be regulated by other DcWRKY genes or self-regulated mechanisms. A lot of studies have also reported that WRKY factors were responsive to various stresses including drought, cold and salinity14,33, that may due to the upstream genes specificity bind the corresponding cis element to regulate the expression of WRKY genes.

cis elements analysis of the promoter regions of carrot WRKY genes.
The micro-segments in different colors were the putative elements sequence. The description of the ten cis elements were in brackets.
Subcellular localization of DcWRKYs
Subcellular location analysis on the combination of WoLF PROST and TargetP showed that most DcWRKY proteins were located in the nucleus (Table S1). To examine the subcellular localization of DcWRKY proteins, the coding sequences of three predicted nucleus-localized WRKYs were fused to the N-terminus of GFP and expression in tobacco cells via biolistic bombardment. The GFP fluorescence was observed only in the nucleus of transformant cells (Fig. 4), indicating that DcWRKY45, DcWRKY11 and DcWRKY80 were localized to the nucleus in vivo.

Subcellular localization of carrot WRKYs.
The DcWRKY-GFP fusion plasmids were transiently expressed in tobacco cells. GFP fuorescence was localized in the nucleus. Bar = 50 μM.
Evolution and distribution of WRKY family factors among different species
The WRKY family factors are commonly found in plants, but are also reported in two nonphotosynthetic organisms, namely, D. discoideum14 and G. lamblia34. We constructed a schematic of the phylogenetic tree in eukaryote evolution. Based on the whole-genome level, the number of WRKYs in each species was counted (Fig. 5). In D. discoideum and G. lamblia, only one WRKY protein was identified in both organisms. Similarly, only two WRKY proteins were identified in the algae Chlamydomonas reinhardtii and Volvox carteri. However, land plants have a relatively large number of WRKY family factors. In addition, species that have a larger genome seem to contain a greater number of WRKY transcription factors except for P. abies. In all species, the densities of WRKY proteins in A. thaliana (0.6050 number/Mb) were the highest, followed by Citrus sinensis (0.2508 number/Mb), Oryza sativa (0.2325 number/Mb) and D. carota (0.1979 number/Mb), which were higher than those in lower plants. The results of evolutionary analysis imply that a sharp expansion occurred in the evolutionary process from low plants to high plants and the density of WRKY proteins increased as the plants evolved. Notably, group I existed in all species, whether in plantae, slime mold, or protozoans, suggesting that this group evolved early and represents the ancestral form. The number of group III in P. abies, S. moellendorffii and P. patens was less than the other higher plants. However, the number of this group was higher in monocots than in eudicots.

Schematic of species phylogenetic relationships.
The distributions of WRKY transcription factors among different species are compared. Each color represents a WRKY subgroup and the colored section represents the proportion of this subgroup.
Identification of orthologous and paralogous WRKY genes in plants
To survey the orthologous and paralogous WRKY genes among different species, we performed a comparative study of nine divergent taxa in lower and higher plants. The genome sequences of alga (C. reinhardtii), moss (P. patens), fern (S. moellendorffii), P. abies, Arabidopsis, carrot, grapevine, apple and rice have been completed. A total of 73 homologous gene groups were obtained by OrthoMCL software, containing 543 paralogous, 452 orthologous and 677 coorthologous gene pairs. Results of the comparison between different species are presented in Table 2. A large number of paralogous gene pairs were found in P. abies (237), followed by apple (102), P. patens (67) and carrot (56). Species with large genome seems to show correspondingly great paralogous genes, except for grape. Grape had only seven paralogous gene pairs. A total of 28/23 and 21/19 orthologous/coorthologous gene pairs were identified in carrot–rice and carrot–Arabidopsis. Numerous orthologous/coorthologous gene pairs were also found between carrot and other dicotyledons. However, a small number of homologous genes were found between the gymnosperm P. abies and other plants, although the former has a relatively large WRKY family (72 WRKY transcription factors) and has the most number of paralogous gene pairs (237 pairs).
Interaction network of DcWRKY proteins in carrot
Pearson correlation coefficients (PCC value) were calculated among carrot proteins based on the orthologs in Arabidopsis to verify the co-expression relationships between DcWRKY factors and other carrot proteins. A total of 38 DcWRKY proteins showed interactions with other proteins in carrot genome and 124 orthologs pairs were identified (Fig. 6). Among them, 75 orthologs pairs showed positive correlations (PCC value > 0) and 35 orthologs pairs showed negative correlations (PCC value < 0). In addition, the PCC values of 14 proteins could not be calculated. Three pairs of DcWRKY proteins, namely, DcWRKY80 (group IIa)/DcWRKY31 (group IIa), DcWRKY81 (group IIa)/DcWRKY50 (group IIb) and DcWRKY31 (group IIa)/DcWRKY47 (group III), showed co-expression relationships with positive correlations. These relationships indicate that the proteins that belonged to the same group may have the same functions in the regulation network. DcWRKY20 protein, which belonged to group IId, showed significant correlations with 17 proteins, indicating that DcWRKY20 probably plays an important role in biological regulation mechanisms by inducing other genes. Moreover, two DcMAPK proteins Dck19436 (DcMAPK1) and Dck06213 (DcMAPK4) respectively showed potential regulation relationships with nine DcWRKY proteins, including DcWRKY20.

