
Abstract
Subtropical China is a centre of speciation and well known for its high biological diversity and endemism. To understand the impact of historical, geographical and ecological factors on the intraspecific lineage divergence of invertebrates, we examined these processes in a semiaquatic bug, Microvelia horvathi (Hemiptera: Veliidae). Three hypotheses were developed using ecological niche models (ENM). We tested these hypotheses using mitochondrial (COI + COII) and nuclear data (ITS1 + 5.8S + ITS2). The phylogenic analysis revealed a shallow divergence in mitochondrial data. Clade I was mostly confined to the northern region and clade II was nearly restricted to the southern region. The historical process of Pleistocene climatic fluctuations during the LGM promoted divergence, along with such geographical barriers as the Wuyi, Nanling and Xuefeng mountains and ecological factors of temperature and vegetation type, contributed to these shallow genetic divergences and helped maintain them. The north-south population differentiation probably occurred during the transition from LIG to LGM, with post-LGM population expansion. The results of genetic data were mostly consistent with the spatial predictions from ENM. Our study emphasizes the multiple effects influencing genetic population differentiation and also contributes to our knowledge of the phylogeography of other aquatic organisms in subtropical China.
Similar content being viewed by others


Introduction
Understanding the historical and environmental factors that influence the genetic structure of populations is important for studying the mechanisms of adaptive divergence and speciation. In the Northern Hemisphere, patterns of contemporary population genetic structure of extant species have been greatly affected by Pleistocene climatic fluctuations, such as range expansion or isolation in glacial refugia1 and geographical features, such as spatial distance and geographical barriers2. Besides these two factors, Very recent environmentally mediated episodes (ecological factors) might also contribute to genetic differentiation3.
The Asian subtropics are a global biodiversity hotspot considered to be one of the most important areas of endemism for many organisms4. This region experienced repeated climate changes during the Pleistocene and also exhibited dramatic geographic heterogeneity5. In China, the subtropical zone ranges from the eastern Tibetan Plateau to the Pacific Ocean and from the Qingling Mountains-Huai River line (34° N) to the tropical south (20° N). Since Pleistocene climatic fluctuations in subtropical China were less extensive than in Western Europe and North America and most areas were not covered by large ice sheets during the late Pleistocene4, it had been argued that homogenizing gene flow during the Pleistocene inhibited divergence among the populations6. However, in a few cases Pleistocene lineage diversification had been documented in some organisms7,8. This raised the question why some population experienced continuous gene flow while others experienced diversification. The study of demographic expansion over time also showed contradictory patterns in subtropical China. Some species, such as the fig-wasp, Ceratosolen sp.9 and the tree, Castanopsis eyrei10, revealed dramatic post-LGM expansion scenarios. Other species showed the opposite, for instance, the green-backed tit, Parus monticolus11 and another semi-aquatic bug, Microvelia dougalsi douglasi12 underwent unusual pre-LGM expansion. These apparently conflicting results suggested that subtropical China underwent complex Pleistocene climate changes throughout the last ice age cycles even though not covered by the ice sheet13, causing different responses by different organisms. In addition, geological tectonic uplift in subtropical China occurred in the late Miocene and Pliocene, resulting in relatively high mountain systems such as the eastern Himalayas and the mountains of South and Southwest China (the Hengduan, Wuyi, Nanling Mts.)14,15. These geographically heterogeneous mountain systems in subtropical China might have caused genetic diversification among populations adapting to the new stable environments and mountainous areas also might have harboured many refugial populations in periods of adverse climatic conditions13, all of which led to the formation of new lineages/taxa and contributed to high genetic diversity.
Phylogeography has benefitted from recent developments in GIS technology, which now allows a more powerful investigation of the geographic components of genetic variation16. Ecological niche models (ENM) projects a model of past climatic conditions to explore the historic distribution of a species and the degree to which its distribution has changed17. This approach to generating a historical distribution is spatially explicit and independent from phylogeographical inference18. The use of ENM to explore past species distributions could supplement the limited invertebrate fossil record in subtropical China and, in combination with molecular data, enhance our understanding of underlying historical events and population dynamics (dispersal barriers, population size changes and localization of putative Pleistocene refugia) through time16.
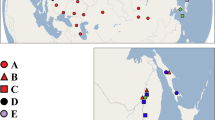
Many previous phylogeographical studies have focused on plant and vertebrate species in subtropical China10,19. To reveal a more complete phylogeographical fauna pattern, common and widespread species need to be investigated, especially invertebrates. To understand the impact of Pleistocene climatic fluctuations, geographical barriers and ecological factors on the phylogeography of invertebrates, we examined these processes on a widespread subtropical semiaquatic bug, Microvelia horvathi Lundblad 1933, which belongs to the familiy Veliidae (Insecta: Hemiptera) and is found in the subtropics of China, Japan and Korea. This species usually lives on the quiet water surfaces of nearshore areas, including rain pools, ditches, swampy ground, or paddy fields20. Its distribution range is mostly endemic to subtropical China and restricted by geographical barriers (the Taihang, Qinling and Hengduan Mts.) (Fig. 1), suggesting that this small semi-aquatic insect might have a relatively low dispersal ability and be sensitive to climate fluctuations. Its distribution range includes a series of east-west mountain ranges (the Xuefeng, Nanling and Wuyi Mts.) (Fig. 1). Thus, gene flow among populations, especially populations on both sides of these mountains, is likely to be obstructed or slowered and genetic differentiation is expected.