Interaction network of WRKY factors in carrot according to the orthologs in Arabidopsis.
PCC: Pearson correlation coefficient. Different graphical represent the subcelluar location of different proteins. Ellipse: Nucleus; Rectangle: Unclear; Triangle: Plastid; Diamond: Vacuole; Hexagon: Unclear.
To assess whether there were interactions between DcWRKY20 and two DcMAPK proteins, the fusion plasmids in the vectors pGADT7 and pGBKT7 were co-transformed into the yeast strain AH109 and the interaction was quantified with X-α-Gal assays. Figure 7 showed that co-expression of the DcWRKY20 with DcMAPK1 or DcMAPK4 resulted in X-α-galactosidase expression activity. The positive control showed that pGBKT7-53 can interact with pGADT7-T and the negative control did not display such expression. These results demonstrated that DcWRKY20 interacted directly with DcMAPK1 and DcMAPK4 in yeast.

Interaction between DcWRKY20 and DcMAPK1/DcMAPK4 in yeast cells.
Transformants grown on DDO, QDO and QDO/X/A. DDO: SD/-Leu/-Trp; QDO: SD/-Ade/-His/-Leu/-Trp; QDO/X/A: QDO with X-a-gal and aureobasidin A.
Expression analysis of DcWRKY genes during root development in carrot
The transcript abundances of DcWRKY genes during development in carrot were analyzed through the calculation of transcriptome data (SRR2177455). All DcWRKY genes were surveyed and 71 DcWRKY genes were found to have the expression. As showed in Fig. 8, the DcWRKY genes showed a broad expression during different developmental stages. Some genes, like DcWRKY6, 8, 24, 31, 88 and 95, showed relatively high expression levels in all stages, while DcWRKY1, 23, 53 and 89 expressed very low. Throughout the Fig. 7, we found that the stage with 25 d had a large difference compared to other three stages, while a similar pattern was observed between 60 d and 90 d.

Transcript abundances of DcWRKY genes at different developmental stages (25 d, 40 d, 60 d and 95 d) in carrot root.
The WRKY genes in Arabidopsis, such as AtWRKY635, AtWRKY4436 and AtWRKY7537, which have been confirmed to play important roles in plant development. According to our homologous analysis and transcriptome data, several DcWRKY genes were selected for qRT-PCR validation. As illustrated in Fig. 9, the expression patterns of those eleven DcWRKY genes had a greater difference at four development stages. Meanwhile, the qRT-PCR analysis and transcriptome data of most genes were consistent. The expression levels of four genes (DcWRKY2, 64, 69 and 88) increased over the process of carrot development. Among them, DcWRKY2 and DcWRKY64 showed significant expression changes at the initial stage and maintained higher expression levels at the later stages, while the expression of two others (DcWRKY69 and DcWRKY88) was highly variable. DcWRKY11 and DcWRKY95 exhibited a decrease pattern of expression at all four stages (Fig. 8) and qRT-PCR verified this result (Fig. 9). The expression levels of DcWRKY6 and DcWRKY8 at the initial stage were significantly decreased, while they showed a significant increase in the later stages.

Expression profiles of DcWRKY genes at different developmental stages (25 d, 40 d, 60 d and 95 d) in carrot root.
Expression profiles of DcWRKY genes under biotic stresses in carrot
In regards to DcWRKY genes response to biotic stresses, fourteen DcWRKY genes which their orthologous genes in Arabidopsis involved in biotic stress were selected to determine the expression profiles after treatment with whitefly and aphids infections, respectively. As shown in Fig. 10, those selected DcWRKY genes were sensitive to biotic stresses. Nine of the tested DcWRKY genes exhibited a high level of accumulation after subjected to aphid stress, especially DcWRKY5, followed by DcWRKY31, 8, 30 and 1. The expressions of DcWRKY8, 28, 30, 31 and 74 were induced after inoculation with whitefly. Among them, the effect whitefly infection on DcWRKY28 and DcWRKY30 response were much stronger than others. DcWRKY8, 30 and 31 were found to be both significantly upregulated in two biotic stresses, whereas weak expressions were observed for DcWRKY80 and DcWRKY90.

Expression profiles of DcWRKY genes under biotic stresses (whitefly and aphid tests) in carrot.
Expression profiles of DcWRKY genes under abiotic stresses in carrot
Previous studies reported that WRKY genes are involved in abiotic stress resistance22,38. Basing on the transcriptome data in Arabidopsis under different abiotic stresses39 and the orthologous genes between DcWRKYs and AtWRKYs (Table 1), we selected 12 DcWRKY genes from each group to analyze their expression patterns under four abiotic stresses (cold, heat, salt and drought) in carrot. Figure 11 shows that the members of group I, DcWRKY27 and DcWRKY30, had only one motif difference in structure but exhibited different responses to abiotic stresses.

Expression patterns of DcWRKY genes under heat, cold, salt and drought stresses.
Samples collected at 0, 1, 2, 4, 8 and 24 h after each treatment.
In heat and cold treatments, DcWRKY27 was evidently upregulated and maintained a high expression level during 24 h, whereas DcWRKY30 was not sensitive to temperature treatment. Moreover, the expression level of DcWRKY27 also increased in salt and drought treatments; the expression level in salt treatment increased by almost 80 times. However, the trend in the expression of DcWRKY30 initially increased and reached a maximum after 2 h and then decreased in salt and drought conditions. Based on the further classification of group II (IIa+IIb, IIc and IId+IIe), we found that the phenomenon of the same subgroup genes that showed a similar expression trend was common in group II, for example, DcWRKY45 (IIa)/DcWRKY1 (IIb), DcWRKY5 (IIc)/DcWRKY11 (IIc) and DcWRKY58 (IId)/DcWRKY88 (IId). The members of group II seemed to be more sensitive to drought and salt stresses. In particular, under salt stress, DcWRKY1 and DcWRKY18 increased by 14- and 12-fold, respectively.
DcWRKY8 and DcWRKY10, which both belonged to group III, had a motif difference. The expression levels of the DcWRKY8 and DcWRKY10 genes slightly changed under cold and heat conditions. Moreover, DcWRKY8 responded rapidly to salt stress response, but both genes had a similar change trend under drought treatment: an initial increase and peak at 2 h followed by a decrease.
Discussion
Increasing studies have confirmed that WRKY transcription factors are involved in plant growth and development, signal transduction and stress response. Studies on functions of WRKY factors were conducted on model plants, such as Arabidopsis15 and rice9, but almost no reports on carrot, an Apiaceae plant, have been found. In this study, 95 genes were identified to encode WRKY transcription factors and they were divided into three groups based on the similarity of structure and motif. Among all motifs, motifs 1, 3 and 5 contained a WRKYGQK sequence. Motifs 3 and 5 were present in group I members, while motif 1 was found in groups II and III. In addition, comparative structural analysis of DcWRKYs revealed that DcWRKYs in the same group shared similar exon–intron structures. The analysis on structures of DcWRKY genes might provide a way to find out which group of WRKY genes might be of a more ancient origin.
A phylogenetic tree of WRKY transcription factors from carrot and the dicotyledonous model plant Arabidopsis was constructed. The result was consistent with domain and zinc finger type classifications of carrot WRKY transcription factors. Basing on the current genomic data, we built a model diagram for the origin and evolution of WRKY family transcription factors. Some species that can represent different branches on the plant’s evolutionary tree were selected to analyze the WRKY family. As illustrated in Fig. 5, group I existed in all species, including the two nonplant species G. lamblia and D. discoideum. However, groups II and III only seemed to be specific for the green plant lineage and expand with the evolution of higher plants17,40. The results expound and support that all the WRKY groups may have evolved prior to the moss lineage. Group I may have originated before the origin of eukaryotes about 1.5 billion years ago14.
The events of gene duplication and loss are the driving forces during species evolution. With genome amplification, the number and density of WRKY factors are greatly increased. The algae C. reinhardtii and V. carteri both contain only two WRKY factors, whereas the moss P. patens and the fern S. moellendorffii have 41 and 19 WRKY members, respectively. Groups II and III appeared before the origin of the moss plant and have been duplicated many times with plant evolution, which accounted for a large proportion in higher plants. In this study, we selected several monocotyledonous and dicotyledonous plants to compare the duplication and evolution of WRKY factors. The duplication of WRKY factors in monocotyledonous and dicotyledonous plants was independent and the duplication and diversification of WRKY factors occurred before the differentiation of monocots and eudicots. The number of group III members in monocotyledonous plants was evidently higher than that in dicotyledonous plants. This result indicates that group III seems to be more active in duplication and may have more function in monocots. Gene replication has been confirmed as one of the reasons that lead to new gene functions41. Therefore, we can infer that WRKY factors were expended during plant evolution. The unique gene duplication events in different species revealed the species specificity in the evolution of WRKY factors.
The homologous WRKY genes were compared to further analyze the genetic relationship. A total of 73 homologous gene groups belonging to nine distinctly divergent groups were identified based on the sequence similarity. The number of homologous genes showed a significant difference among different species, which may be influenced by the genetic distance between the analyzed species. Numerous homologous gene pairs were found between carrot and other dicotyledons because of their close relationship. However, few homologous gene pairs were identified between carrot and other monocotyledonous and lower plants. This study also shows that species with large genome indicates more paralogous genes. This phenomenon may be contributed by the plant’s whole genome duplication (WGD). The WGD event is an important evolutionary feature of plant genome, which can explain the results of gene duplication or loss42,43. Previous studies showed that Arabidopsis has undergone three WGD and rice also has undergone at least one WGD event5,44. Furthermore, the number of paralogous genes in grapevine is much less than that in other higher species, which could be due to grapevines’ failure to undergo any WGD event45. Carrot may also have experienced one or more rounds of WGD events, which could have led to the DcWRKY factors being expended.