Sample size and halotype frequencies of two major clades of M. horvathi, designated clade I and II according to the molecular phylogeny in Fig. 3a.
Frequencies of clade I and II haplotypes in each population are showed by the pie graph. Names of mountain ranges in the map were labelled. Figure was generated in ArcGIS 10 (Environmental Systems Research Institute).
In this study, we first generated potential distribution maps of M. horvathi in the present, the LGM and the LIG. ENM projected that M. horvathi potential spaces contracted and were ultimately isolated in two mainly separated refugia (northern and southern refugia) where they underwent major divergence (Fig. 2, middle panel) during the LGM period. After that, the populations expanded from the separated refugia and came into contact with gene exchange, resulting in a largely continuous range in subtropical China exhibited in our current predicted space (Fig. 2, left panel). Based on the result of ENM, we derived three hypotheses: If the populations within these two refugia still had strong gene exchange with each other through secondary contact, counteracting the accumulated major divergence during the LGM, molecular data should reveal a relatively homogenizing genetic pattern between the northern and southern populations in this scenario (Hypothesis 1). Otherwise, if geographical barriers or ecological factors inhibited or slowed down the homogenizing gene flow between northern and southern populations, in this scenario a shallow genetic divergence pattern should be revealed between northern and southern populations in the molecular data (Hypothesis 2). Finally, since the ENM showed unsuitable LGM climate conditions and suitable current climate conditions (Fig. 2), we expected analyses of demographic history (mtDNA) to show that population expansion followed the same pattern (Hypothesis 3).

Hindcasting the current niche model (left panel) onto the LIG (right panel) and LGM (middle panel) periods in East Asia using Maxent.
Niche model results were modified in ArcGIS 10 (Environmental Systems Research Institute).
Results
Paleoclimate niche modeling reconstruction
A high AUC value was obtained from the current potential distribution (AUC = 0.987), indicating good predictive model performance. The current niche-predicted distribution of this species was generally similar to its actual distribution, with a potentially continuous range in subtropical China and a fragmentary range in southern Japan and Korea (Fig. 2). When projecting the current niche into historical climate conditions, the suitable climate space continued to stay in situ during the LIG period, but expanded moderately toward southwestern China and most parts of Japan and Korea (Fig. 2). When the ice age arrived (LGM period), under the CCSM model, the species’ potential range contracted greatly and was mainly located in two segregated refugia (the Sichuan Basin space and a space from northern Vietnam to northern Guangxi) (Fig. 2).
Genetic polymorphism and phylogenetic analysis
For the mitochondrial data, 1405 bp of protein-coding regions of mitochondrial genome were obtained from 250 individuals, including sections of the COI (726 bp) and COII (679 bp) genes. Ninety unique haplotypes were derived among all individuals. The 96 polymorphic sites included 67 singleton variable and 29 parsimony informative sites. The nucleotide diversities and haplotype diversities ranged from 0.00057 to 0.00360 with an average of 0.00262 and 0.378 to 1.000 with an average of 0.845, respectively (Table 1). NJ analyses produced tree topology based on combined mitochondrial sequences (COI + COII) (Fig. 3a). The whole haplotypes were shallowly separated into two supported major clades, clade I and clade II. The haplotype frequencies of clade I and clade II were shown in a pie graph revealing that the geographical distributions of the two monophyletic clades were uneven (Fig. 1). Most haplotypes of clade I were located widely in the northern region of subtropical China, whereas most haplotypes of clade II were commonly found in the narrower southern region (Fig. 1). Interestingly, the intermediate populations (ZJTS, JXLN and GZYH) living between the northern and southern regions simultaneously possessed the haplotype diversities of both lineages. The network analysis based on all ninety haplotypes showed the same pattern, with two well-defined independent haplogroups (Figs 3b and 4a). In the haplotype network (Fig. 4a), h1, h12, h15 and h25 were the four most frequent haplotypes, characterizing 37.6%, 6%, 3.2% and 9.6% of individuals respectively. The h1 ancestral haplotype was mostly found in the northern region, whereas h12, h15 and h25 were the ancestral haplotypes found in the southern region (Table 1).

(a) Phylogenetic NJ tree analyses of all ninety haloptypes based on mitochondrial markers (COI + COII). Branches of above 50% bootstrap support are showed in number. (b) The Neighbor-Net algorithm. Clade I and clade II are shown in blue (at the top) and green (at the bottom), respectively. (c) Historical demographic trends of the whole data represented by Bayesian skyline plot (BSP) based on mitochondrial data. X axis is the time scale before present. Y axis is the estimated effective population size. Estimates of means are joined by a solid line. LGM represents Last Glacial Maximum.

Median joining haplotypes network constructed using Network.
Haplotype circle size denotes the number of sampled individuals. Numbers of base pair changes (no number = 1 bp) are given. (a) Based on mitochondrial data. (b) Based on nuclear data.
For the nuclear sequencing data, 1039 bp fragments were successfully obtained from 237 individuals, including ITS1, 5.8S and ITS2 genes. 19 unique haplotypes were derived among all individuals. The 19 polymorphic sites included 16 singleton variable and 3 parsimony informative sites. The nucleotide diversities and haplotype diversities ranged from 0 to 0.00123 with an average of 0.00055 and 0.000 to 0.833 with an average of 0.478, respectively (Table S1). The mitochondrial DNA structure of lineage differentiation in M. horvathi was not apparent in the nuclear data (Fig. 4b).