Previous studies founded that WRKY transcription factors have complex regulatory networks in response to biotic and abiotic stresses. Among the networks, the WRKY factors are controlled by different levels, including direct positive or negative control by WRKY factors, regulation via other transcription factors or proteins and the small-RNA-WRKY interactome21. In this study, an interaction network between DcWRKYs and other carrot proteins was constructed. Thirty-eight DcWRKY proteins showed co-expression relationships with other proteins in carrot genome, indicating that these DcWRKYs interact with other proteins to modulate stress resistance. The DcWRKY20, was identified in a yeast-two-hybrid system that interacted directly with two mitogen-activated protein kinases DcMAPK1 and DcMAPK4. In Arabidopsis, MAPKs are important regulating factors and act by phosphorylating transcription factors, which subsequently activates transcription of other genes, including WRKY genes46,47. In Arabidopsis, WRKY33 was subject to post-translational modification by MAPK4 that was involved in pathogens and SA-mediated responses48. The phosphorylation of OsWRKY30 by MAPKs was crucial in order for OsWRKY30 to perform its biological function in rice49. Here we found that DcWRKY20 can interact with MAPKs, speculating that DcWRKY20 activity may be regulated by post-translational modifications with MAPKs. We also confirmed the W-box binding activity of several DcWRKY factors by yeast one-hybrid system. However, no obviously interactions of these DcWRKYs and the W-box element were observed in yeast cells (data not shown), suggesting that they may not have direct control relationship. Also the absence of interaction with W-box in yeast may be due to the absence of required co-factors, such as phosphorylation11, zinc-ions50.
Evidence has demonstrated that WRKY transcription factor are involved in plant growth and development. The WRKY transcription factor in rice, OsWRKY78, can regulates stem elongation and seed development51. Overexpression OsWRKY31 gene enhances disease resistance and affects root growth and auxin response in transgenic rice plants52. GhWRKY15, a member of the WRKY family identified from cotton, is involved in disease resistance and plant development53. Expression analyses in carrot root development helped to screen WRKY genes which may be involved in carrot root development. The DcWRKY3, DcWRKY8 DcWRKY24, DcWRKY64 and DcWRKY88, were abundantly expressed at all four stages of development in carrot root. Notably, the orthologous genes of DcWRKY3 and DcWRKY8 in Arabidopsis, AtWRKY6 (AT1G62300.1) and AtWRKY53 (AT4G23810.1), which have been confirmed to play an important role in leaf development35,54. This result suggested that DcWRKY3 and DcWRKY8 may have similar functions to their orthologous Arabidopsis genes during plant development.
Transcriptome analysis revealed that a large number of drought, cold and high-salinity stress-responsive genes, including numerous WRKY genes, were identified in Arabidopsis39. The comparative analysis of DcWRKY genes with their homologous AtWRKY genes helped to predict the potential functions of DcWRKY proteins. Following homologous gene annotations in Arabidopsis, we deduced the functional roles of DcWRKYs. A qRT-PCR experiment showed that the expression profile of DcWRKY genes agreed well with the WRKY genes in Arabidopsis. The two Arabidopsis WRKY genes AtWRKY33 (AT2G38470.1) and AtWRKY40 (AT1G80840.1) were induced by biotic and abiotic stresses55,56. Their orthologous genes in carrot are DcWRKY30 and DcWRKY31, which were also found to be evidently upregulated under drought, salt and pathogenic stresses. As plants evolved from lower to higher, plants have established a series of mechanisms of plant growth and development, metabolic regulation and stress response. The rapid expansion of the WRKY gene family may be a way to meet the requirements for these pathways. Most stress-resistance traits are often controlled by multiple genes57,58. The expression patterns of several DcWRKY genes significantly changed, indicating that these genes all appeared to be involved in biotic and abiotic stress response. We expect that future research will reveal the accurate regulation mechanisms of WRKY genes in signaling pathways and stress responses.
Materials and Methods
Identification of putative WRKY genes in carrot
The genome sequences of carrot (Daucus carota L. cv. Kuroda) were downloaded from the Carrot Genome Project web site (http://apiaceae.njau.edu.cn/carrthe otdb/index.php)30 and the transcriptome sequences were downloaded from NCBI SRA database59 and Carrot Genome Project web site30. The hidden Markov model (HMM) of the WRKY domain (PF03106) was downloaded from the Sanger database (http://pfam.sanger.ac.uk/family/WRKY). All the putative carrot DcWRKY factors were obtained via screening carrot genome sequences and transcriptome sequences by HMMER 3.0 software (http://hmmer.janelia.org/) using default parameters. The sequences were then submitted to the NCBI database (http://ncbi.nlm.nih.gov/) to search for WRKY domain. All the nonredundant gene sequences encoding complete WRKY domains were considered as putative WRKY genes.