Phylogeographic structure and hierarchical partitioning of genetic structure
SAMOVA showed a maximum value in FCT of K = 2 (Fig. 5). The two groups corresponded genetically and geographically (i.e. north and south populations) in subtropical China. Zones of genetic discontinuities identified by Barrier 2.2 showed the potential geographical barriers associated with the genetic abruption (Fig. S1). The bold lines mainly divided the entire sampling region into a northern and a southern subregion, reflecting significant genetic/geographical isolation between them. Isolations between the northern and southern subregions (Fig. S1) were mostly consistent with the results of SAMOVA. The nuclear sequencing data showed much lower genetic diversities than the mitochondrial data and revealed no distinct phylogeographical structure pattern (Fig. 4b). Therefore SAMOVA and Barrier analysis were not implemented in nuclear data.

Values of fixation indices (F) as a function of number of groups (K) based on mitochondrial data.
FCT is the differentiation between groups, FST is the differentiation between populations among groups and FSC is the differentiation between populations within groups.
AMOVA analysis showed a relatively significant genetic variance between the northern and southern groups in mitochondrial DNA data (48.45%, P < 0.05) (Table S2). However, the same analyses of nuclear DNA data attributed most genetic variance (83.69%) to differentiation among individuals within populations (Table S2). The Mantel tests revealed a significant positive relationship in the mitochondrial DNA (r = 0.5590, P < 0.001), but not in nuclear DNA (r = −0.0478, P = 0.3120) (Fig. S2a,b).
Historical demographic changes in mitochondrial data
The negative values of Tajima’s D, Fu and Li’s D* and the significance of Fu’s Fs (Table 2) suggested a scenario of recent demographic expansion in the northern and southern populations. For the whole dataset, the negative values of D, D*, together with the significance of Fs (Table 2) also suggested an overall past population expansion. Divergence time between clade I and clade II from the results of BEAST was moderately in keeping with the result of IMa2, which was estimated to be 0.051 Ma (95% HPD: 0.073–0.031 Ma) and 0.022 Ma (95% HPD: 0.033–0.016 Ma) separately (i.e. population divergence occurred during the LIG to LGM transition). From t = τ/2u, we estimated the expansion time ranged from approximately 12.75–25.50 Ka and the Bayesian skyline demographic reconstructions showed a pattern of population growth through time in which the plots started from 15 Ka (i.e., population expansion occurred in the post-LGM period) (Fig. 3c).
Ecological vicariance analyses
SEEVA analysis compared eight environmental variables between the sister lineages for M. horvathi. Three of eight comparisons were significant (Table S3). The phylogenetic splits between the north and south lineages showed strong divergence in terms of annual mean temperature (BIO1, D = 0.73*), minimum temperature of the coldest month (BIO6, D = 1.0*) and vegetation (D = 0.82*) (Fig. 6). Although M. horvathi depended on the water habitat for survival, the split between the north and south lineages only showed a slight divergence in annual precipitation (BIO12, D = 0.40) and precipitation in the driest month (BIO14, D = 0.41). The PCA-env results suggested the two different lineages might not occupy identical habitat, but the differences could simply be due to the differential availability of habitat in the different regions that they occupied (Fig. S3).

The results of the spatial evolutionary and ecological vicariance analysis (SEEVA) of M. horvathi using annual mean temperature (BIO1), min temperature of coldest month (BIO6) and vegetation type as four qualitative or quantitative section states.
The blue scale of the histograms represents the four different states. Vegetation: A, temperate deciduous forest; B, evergreen broad leaved forest; C, monsoon rain forest/evergreen broad leaved forest; D, island monsoon rain forest.
Discussion
In this paper, we applied ENM to develop three hypotheses about genetic structure and population demography and tested the hypotheses by analyzing two genetic data sets in a phylogeographical framework. Below, we discuss these hypotheses in the light of our a priori expectations and the observed results.
A shallow divergence pattern between northern and southern populations was revealed by mitochondrial DNA, but no such pattern was found in nuclear DNA, which instead showed a widely shared ancestral haplotype (Fig. 4). Such discordances between mitochondrial and nuclear data might result from a variety of factors, such as incomplete lineage sorting of ancestral polymorphism, potentially sex-biased dispersal or the different evolutionary rates of different markers10. Firstly, if incomplete lineage sorting leads to the observed mitochondrial DNA pattern, we would anticipate a less shallow divergence pattern in the nuclear DNA, since the relatively larger effective population size would result in a longer sorting time21. However, analysis of the nuclear DNA data did not reveal any genetic divergence in the populations, suggesting that incomplete lineage sorting was not likely to be the main reason. Secondly, since sex-biased dispersive behavior influencing the phylogeographical structure had been studied in many animals22, we hypothesized that if females possessed native philopatry and males displayed high dispersal capacity, it would show that a significant IBD pattern detected in the mitochondrial DNA and a nonsignificant IBD pattern in nuclear DNA23. Interestingly, this hypothesis was in accordance with our IBD results (Fig. S2), which did not reject the possibility that a higher male dispersal ability was responsible for incongruence between mitochondrial DNA and nuclear DNA. However, there were no direct evidences in the literature for M. horvathi to testify whether the different sexes showed different dispersal capacity, which should be studied further through ecological and biological observations or experiments in the future. Thirdly, the reconstructed palaeodistribution from ENM prediction might show that the discordance patterns observed in mitochondrial DNA and nuclear DNA were most likely explained by the different evolutionary rates of these two markers. The ENM indicated a potentially largely continuous range in subtropical China during the LIG period with relatively high temperature than now24, but in the LGM period, the most suitable habitats contracted into two main refugia (the Sichuan Basin space and a space from northern Vietnam to northern Guangxi). Under the CCSM model, we suggested that the glacially-induced population isolation was sustained only long enough for divergence in mitochondrial DNA, which possesses a relatively fast mutation rate and in a small effective population size, but not enough for divergence in the nuclear gene (ITS), with a slower evolutionary rate and a larger effective population size. Based on the molecular evidence, the divergence in mtDNA accumulated during the LGM had not entirely disappeared though the northern and southern populations came into contact with gene exchange during the LGM to current transition. Geographical barriers and ecological factors likely inhibited or slowed down the homogenizing gene flow between the northern and southern populations, which shaped the currently shallow divergence pattern. This accorded with Hypothesis 2 above.