Sequence alignments and phylogenetic analysis
The database of the Arabidopsis WRKY family factors was downloaded from the plant transcription factor database (http://www.arabidopsis.org/). The WRKY family databases of other species were downloaded from the plant transcription factor database (http://planttfdb.cbi.pku.edu.cn/)3. All WRKY factors were classified into subgroups based on the sequence alignments of WRKY proteins in carrot and Arabidopsis using ClustalW with default parameters60. The domain region of the WRKY proteins from carrot and Arabidopsis were used to construct a phylogenetic tree with MEGA5.0 using the neighbor-joining method with 1000 bootstrap replicates61.
Characterization of conserved motif distributions and structures of DcWRKY factors
Analysis of the exon-intron organization of DcWRKY genes was performed by comparing coding sequences with their corresponding genomic sequences using the GSDS software (http://gsds.cbi.pku.edu.cn)62. Conserved motifs for each deduced DcWRKY amino acid sequence were analyzed by MEME Suite (version4.9.0; http://meme.nbcr.net/meme/)63. The parameters were set as follows: maximum number, 20; minimum width, 10; and maximum width, 50. The compositions as well as physical and chemical characterizations of deduced DcWRKY proteins were analyzed by the Protparam tool (http://web.expasy.org/protparam) and the Sequence Manipulation Suite (http://www.bio-soft.net/sms/). The subcellular locations were predicted using WoLF PSORT (http://wolfpsort.org)64 and TargetP1.1 server (http://www.cbs.dtu.dk/services/TargetP)65. The upstream 1,500 bp regions of all DcWRKY genes were analyzed to determine the cis-regulatory elements by using the plant database PlantCARE (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/)66.
Subcellular localization
To test the predicted nuclear localization patterns, the full length of three DcWRKY genes (DcWRKY11, DcWRKY45, DcWRKY80) without the stop codon were amplified with primers as shown in Table S2. The PCR products were inserted into a GFP-fusion expression vector PA7 and transferred to tobacco leaves to determine the subcellular localization. Empty vector 35S::GFP was used as control. Fluorescence images of GFP fusion proteins were observed using the LSM780 confocal microscopy imaging system (Zeiss, Germany).
Identification of orthologous and paralogous WRKY factors
The orthologous, paralogous and coorthologous WRKY factors in alga (C. reinhardtii), moss (P. patens), fern (S. moellendorffii), Picea abies, Arabidopsis, carrot, grape, apple and rice were identified using OrthoMCL (http://orthomcl.org/orthomcl/) with default settings67. The interaction network associated with the Arabidopsis WRKY orthologous factors in carrot was constructed using the Arabidopsis Interactions Viewer and cytoscape software68.
Yeast two-hybrid assay
For the yeast two-hybrid assay, the DcWRKY20 cDNA fragment was cloned into the pGBKT7 vector and the DcMAPK1 and DcMAPK4 cDNA fragments were cloned into the pGADT7 vector, respectively. Unique amplified primers for cloning these three genes were represented in Table S2. These vectors were co-transformed into the yeast AH109 strain and the transformants were identified with X-α-gal according to a manufacture’s protocol (Clontech, Takara, Dalian, China). Yeast co-transformation with pGBK7-53 and pGADT7-T was used as a positive interaction control and co-transformation with pGBKT7-Lam and pGADT7-T was used as a negative control.
Transcript abundance analysis
Transcript abundance analysis in carrot root development was based on transcriptome data (SRR2177455). Transcript abundance analysis of DcWRKY genes were estimated by calculating read density as ‘reads per kilobase of exon model per million mapped reads’ (RPKM)69. The heatmap was performed by Multiexperiment Viewer software (http://www.tm4.org/)70.
Plant materials, abiotic and biotic treatments and qRT-PCR
Experimental samples were obtained from two-month-old ‘Kurodagosun’ carrot seedlings, which were grown in pots in a controlled-environment growth chamber. The temperature was set at 4 °C for the cold treatment and 38 °C for the heat treatment. Salt and drought treatments were carried out by irrigating with 200 mM NaCl and 20% PEG6000, respectively. Seedlings irrigated with sterile water were used as blank control. For plant biotic damage, whitefly was mechanically inoculated on the carrot leaves by rubbing the leaf with the virus and adult aphids settled on carrot plants for the aphid tests. After 7 days, infective-stage leaves were collected and used for bioassays. All samples were immediately frozen in liquid nitrogen and stored at −70 °C.
Total RNAs were extracted from the samples using the total RNA kit (Tiangen, Beijing, China) and then reverse transcribed into cDNAs using the PrimeScript RT reagent kit (TaKaRa, Dalian, China). Three independent PCR reactions were carried out for each gene using the MyiQ Single-Color RT-PCR detection system (Bio-Rad, Hercules, CA, USA). The PCR amplification profile was as follows: 95 °C for 30 s and 40 cycles of 94 °C for 5 s and 60 °C for 30 s, at last a melting curve (65–95 °C, at increments of 0.5 °C) was generated to check the amplification specificity. The relative gene expression was calculated with the 2−ΔΔCT method71. All primers used are shown in Table S2. The DcTUB gene was used as an internal control to normalize the expression of DcWRKY genes.