It has been also demonstrated that mountains, as geographical barriers, played a key role in speciation and lineage differentiation by obstructing or slower migration/gene flow between populations25. Many previous studies had paid close attention to the effects of very large mountain systems such as the Hengduan and Qinling Mts., but less attention had been paid to the effect of the smaller mountain systems in subtropical China. In the Miocene and Pliocene, intensive tectonic activity had already begun in subtropical China15. Today, the north-south fauna dividing line in subtropical China was associated with a dramatic series of east-west trending mountain ridges reaching elevations of over 2000 m above sea level and differences between valleys and ridges extending 500 m above sea level in elevation. The Wuyi, Nanling and Xuefeng Mts were characteristic in this respect. The Wuyi and Nanling Mts. acted as protective barriers against the inflow of cold air from the northwest and retained warm moist air originating from the sea, resulting in a humid climate with high rainfall26. The Xuefeng Mountains ran northeast to southwest and acted as a protective barrier against different climate environments on the eastern edge of the Guizhou Plateau. These three east-west trending mountains were an important faunal dividing line between the Palaearctic Realm and the Oriental Realm27. We suggested that gene flow between the northern and southern populations was likely to be influenced by these mountains. Our molecular data confirmed hypothesis 2, showing that M. horvathi was genetically and geographically divided into north-south clades (Fig. 1, clade I and clade II). The haplotypes of clade I were widespread in the north and the haplotypes of clade II were mainly restricted to the narrow south areas. For example, some haplotypes were confined to the north or south, a haplotype h1 pattern which was found in 21 populations mostly distributed in the north of the mountains. A similar north-south geographical differentiation had been observed in previous studies, including the Chinese canopy tree Eurycorymbus cavaleriei28 and the small freshwater fish Squalidus argentatus29. Therefore, we speculated that the Wuyi, Nanling and Xuefeng Mts. might have acted as geographical barriers preventing or slowing down gene flow between the northern and southern populations, resulting in lineage differentiation especially during the inter-glacial periods. Further, these mountains areas commonly identified as biodiversity hotspots30 and acted as important refugia during Pleistocene glacial periods because of the extremely topographical complexity which could provide different suitable stable microclimates for many species to survive during the glacial period, harboring high diversities of haplotypes and lineages. Our molecular data supported this idea; the GZYH, GXME, JXLN, GDBL, ZJTS and FJSW populations of M. horvathi were entirely located in the Wuyi, Nanling and Xuefeng Mts. and occupied very high haplotype diversities relative to the other populations (e.g. HdGZYH = 0.844, HdGXME = 0.900, HdJXLN = 0.972, HdFJSW = 0.917) and all these populations also showed haplotype diversities both from clade I and clade II (Fig. 1). Thus, we suggested that these mountain areas were refugia for M. horvathi during the glacial period. In the LGM prediction under the CCSM model, one refugium was identified in northern Guangxi extending to northern Guangdong (areas of Nanling and Xuefeng Mts.) (Fig. 2). In conclusion, the Wuyi, Nanling and Xuefeng mountains not only as geographical barriers, obstructing or slowering gene flow between northern and southern populations and promoting to lineage differentiation, but also as potentially suitable areas during the glacial period, preserving the very high genetic diversities and diverse genetic lineages.
The geographical distribution of species was also affected by both historical and present-day ecological factors. Current ecological factors were clearly important in maintaining divergence of the genetic lineages of M. horvathi. This conclusion was based on the relatively strong coincidence between the northern and southern structural groups and zoogeographical fauna in subtropical China27, which revealed that the two groups occupied different climatic regimes (the Palaearctic Realm and Oriental Realm). In this case, the effects of ecological factors might be much more prominent for lineage differentiation. The Palaearctic Realm and Oriental Realm in subtropical China showed rather prominent difference in many environmental variables, such as temperature, precipitation, elevation and vegetation types. In the Palaearctic Realm of subtropical China, M. horvathi inhabited temperate deciduous forest and evergreen broad-leaved forest. In contrast, in the Oriental Realm, it mostly inhabited monsoon rain forest, evergreen broad-leaved forest and island monsoon rain forest, which were warmer and had more precipitation during the year. Among many present-day ecological factors, SEEVA analysis (Table S3, Fig. 6) showed that temperature and vegetation type greatly contributed to the phylogenetic split between the north and south lineages. The cooler northern region and warmer southern region might have easily affected the physiological activity of M. horvathi, gradually shaping the different temperature tolerance in the different lineages. However, the precipitation had less influence on the north-south lineage differentiation in subtropical China than expected, given that M. horvathi was a semi-aquatic species dependent on a water habitat for survival. It was reasonable to assume that M. horvathi, as a very small insect, only needed minor water microhabitats and that large-scale precipitation might probably destroy microhabitats including small rain pools, ditches and swampy ground. Therefore, the main ecological constraint on local lineages could be temperature, with the genetic differentiation resulting from Pleistocene climatic fluctuations being more or less maintained. However, to clarify how different lineages of M. horvathi adapted to local ecological niches, we still need to further explore the evidence, including studies of the function- related protein-coding genes.
Estimating divergence time derived from the mitochondrial data in BEAST and IMa2 indicated that diversification of M. horvathi was fairly recent. The north-south differentiation probably occurred during the transition from the LIG to the LGM. In the MJ haplotypes network, four starlike shapes in the south region and one relatively large starlike shape in the north region (Fig. 4a) suggested that M. horvathi experienced more population expansion events in the south region than in the north region. The molecular result of neutrality tests (Table 2) and the Bayesian skyline plot (BSP) (Fig. 3c) all indicated that historical population expansion happened after the LGM period. These results were in line with the spatial prediction of ENM (Hypothesis 3) (Fig. 2). From the LIG to LGM period, when the weather gradually became increasingly dryer and colder, potential spaces for M. horvathi contracted and were gradually isolated into two largely separate northern and southern refugia (Fig. 2), after which populations in the isolated refugia had less gene exchange and accumulated major divergence. After the LGM period, when the weather turned warm and moist, the two formerly separate groups expanded and a significantly suitable range increased into the current predicted space (Fig. 2). These two independent hindcasting methods (phylogeograpic analysis and ENM) achieved a similar historical demographic change pattern. Interestingly, this result was contradictory to the previous phylogeographic studies in Microvelia douglasi douglasi12, although these two species both belong to the same genus. We speculated that the main reason for the contradictory patterns might be differences in the ecologic requirements of these two species. M. douglasi douglasi was widely distributed in the east and southeastern Asia today, but M. horvathi was mainly confined to subtropical China. The wide distributional range meant that M. douglasi douglasi exhibited much higher genetic diversity, with the potential to adapt to diverse environments easily. Secondly, in the potential LGM distribution predicted by ENM, M. dougalsi douglasi inhabited connected and relatively large areas in subtropical China and tropical Asia including Vietnam and Indonesia, while M. horvathi only inhabited segregated, narrow and discontinuous areas in subtropical China (Fig. 2). Therefore, we proposed that M. horvathi might be less adapted to the whole range of local environments in subtropical China than M. douglasi dougalsi, with relatively lower ecological tolerance and was particularly sensitive to population changes driven by Pleistocene climatic fluctuations.
Methods
Sample collection and DNA sequencing
We selected 31 sampling sites for M. horvathi across the natural distributional area, which largely covered the whole distributional range of this species in China (Fig. 1, Fig. S1). Distribution maps were generated by the software ArcGIS 10 (ESRI, Redlands, CA, USA). In each population, three to ten (mean of eight) mature males or females were collected. All the materials were deposited in the College of Life Sciences at Nankai University (Tianjin, China). Genomic DNA was extracted from the entire body excluding the genitalia. A total of 250 individuals were sequenced for mitochondrial markers (COI + COII) and nuclear markers (ITS1 + 5.8S + ITS2). Polymerase chain reactions (PCR) were performed using specific primers following Ye et al. (2014). The PCR procedure for COI, COII and nuclear markers (ITS1 + 5.8S + ITS2) included an initial denaturation at 94 °C for 2 minutes, followed by 31–33 cycles of 30 seconds at 92 °C, 30 seconds at 48–52 °C and 1 minute at 72 °C, ending with a final extension at 72 °C for 8 minutes. The chromatograms of sequences were unimodal, which excluded pseudogene disturbance. Sequences were visually proofread and aligned in Bioedit 7.1 software31.
Paleoclimate niche modeling reconstruction
A total of 47 occurrence records were obtained for niche modeling, including 33 records from the alcohol preserved specimens and 14 records from pinned specimens in Nankai University or the published literature. We chose the seven bioclimatic variables from the WorldClim database (http://www.worldclim.org/) that were thought to most likely restrict semi-aquatic bugs distribution12, namely, annual mean temperature (BIO1), mean diurnal temperature range (BIO2), maximum temperature of the warmest month (BIO5), minimum temperature of the coldest month (BIO6), annual mean precipitation (BIO12), precipitation of the wettest month (BIO13) and precipitation of the driest month (BIO14). All the variables were derived from the WorldClim data center at a resolution of 2.5-arc. We used maximum entropy implemented in Maxent (version 3.3.3 k32) to estimate niches in environmental dimensions. Analysis was run using default program conditions. Area Under Curve (AUC) of the Receiver Operating Characteristic (ROC) plot was used for model evaluation. For hindcasting the effect of Pleistocene climatic fluctuations, the current native niche models were calibrated using the above environmental variables and then transferred onto the reconstructed climatic conditions during the LGM and LIG periods. The Community Climate System Model 3 (CCSM 3) for LGM conditions was used.