Additional Information
How to cite this article: Li, M.-Y. et al. Genomic identification of WRKY transcription factors in carrot (Daucus carota) and analysis of evolution and homologous groups for plants. Sci. Rep. 6, 23101; doi: 10.1038/srep23101 (2016).
References
Valliyodan, B. & Nguyen, H. T. Understanding regulatory networks and engineering for enhanced drought tolerance in plants. Curr Opin Plant Biol 9, 189–195 (2006).
Hobert, O. Gene regulation by transcription factors and microRNAs. Science 319, 1785–1786 (2008).
Jin, J. P., Zhang, H., Kong, L., Gao, G. & Luo, J. C. PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors. Nucl Acids Res, 42, D1182–D1187 (2014).
Riechmann, J. L. et al. Arabidopsis transcription factors: genome-wide comparative analysis among eukaryotes. Science 290, 2105–2110 (2000).
Goff, S. A. et al. A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 296, 92–100 (2002).
Eulgem, T., Rushton, P. J., Robatzek, S. & Somssich, I. E. The WRKY superfamily of plant transcription factors. Trends Plant Sci 5, 199–206 (2000).
van Verk, M. C., Pappaioannou, D., Neeleman, L., Bol, J. F. & Linthorst, H. J. A novel WRKY transcription factor is required for induction of PR-1a gene expression by salicylic acid and bacterial elicitors. Plant physiol 146, 1983–1995 (2008).
Guo, C. L. et al. Evolution and expression analysis of the grape (Vitis vinifera L.) WRKY gene family. J Exp Bot 65, 1513–1528 (2014).
Wu, K. L., Guo, Z. J., Wang, H. H. & Li, J. The WRKY family of transcription factors in rice and Arabidopsis and their origins. DNA Res 12, 9–26 (2005).
Ishiguro, S. & Nakamura, K. Characterization of a cDNA encoding a novel DNA-binding protein, SPF1, that recognizes SP8 sequences in the 5′ upstream regions of genes coding for sporamin and β-amylase from sweet potato. Mol Gen Genet 244, 563–571 (1994).
Rushton, P. J. et al. Interaction of elicitor-induced DNA-binding proteins with elicitor response elements in the promoters of parsley PR1 genes. EMBO J 15, 5690–5700 (1996).
Dong, J. X., Chen, C. H. & Chen, Z. X. Expression profiles of the Arabidopsis WRKY gene superfamily during plant defense response. Plant Mol Biol 51, 21–37 (2003).
Qiu, Y. P., Jing, S. J., Fu, J., Li, L. & Yu, D. Q. Cloning and analysis of expression profile of 13WRKY genes in rice. Chinese Sci Bull 49, 2159–2168 (2004).
Ülker, B. & Somssich, I. E. WRKY transcription factors: from DNA binding towards biological function. Curr Opin Plant Biol 7, 491–498 (2004).
Wang, Q. S. et al. WRKY gene family evolution in Arabidopsis thaliana. Genetica 139, 973–983 (2011).
Rensing, S. A. et al. The Physcomitrella genome reveals evolutionary insights into the conquest of land by plants. Science 319, 64–69 (2008).
Zhang, Y. J. & Wang, L. J. The WRKY transcription factor superfamily: its origin in eukaryotes and expansion in plants. BMC Evol Biol 5, 1 (2005).
Eulgem, T. & Somssich, I. E. Networks of WRKY transcription factors in defense signaling. Curr Opin Plant Biol 10, 366–371 (2007).
Jiang, W. B. & Yu, D. Q. Arabidopsis WRKY2 transcription factor mediates seed germination and postgermination arrest of development by abscisic acid. BMC Plant Biol 9, 96 (2009).
De Geyter, N., Gholami, A., Goormachtig, S. & Goossens, A. Transcriptional machineries in jasmonate-elicited plant secondary metabolism. Trends Plant Sci 17, 349–359 (2012).
Pandey, S. P. & Somssich, I. E. The role of WRKY transcription factors in plant immunity. Plant physiol 150, 1648–1655 (2009).
Chen, L. G. et al. The role of WRKY transcription factors in plant abiotic stresses. BBA-Gene Regul Mech 1819, 120–128 (2012).
Ren, X. Z. et al. ABO3, a WRKY transcription factor, mediates plant responses to abscisic acid and drought tolerance in Arabidopsis. Plant J 63, 417–429 (2010).
Niu, C. F. et al. Wheat WRKY genes TaWRKY2 and TaWRKY19 regulate abiotic stress tolerance in transgenic Arabidopsis plants. Plant Cell Environ 35, 1156–1170 (2012).