Genetic polymorphism and phylogenetic analysis
We estimated genetic diversities by the number of polymorphic sites (S), haplotype distribution (Hap), number of haplotypes (Nhap), haplotype diversity (Hd) and nucleotide diversity (π), which were all calculated in DNASP 4.033. We used the uncorrected p distances to reconstruct an unrooted NJ tree to elucidate the phylogenetic relationships among mitochondrial haplotypes of M. horvathi using PAUP* 4.0b1034. Statistical support for phylogenetic grouping was assessed by 1000 bootstrap replicates. To further investigate the relationships among unique haplotypes, we constructed unrooted networks using two methods: the neighbor-net algorithm with SplitsTree 4.635 and a median-joining method36 constructed with default settings in Network 4.6.1.3 (Fluxus Technology, Suffolk, UK).
Phylogeographic structure and hierarchical partitioning of genetic structure
The SAMOVA 1.037 program was used to simulate 100 simulated annealing processes for K = 2 to 10. The potential geographical zones associated with genetic discontinuities across the entire sample region were further investigated using Monmonier’s algorithm implemented in Barrier 2.238. Mitochondrial DNA identified two major clades, which were apparently consistent with a north-south geographic partition (Figs 1 and 3a). We assessed the hierarchical partitioning of genetic structure between the two groups (north and south) resulting from SAMOVA analysis in each marker using analysis of molecular variance (AMOVA) in Arlequin 3.539. The statistical significance of variance components was assessed using 10000 random permutations. Additionally, for testing an isolation-by-distance model, the Mantel test was performed in IBDWS40 for each genetic marker using 1000 randomizations.
Historical demographic changes based on mitochondrial data
We used three neutrality tests, the Tajima’s D, Fu and Li’s D* and Fu’s Fs tests, implemented in DNASP 4.033 or Arlequin 3.539, to detect deviations from the mutation-drift equilibrium that would be indicative of changes in historical demography and natural selection. We used BEAST 1.8.241 to estimate divergence time for the mitochondrial lineages obtained. The best fit nucleotide substitution model GTR + G + I was estimated by Modeltest 3.742. Divergence time was estimated with an uncorrelated lognormal relaxed clock with the mutation rate 0.4–0.8%/Myr for M. horvathi43. We adopted one year as seven generation for M. horvathi44, using a coalescent tree prior model with the chains run for 100 million generations, checked to make sure the ESS was more than 200 and then discarded the first 10% as burn-in. We also estimated divergence time using the isolation-with-migration model implemented in IMa245. The HKY model and 0.25 as inheritance scalar were used. We set the upper prior bounds to 80 for the population size (q), 32 for the divergence time (t) and 0.125 for the migration rate (m). Final runs consisted of 1 × 108 steps with a burn-in of 1 × 107 steps, with the lowest ESS among the parameters at least above 50.
Then two methods were used to estimate population expansion time. One approach used t = τ/2u (where τ is the crest of mismatch distribution, u is the mutation rate per generation for the whole sequence and t is the expansion time in number of generations; the value u was calculated using the formula u = μk (μ is the mutation rate per nucleotide and k is the number of nucleotides). The other approach used a Bayesian coalescent-based method (Bayesian Skyline Plot, BSP) in BEAST 1.8.2. A relaxed uncorrelated lognormal molecular clock was applied. We ran the chains for 100 million generations and checked to make sure the ESS was more than 200, after which the first 10% were discarded as “burn-in”.
Ecological vicariance analyses
We used two approaches to test ecological divergences between two phylogenetic lineages of M. horvathi. First, we tested the correlations between the ecological divergences and the phylogenetic splits using spatial evolutionary and ecological vicariance analysis (SEEVA46). We extracted seven environmental variables (BIO1, BIO5, BIO6, BIO12, BIO13, BIO14 and Elevation) for each sampling site in ArcGIS (ESRI, Redlands, CA, USA) and primary GIS data layers for temperature and precipitation were obtained from the WorldClim data (http://www.worldclim.org/). Quantitative variables (temperature, elevation and precipitation) were scored as ordered continuous data, while the qualitative variables (vegetation type) were treated as non-ordered categorical data. We divided these variables into four states according to the default setting in SEEVA. We investigated the correlations between ecological shifts and the phylogenetic splits using Fisher’s exact tests and divergence indices (D). Second we used PCA-env function in the R script47 to measure niche overlaps in gridded environmental space between the two lineages48. Observed densities for each region were corrected in light of the availability of environmental space using kernel density functions. Niche overlap was measured along gradients of a multivariate analysis (i.e. principal component analysis, PCA) and the statistical tests of niche equivalency and similarity45 were computed from the density estimations of taxa in environmental space. Similarity was quantified using Schoener’s D, with values ranging from 0 to 1, or from more to less similar respectively.
Additional Information
How to cite this article: Ye, Z. et al. Phylogeography of a semi-aquatic bug, Microvelia horvathi (Hemiptera: Veliidae): an evaluation of historical, geographical and ecological factors. Sci. Rep. 6, 21932; doi: 10.1038/srep21932 (2016).
References
Hewitt, G. M. The genetic legacy of the quaternary ice ages. Nature 405, 907–913 (2000).
Hirao, T., Kubota, Y. & Murakami, M. Geographical patterns of butterfly species diversity in the subtropical Ryukyu Islands: the importance of a unidirectional filter between two source islands. J. Biogeogr. 42, 1418–1430. (2015).
Pease, K. M. et al. Landscape genetics of California mule deer (Odocoileus hemionus): the role of ecological and historical factors in generating differentiation. Mol. Ecol. 18, 1848–1862 (2009).
Edwards M. E. et al. Pollen-based biomes for Beringia 18000, 6000 and 0 14C yr bp. J. Biogeogr. 27, 621–634 (2000).
Dong, L. et al. Phylogeography of silver pheasant (Lophura nycthemera L.) across China: aggregate effects of refugia, introgression and riverine barriers. Mol. Ecol. 22, 3376–3390 (2013).
Song, G. et al. Gene flow maintains genetic diversity and colonization potential in recently range-expanded populations of an oriental bird, the light-vented bulbul (Pycnonotus sinensis, Aves: Pycnonotidae). Divers. Distrib. 19, 1248–1262 (2013).
Meng, H. Z., Li, X. C. & Qiao, P. H. Population structure, historical biogeography and demographic history of the alpine toad Scutiger ningshanensis in the Tsinling mountains of central China. PLoS ONE 9, e100729 (2014).
Wang, Y. H. et al. Molecular phylogeography and ecological niche modelling of a widespread herbaceous climber, Tetrastigma hemsleyanum (Vitaceae): insights into Plio-Pleistocene range dynamics of evergreen forest in subtropical China. New Phytol. 206, 852–867 (2015).
Lin, R. C., Yeung, C. K. & Li, S. H. Drastic post-LGM expansion and lack of historical genetic structure of a subtropical fig-pollinating wasp (Ceratosolen sp. 1) of Ficus septica in Taiwan. Mol. Ecol. 17, 5008–5022 (2008).
Shi, M. M., Michalski, S. G., Welk, E., Chen, X. Y. & Durka, W. Phylogeography of a widespread Asian subtropical tree: genetic east-west differentiation and climate envelope modelling suggest multiple glacial refugia. J. Biogeogr. 41, 1710–1720 (2014).
Wang, W. et al. Glacial expansion and diversification of an East Asian montane bird, the green-backed tit (Parus monticolus). J. Biogeogr. 40, 1156–1169 (2012).
Ye, Z., Zhu, G. P., Chen, P. P., Zhang, D. L. & Bu, W. J. Molecular data and ecological niche modelling reveal the Pleistocene history of a semi-aquatic bug (Microvelia douglasi douglasi) in East Asia. Mol. Ecol. 23, 3080–3096 (2014).
Qiu, Y. X., Fu, C. X. & Comes, H. P. Plant molecular phylogeography in China and adjacent regions: tracing the genetic imprints of Quaternary climate and environmental change in the world’s most diverse temperate flora. Mol. Phylogenet. Evol. 59, 225–244 (2011).
Quade, J., Cerling, T. E. & Bowman, J. R. Development of Asian monsoon revealed by marked ecological shift during the latest Miocene in northern Pakistan. Nature 342, 163 (1989).
Webb, T. & Bartlein, P. J. Global changes during the last 3 million years: climatic controls and biotic responses. Annu. Rev. Ecol. Syst. 23, 141–173 (1992).
Richards, C. L., Carstens, B. C. & Knowles, L. L. Distribution modelling and statistical phylogeography: an integrative framework for generating and testing alternative biogeographical hypotheses. J. Biogeogr. 34, 1833–1845 (2007).
Graham, C. H., Ferrier, S., Huettman, F., Moritz, C. & Peterson, A. T. New developments in museum-based informatics and applications in biodiversity analysis. Trends Ecol. Evol. 19, 497–503 (2004).
Collevatti, R. G. et al. A coupled phylogeographical and species distribution modeling approach recovers the demographical history of a Neotropical seasonally dry forest tree species. Mol. Ecol. 21, 5845–5863 (2012).
Dai, C. Y. et al. Profound climatic effects on two east Asian black throated tits (Ave: Aegithalidae), revealed by ecological niche models and phylogeographic analysis. PLoS ONE. 6, e29329 (2011).
Andersen, N. M. The semiaquatic bugs (Hemiptera, Gerromorpha). Phylogeny, adaptations, biogeography and classification. Entomonograph 3, 1–455 (1982).
Sefc, K. M., Payne, R. B. & Sorenson, M. D. Genetic continuity of brood-parasitic indigobird species. Mol. Ecol. 14, 1407–1419 (2005).
López-Uribe, M. M., Zamudio, K. R. & Cardoso, C. F. Climate, physiological tolerance and sex-biased dispersal shape genetic structure of Neotropical orchid bees. Mol. Ecol. 23, 1874–1890 (2014).
Cooper, S. J. B., Harvey, M. S., Saint, K. M. & Main, B. Y. Deep phylogeographic structuring of population of the trapdoor spide Moggridgea tingle (Migidae) from southwestern Australia: evidence for long-term refugia within refugia. Mol. Ecol. 20, 3219–3236 (2011).
Zhao, J. D., Shi, Y. F. & Wang, J. Comparison between Quaternary Glaciations in China and the Marine Oxygen Isotope Stage (MIS): an improved schema, Acta Geogr. Sin. 66, 867–884 (2011).
Fjeldså, J., Bowie, R. C. K. & Rahbek, C. The role of mountain ranges in the diversification of birds. Annu. Rev. Ecol. Evol. Syst. 43, 249–256 (2012).