Zhou, Q. Y. et al. Soybean WRKY-type transcription factor genes, GmWRKY13, GmWRKY21 and GmWRKY54, confer differential tolerance to abiotic stresses in transgenic Arabidopsis plants. Plant Biotechnol J 6, 486–503 (2008).
Pandey, S. P., Roccaro, M., Schön, M., Logemann, E. & Somssich, I. E. Transcriptional reprogramming regulated by WRKY18 and WRKY40 facilitates powdery mildew infection of Arabidopsis. Plant J 64, 912–923 (2010).
Giacomelli, J. I., Weigel, D., Chan, R. L. & Manavella, P. A. Role of recently evolved miRNA regulation of sunflower HaWRKY6 in response to temperature damage. New Phytol 195, 766–773 (2012).
Maeda, K., Kimura, S., Demura, T., Takeda, J. & Ozeki, Y. DcMYB1 acts as a transcriptional activator of the carrot phenylalanine ammonia-lyase gene (DcPAL1) in response to elicitor treatment, UV-B irradiation and the dilution effect. Plant Mol Biol 59, 739–752 (2005).
Guan, Y., Ren, H., Xie, H., Ma, Z. & Chen, F. Identification and characterization of bZIP-type transcription factors involved in carrot (Daucus carota L.) somatic embryogenesis. Plant J 60, 207–217 (2009).
Xu, Z. S., Tan, H. W., Wang, F., Hou, X. L. & Xiong, A. S. CarrotDB: a genomic and transcriptomic database for carrot. Database 5, 1929–1245 (2014).
Chen, C. H. & Chen, Z. X. Potentiation of developmentally regulated plant defense response by AtWRKY18, a pathogen-induced Arabidopsis transcription factor. Plant Physiol 129, 706–716 (2002).
Miao, Y., Laun, T. M., Smykowski, A. & Zentgraf, U. Arabidopsis MEKK1 can take a short cut: it can directly interact with senescence-related WRKY53 transcription factor on the protein level and can bind to its promoter. Plant Mol Biol 65, 63–76 (2007).
Seki, M. et al. Monitoring the expression profiles of 7000 Arabidopsis genes under drought, cold and high-salinity stresses using a full-length cDNA microarray. Plant J 31, 279–292 (2002).
Pan, Y. J., Cho, C. C., Kao, Y. Y. & Sun, C. H. A novel WRKY-like protein involved in transcriptional activation of cyst wall protein genes in Giardia lamblia. J Biol Chem 284, 17975–17988 (2009).
Robatzek, S. & Somssich, I. E. A new member of the Arabidopsis WRKY transcription factor family, AtWRKY6, is associated with both senescence- and defence-related processes. Plant J 28, 123–133 (2001).
Johnson, C. S., Ben, K. & Smyth, D. R. TRANSPARENT TESTA GLABRA2, a trichome and seed coat development gene of Arabidopsis, encodes a WRKY transcription factor. Plant cell 14, 1359–1375 (2002).
Devaiah, B. N. & Raghothama, K. G. WRKY75 transcription factor is a modulator of phosphate acquisition and root development in Arabidopsis. Plant Physiol 143, 1789–1801 (2007).
Cheong, Y. H. et al. Transcriptional profiling reveals novel interactions between wounding, pathogen, abiotic stress and hormonal responses in Arabidopsis. Plant Physiol 129, 661–677 (2002).
Matsui, A. et al. Arabidopsis transcriptome analysis under drought, cold, high-salinity and ABA treatment conditions using a tiling array. Plant Cell Physiol 49, 1135–1149 (2008).
Brand, L. H., Fischer, N. M., Harter, K., Kohlbacher, O. & Wanke, D. Elucidating the evolutionary conserved DNA-binding specificities of WRKY transcription factors by molecular dynamics and in vitro binding assays. Nucleic Acids Res 41, 9764–9778 (2013).
Yang, X. & Tuskan, G. A. Divergence of the Dof gene families in poplar, Arabidopsis and rice suggests multiple modes of gene evolution after duplication. Plant Physiol 142, 820–830 (2006).
Van de Peer, Y., Maere, S. & Meyer, A. The evolutionary significance of ancient genome duplications. Nat Rev Genet 10, 725–732 (2009).
Kaessmann, H. Origins, evolution and phenotypic impact of new genes. Genome Res 20, 1313–1326 (2010).
Bowers, J. E., Chapman, B. A., Rong, J. & Paterson, A. H. Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 422, 433–438 (2003).
Tang, H. et al. Synteny and collinearity in plant genomes. Science 320, 486–488 (2008).
Zhang, S. & Klessig, D. F. MAPK cascades in plant defense signaling. Trends Plant Sci 6, 520–527 (2001).
Cvetkovska, M., Rampitsch, C., Bykova, N. & Xing, T. Genomic analysis of MAP kinase cascades in Arabidopsis defense responses. Plant Mol Biol Rep 23, 331–343 (2005).
Zheng, Z. Y., Qamar, S. A., Chen, Z. X. & Mengiste, T. Arabidopsis WRKY33 transcription factor is required for resistance to necrotrophic fungal pathogens. Plant J 48, 592–605 (2006).
Shen, H. S. et al. OsWRKY30 is activated by MAP kinases to confer drought tolerance in rice. Plant Mol Biol. 80, 241–253 (2012).
Pater, S., De, Greco, V., Pham, K., Memelink, J. & Kijne, J. Characterization of a zinc-dependent transcriptional activator from Arabidopsis. Nucleic Acids Res 24, 4624–4631 (1996).
Zhang, C. Q. et al. The WRKY transcription factor OsWRKY78 regulates stem elongation and seed development in rice. Planta 234, 541–554 (2011).
Zhang, J., Peng, Y. L. & Guo, Z. J. Constitutive expression of pathogen-inducible OsWRKY31 enhances disease resistance and affects root growth and auxin response in transgenic rice plants. Cell Res 18, 508–521 (2008).
Yu, F. et al. GhWRKY15, a member of the WRKY transcription factor family identified from cotton (Gossypium hirsutum L.), is involved in disease resistance and plant development. BMC Plant Biol 12, 144 (2012).
Miao, Y., Laun, T., Zimmermann, P. & Zentgraf, U. Targets of the WRKY53 transcription factor and its role during leaf senescence in Arabidopsis. Plant Mol Bioly 55, 853–867 (2004).
Li, S. J., Fu, Q. T., Chen, L. G., Huang, W. D. & Yu, D. Q. Arabidopsis thaliana WRKY25, WRKY26 and WRKY33 coordinate induction of plant thermotolerance. Planta 233, 1237–1252 (2011).
Van Aken, O., Zhang, B., Law, S., Narsai, R. & Whelan, J. AtWRKY40 and AtWRKY63 modulate the expression of stress-responsive nuclear genes encoding mitochondrial and chloroplast proteins. Plant Physiol 162, 254–271 (2013).
Fujita, M. et al. Crosstalk between abiotic and biotic stress responses: a current view from the points of convergence in the stress signaling networks. Curr Opin Plant Biol 9, 436–442 (2006).
Yamaguchi-Shinozaki, K. & Shinozaki, K. Transcriptional regulatory networks in cellular responses and tolerance to dehydration and cold stresses. Annu Rev Plant Biol 57, 781–803 (2006).
Iorizzo, M. et al. De novo assembly and characterization of the carrot transcriptome reveals novel genes, new markers and genetic diversity. BMC Genomics 12, 389 (2011).
Thompson, J. D., Higgins, D. G. & Gibson, T. J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22, 4673–4680 (1994).
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance and maximum parsimony methods. Mol Biol Evol 28, 2731–2739 (2011).
Hu, B. et al. GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics (Oxford, England ) 31, 1296–1297 (2015).
Bailey, T. L. et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37, W202–W208 (2009).
Horton, P. et al. WoLF PSORT: protein localization predictor. Nucleic Acids Res 35, W585–W587 (2007).
Emanuelsson, O., Brunak, S., von Heijne, G. & Nielsen, H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc 2, 953–971 (2007).
Postel, D., Vanlemmens, P., Gode, P., Ronco, G. & Villa, P. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res 30, 325–327 (2002).
Li, L., Stoeckert, C. J. & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13, 2178–2189 (2003).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504 (2003).
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5, 621–628 (2008).
Saeed, A. et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34, 374 (2003).
Pfaffl, M. W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29, e45 (2001).
Acknowledgements
The research was supported by Jiangsu Natural Science Foundation (BK20130027); New Century Excellent Talents in University (NCET-11-0670); Priority Academic Program Development of Jiangsu Higher Education Institutions Project.
Author information
Authors and Affiliations
Contributions
A.S.X. and M.Y.L. conceived and designed the experiments. M.Y.L., Z.S.X., C.T., Y.H. and W.F. performed the experiments. M.Y.L., Z.S.X. and A.S.X. analyzed the data. A.S.X. contributed reagents/materials/analysis tools. M.Y.L. wrote the paper. M.Y.L. and A.S.X. revised the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Li, MY., Xu, ZS., Tian, C. et al. Genomic identification of WRKY transcription factors in carrot (Daucus carota) and analysis of evolution and homologous groups for plants. Sci Rep 6, 23101 (2016). https://doi.org/10.1038/srep23101
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep23101
This article is cited by
-
Mechanisms of salinity tolerance and their possible application in the breeding of vegetables
BMC Plant Biology (2023)
-
WRKY transcription factors: evolution, regulation, and functional diversity in plants
Protoplasma (2023)
-
Transcriptome sequencing and differential expression analysis of natural and BTH-treated wound healing in potato tubers (Solanum tuberosum L.)
BMC Genomics (2022)
-
Identification and Expression Analysis of the SBP-box Gene Family Related to Abiotic Stress in Tea Plant (Camellia sinensis (L.) O. Kuntze)
Plant Molecular Biology Reporter (2022)
-
Dynamic differential evolution schemes of WRKY transcription factors in domesticated and wild rice
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