Xue, L. F., Wang, C. L. & Shen, S. H. The fine distribution of climate resources and zoning study of Nanling in northern Guangdong province. Chin. J. Agrometeorol. 32, 178–183 (2011).
Holt, B. G. et al. An update of Wallace’s zoogeographic regions of the world. Science 339, 74 (2013).
Wang, J., Gao, P. X., Kang, M., Lowe, A. J. & Huang, H. Refugia within refugia: the case study of a canopy tree (Eurycorymbus cavaleriei) in subtropical China. J. Biogeogr. 36, 2156–2164 (2009).
Yang, J. Q. et al. Phylogeographical analysis on Squalidus argentatus recapitulates historical landscapes and drainage evolution on the island of Taiwan and mainland China. Int. J. Mol. Sci. 13, 1405–1425 (2012).
Myers, N. et al. Biodiversity hotspots for conservation priorities. Nature 403, 853–858 (2000).
Hall, T. BioEdit version 7.1.7. (2012) Available at: http://www.mbio.ncsu.edu/bioedit/bioedit.html. (Accessed: 25th November 2012).
Phillips, S. J., Anderson, R. P. & Schapire, R. E. Maximum entropy modeling of species geographic distributions. Ecol. Modell. 190, 231–259 (2006).
Rozas, J., Sánchez-DelBarrio, J. C. & Messeguer, X. & Rozas, R. dnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19, 2496–2497 (2003).
Swofford, D. L. PAUP*. Phylogenetic Analysis Using Parsimony (*and other methods). Version 4.0b10 (Sinauer Associates, Sunderland, MA, 2002).
Huson, D. H. & Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267 (2006).
Bandelt, H. J., Forster, P. & Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 16, 37–48 (1999).
Dupanloup, I., Schneider, S. & Excoffier, L. A simulated annealing approach to define the genetic structure of populations. Mol. Ecol. 11, 2571–2581 (2002).
Manni, F., Guérard, E. & Heyer, E. Geographic patterns of (genetic, morphologic, linguistic) variation: how barriers can be detected by “Monmonier’s algorithm”. Hum. Biol. 76, 173–190 (2004).
Excoffier, L. & Lischer, H. E. L. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567 (2010).
Jensen, J. L., Bohonak, A. J. & Kelley, S. T. Isolation by distance, web service. BMC Genet. 6, 13 (2005).
Drummond, A. J. & Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214 (2007).
Posada, D. & Crandall, K. A. Modeltest: testing the model of DNA substitution. Bioinformatics 14, 817–818 (1998).
Damgaard, J. & Zettel, H. Genetic diversity, species phylogeny and historical biogeography of the Aquarius paludum group (Heteroptera: Gerridae). Insect Syst. Evol. 34, 313–328 (2003).
Xiao, T. G., Hu, S. H. & Chen, C. M. Studys on Microvelia horvathi Lundblad (Hemiptera: Veliidae): I. morphology and bionomics. J. Hunan Agr. Univ. 2, 49–56 (1987).
Hey, J. Isolation with migration models for more than two populations. Mol. Biol. Evol. 27, 905–920 (2010).
Struwe, L., Smouse, P. R., Heiberg, E., Haag, S. & Lathrop, R. G. Spatial evolutionary and ecological vicariance analysis (SEEVA), a novel approach to biogeography and speciation research, with an example from Brazilian Gentianaceae. J. Biogeogr. 38, 1841–1854 (2011).
Broennimann, O. et al. Measuring ecological niche overlap from occurrence and spatial environmental data. Global Ecol. Biogeogr. 21, 481–497 (2012).
Warren, D. L., Glor, R. E. & Turelli, M. Environmental niche equivalency versus conservatism: quantitative approaches to niche evolution. Evolution 62, 2868–2883 (2008).
Acknowledgements
Funding for this research was provided by the National Natural Science Foundation of China (Nos 31430079, J1210005, 31501853), Specialized Research Fund for the Doctoral Program of Higher Education of China (No. 20100031110026) and Postdoctoral Science Foundation of China (2015M570225).
Author information
Authors and Affiliations
Contributions
Z.Y. analyzed the molecular data and performed the research; G.-P.Z. contributed to ecological niche modeling analysis; J.D. and X.C. participated in discussions and P.-P.C. and W.-J.B. designed the research. The manuscript was written by Z.Y. and W.-J.B.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Ye, Z., Zhu, G., Damgaard, J. et al. Phylogeography of a semi-aquatic bug, Microvelia horvathi (Hemiptera: Veliidae): an evaluation of historical, geographical and ecological factors. Sci Rep 6, 21932 (2016). https://doi.org/10.1038/srep21932
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep21932
This article is cited by
-
Behavioral patterns that determine the mating rates in a wing dimorphic riffle bug, Microvelia horvathi Lundblad, 1933 (Hemiptera: Heteroptera: Veliidae)
Journal of Ethology (2022)
-
Phylogeography and population genetic structure of red muntjacs: evidence of enigmatic Himalayan red muntjac from India
BMC Ecology and Evolution (2021)
-
Bridgehead effect and multiple introductions shape the global invasion history of a termite
Communications Biology (2021)
-
Genetic divergence, population differentiation and phylogeography of the cicada Subpsaltria yangi based on molecular and acoustic data: an example of the early stage of speciation?
BMC Evolutionary Biology (2019)
-
Evolutionary history of a beautiful damselfly, Matrona basilaris, revealed by phylogeographic analyses: the first study of an odonate species in mainland China
Heredity (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
